Características genómicas de los cánceres infantiles (PDQ®) : Tratamiento - información para profesionales de salud [NCI]
Leucemias
Leucemia linfoblástica aguda
Características genómicas de la leucemia linfoblástica aguda infantil
Se han investigado a fondo las características genómicas de la leucemia linfoblástica aguda (LLA) infantil y se definieron múltiples subtipos diferenciados a partir de la caracterización citogenética y molecular; cada subtipo con su propio perfil de características clínicas y pronósticas.[
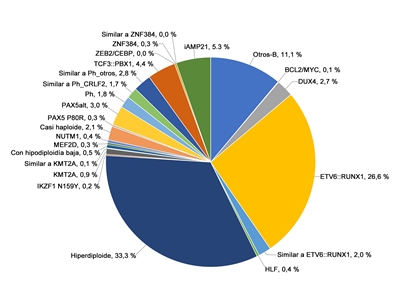
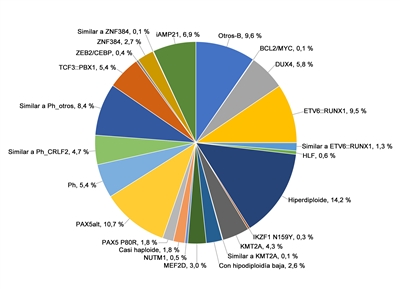
En esta sección, los porcentajes de los subtipos genómicos entre todos los casos de LLA-B y LLA-T provienen, sobre todo, de un informe que describe la caracterización genómica de pacientes tratados en varios ensayos clínicos del Children's Oncology Group (COG) y el St. Jude Children's Research Hospital (SJCRH). Se presentan los porcentajes por subtipo para los pacientes con LLA-B de riesgo estándar y riesgo alto según el NCI (hasta los 18 años).[
Características citogenéticas y genómicas de la leucemia linfoblástica aguda de células B
La LLA-B se clasifica de acuerdo a las siguientes alteraciones genómicas: 1) fusiones génicas que producen factores de transcripción con actividad anómala, 2) ganancias y pérdidas cromosómicas (por ejemplo, hiperdiploidía o hipodiploidía) y 3) alteraciones que producen la activación de genes de tirosina–cinasas.[
El panorama genómico de la LLA-B se caracteriza por una serie de alteraciones genómicas que interrumpen el desarrollo normal de las células B y, en algunos casos, por variantes de genes que proporcionan una señal de proliferación (por ejemplo, variantes activadoras de los genes de la familia RAS o variantes y translocaciones que producen señalización mediante una vía de cinasa). Las alteraciones genómicas que interrumpen el desarrollo de las células B son, entre otras, translocaciones (por ejemplo, las fusiones TCF3::PBX1 y ETV6::RUNX1), variantes de un solo nucleótido (por ejemplo, de IKZF1 y PAX5), y deleciones intragénicas o intergénicas (por ejemplo, de IKZF1, PAX5, EBF y ERG).[
Las alteraciones genómicas de la LLA-B no suelen ocurrir al azar, más bien se agrupan en los subtipos demarcados por sus características biológicas y perfiles de expresión génica. Los casos con translocaciones cromosómicas recurrentes (por ejemplo, las fusiones TCF3::PBX1 y ETV6::RUNX1, y la LLA con reordenamiento de KMT2A) exhiben características biológicas distintivas que ilustran este punto, al igual que los siguientes ejemplos de alteraciones genómicas específicas dentro de subtipos biológicos únicos:
- Las deleciones y variantes de IKZF1 se observan con mayor frecuencia en los casos de LLA con BCR::ABL1 y LLA similar a BCR::ABL1.[
4 ,5 ] - Las deleciones intragénicas de ERG se presentan en un subtipo diferenciado caracterizado por reordenamientos del gen DUX4.[
6 ,7 ] - Las variantes de TP53, a menudo de la línea germinal, son muy frecuentes en pacientes de LLA con hipodiploidía baja de 32 a 39 cromosomas.[
8 ] Las variantes de TP53 son infrecuentes en otros pacientes de LLA-B.
Las variantes de un solo nucleótido activadoras de genes de cinasas son infrecuentes en la LLA-B de riesgo alto. Los genes de las cinasas JAK son los genes de cinasas que se encuentran alterados con mayor frecuencia. Por lo general, estas variantes se observan en los pacientes con LLA similar a BCR::ABL1 que tienen anomalías en CRLF2, aunque también se observan variantes de JAK2 en cerca del 25 % de los niños con síndrome de Down y LLA, que se presenta solo en casos con reordenamientos génicos de CRLF2.[
La comprensión de las características genómicas de la LLA-B en el momento de la recaída está menos avanzada que la comprensión de las características genómicas de la LLA en el momento del diagnóstico. A menudo, la LLA infantil es policlonal en el momento del diagnóstico y, por influencia selectiva del tratamiento, algunos clones se extinguen mientras que surgen clones nuevos con perfiles genómicos diferenciados.[
Se ha observado que varias anomalías cromosómicas recurrentes tienen importancia pronóstica; en especial, para la LLA-B. Algunas anomalías cromosómicas se relacionan con desenlaces más favorables, como las trisomías de pronóstico favorable (51–65 cromosomas) y la fusión ETV6::RUNX1.[
En reconocimiento de la importancia clínica de muchas de estas alteraciones genómicas, en la quinta edición de la revisión de la Classification of Haematolymphoid Tumours se enumeran las siguientes entidades para la LLA-B:[
- Leucemia o linfoma linfoblástico-B, sin otra indicación (SAI).
- Leucemia o linfoma linfoblástico-B con hiperdiploidía alta.
- Leucemia o linfoma linfoblástico-B con hipodiploidía.
- Leucemia o linfoma linfoblástico-B con iAMP21.
- Leucemia o linfoma linfoblástico-B con la fusión BCR::ABL1.
- Leucemia o linfoma linfoblástico-B con características similares a BCR::ABL1.
- Leucemia o linfoma linfoblástico-B con reordenamientos en KMT2A.
- Leucemia o linfoma linfoblástico-B con la fusión ETV6::RUNX1.
- Leucemia o linfoma linfoblástico-B con características similares a ETV6::RUNX1.
- Leucemia o linfoma linfoblástico-B con la fusión TCF3::PBX1.
- Leucemia o linfoma linfoblástico-B con la fusión IGH::IL3.
- Leucemia o linfoma linfoblástico-B con la fusión TCF3::HLF.
- Leucemia o linfoma linfoblástico-B con otras anomalías genéticas definidas.
La categoría de LLA-B con otras anomalías genéticas definidas incluye posibles entidades nuevas, como LLA-B con reordenamientos en DUX4, MEF2D, ZNF384 o NUTM1; LLA-B con fusiones IG::MYC; y LLA-B con anomalías en PAX5alt o PAX5 p.P80R (NP_057953.1).
Estas y otras anomalías cromosómicas y genómicas de la LLA infantil se describen a continuación.
- Número de cromosomas.
- Hiperdiploidía alta (51–65 cromosomas).
En los pacientes pediátricos con LLA-B, la hiperdiploidía alta (presencia de 51 a 65 cromosomas por célula o un índice de DNA superior a 1,16), se presenta en alrededor del 33 % de los casos de riesgo estándar del NCI y del 14 % de los casos de riesgo alto del NCI.[
1 ,20 ] Es posible evaluar la hiperdiploidía por la medición del contenido de DNA en las células (índice de DNA) o por cariotipado. En los casos que exhiben un cariotipo normal o en los que el análisis citogenético estándar fue insatisfactorio, la hibridación fluorescente in situ (FISH) de interfase a veces permite detectar una hiperdiploidía oculta.La hiperdiploidía alta por lo general se presenta en los casos con factores clínicos de pronóstico favorable (pacientes de 1 a <10 años con recuento de glóbulos blancos [GB] bajo) y es, por sí sola, un factor independiente de pronóstico favorable.[
20 ,21 ,22 ] En un estudio, dentro del intervalo hiperdiploide de 51 a 65 cromosomas, los pacientes con números modales más altos (58–66), presentaron el mejor pronóstico.[22 ] Las células leucémicas hiperdiploides son especialmente susceptibles a la apoptosis y acumulan concentraciones más altas de metotrexato y sus metabolitos activos de poliglutamato,[23 ] lo que quizás explique el desenlace favorable que se observa con frecuencia en estos casos.Aunque el desenlace general de los pacientes con hiperdiploidía alta se considera favorable, se ha observado que factores como la edad, el recuento de GB, las trisomías específicas y la respuesta temprana al tratamiento modifican su importancia pronóstica.[
24 ,25 ,26 ]La importancia pronóstica de las trisomías cromosómicas específicas entre los niños con LLA-B hiperdiploide se ha descrito en múltiples informes.
- En un estudio en el que se combinó la experiencia del Children's Cancer Group y del Pediatric Oncology Group (POG) se observó que los pacientes con trisomías de los cromosomas 4, 10 y 17 (trisomías triples) tienen un desenlace particularmente favorable.[
27 ]; [17 ][Nivel de evidencia B4] - En un informe en el que se usaron los datos del POG se encontró que los pacientes de riesgo estándar del NCI que exhiben trisomías 4 y 10 tienen un pronóstico excelente, independientemente del estado del cromosoma 17.[
28 ] En la actualidad, los protocolos del COG usan trisomías dobles de los cromosomas 4 y 10 para definir la hiperdiploidía favorable. - En un análisis retrospectivo se evaluó a los pacientes tratados en 2 ensayos UKALL consecutivos con el fin de identificar y validar un perfil para predecir el desenlace de la LLA-B con hiperdiploidía alta. Los investigadores definieron un grupo de riesgo bajo (alrededor del 80 % de los pacientes con hiperdiploidía alta) que se relacionó con un pronóstico más favorable. Los pacientes de riesgo bajo tenían trisomías de los cromosomas 17 y 18 o trisomía de uno de estos cromosomas con ausencia de trisomías de los cromosomas 5 y 20. Los demás pacientes se definieron como de riesgo alto y tuvieron un desenlace menos favorable. La ERM al final de la inducción y las alteraciones del número de copias (como la deleción de IKZF1) fueron significativas para el pronóstico dentro de cada grupo de riesgo hiperdiploide.[
29 ]
Es posible que se encuentren translocaciones cromosómicas en combinación con hiperdiploidía alta; en estos casos, la clasificación de riesgo más apropiada para los pacientes se basa en la importancia pronóstica de la translocación. Por ejemplo, en un estudio, el 8 % de los pacientes con la fusión BCR::ABL1 también presentaban hiperdiploidía alta,[
30 ] y el desenlace de estos pacientes (tratados sin inhibidores de tirosina–cinasas) fue inferior al que se observó en los pacientes con hiperdiploidía alta negativos para BCR::ABL1.Algunos pacientes con LLA hiperdiploide tienen un clon hipodiploide que se ha duplicado (hipodiploidía oculta).[
31 ] Las tecnologías moleculares, como las micromatrices de polimorfismos de un solo nucleótido que se usan para detectar la pérdida de heterocigosidad generalizada, se usan para identificar pacientes con hipodiploidía oculta.[31 ] Estos casos se pueden interpretar de acuerdo con el perfil de ganancias y pérdidas de cromosomas específicos (hiperdiploidía con 2 o 4 copias de cromosomas en lugar de 3 copias). Estos pacientes tienen un desenlace desfavorable, similar al de aquellos con hipodiploidía.[32 ]La casi triploidía (68–80 cromosomas) y la casi tetraploidía (>80 cromosomas) son mucho menos comunes y son biológicamente diferentes de la hiperdiploidía alta.[
33 ] A diferencia de la hiperdiploidía alta, una gran proporción de casos con casi tetraploidía albergan una fusión ETV6::RUNX1 críptica.[33 ,34 ,35 ] Antes se consideraba que la casi triploidía y tetraploidía estaban relacionadas con un pronóstico desfavorable, pero en estudios posteriores se indicó que es posible que no sea el caso.[33 ,35 ]El panorama genómico de la LLA hiperdiploide se caracteriza por variantes de los genes de la vía de receptores de tirosina–cinasas (RTK)/RAS en alrededor de la mitad de los casos. Los genes que codifican modificadores de histonas también se presentan de manera recurrente en una minoría de los casos. En el análisis de los perfiles de variantes se observa que las ganancias cromosómicas son episodios iniciales en la patogénesis de la LLA hiperdiploide y quizás se presente in utero, mientras que las variantes de los genes de la vía RTK/RAS son eventos tardíos en la leucemogénesis y por lo general son subclonales.[
1 ,36 ] - En un estudio en el que se combinó la experiencia del Children's Cancer Group y del Pediatric Oncology Group (POG) se observó que los pacientes con trisomías de los cromosomas 4, 10 y 17 (trisomías triples) tienen un desenlace particularmente favorable.[
- Hipodiploidía (<44 cromosomas).
Los casos de LLA-B con un número de cromosomas menor que lo normal se subdividen de varias formas; en un informe se estratifican a partir del número modal de cromosomas en los siguientes cuatro grupos:[
32 ]- Casi haploide: 24 a 29 cromosomas (n = 46).
- Hipodiploidía baja: 33 a 39 cromosomas (n = 26).
- Hipodiploidía alta: 40 a 43 cromosomas (n = 13).
- Casi diploide: 44 cromosomas (n = 54).
En los pacientes pediátricos con LLA-B, los casos casi haploides representan alrededor del 2 % de los casos de riesgo estándar del NCI y del 2 % de los casos de riesgo alto del NCI.[
1 ]En los pacientes pediátricos con LLA-B, los casos de hipodiploidía baja representan alrededor del 0,5 % de los casos de riesgo estándar del NCI y del 2,6 % de los casos de riesgo alto del NCI.[
1 ]La mayoría de pacientes con hipodiploidía se ubican en el grupo casi haploide o en el grupo de hipodiploidía baja; ambos grupos tienen un riesgo elevado de fracaso del tratamiento en comparación con los casos sin hipodiploidía.[
32 ,37 ] Los pacientes con menos de 44 cromosomas en sus células leucémicas tienen un desenlace más precario que aquellos con 44 a 45 cromosomas.[32 ] En varios estudios se observó que los pacientes con enfermedad residual mínima (ERM) alta (≥0,01 %) después de la inducción evolucionan mal, con tasas de supervivencia sin complicaciones (SSC) a 5 años que oscilan entre el 25 % y el 47 %. Aunque la evolución es mejor para los pacientes con hipodiploidía que presentan una ERM baja después de la inducción (tasas de SSC a 5 años, 64–75 %), sus desenlaces siguen siendo inferiores a los de la mayoría de niños con otros tipos de LLA.[38 ,39 ,40 ]Las alteraciones genómicas recurrentes de la LLA casi haploide y con hipodiploidía baja son diferentes entre sí y de las de otros tipos de LLA.[
8 ] En la LLA casi haploide, son comunes las alteraciones que afectan la señalización RTK, la señalización RAS y el gen IKZF3.[41 ] En la LLA con hipodiploidía baja, son comunes las alteraciones genéticas que afectan los genes TP53, RB1 y IKZF2. Es importante destacar que las alteraciones de TP53, que se observan en la LLA con hipodiploidía baja, también están presentes en las células no tumorales en alrededor del 40 % de los casos; esto indica que estas variantes son de la línea germinal y que la LLA con hipodiploidía baja representa, en algunos casos, una manifestación del síndrome de Li-Fraumeni.[8 ] Cerca de dos tercios de los pacientes de LLA con variantes patógenas de la línea germinal en TP53 tienen LLA hipodiploide.[42 ]
- Hiperdiploidía alta (51–65 cromosomas).
- Translocaciones cromosómicas y ganancias o deleciones de segmentos cromosómicos.
- Fusión ETV6::RUNX1 (t(12;21)(p13.2;q22.1)).
En los pacientes pediátricos con LLA-B, la fusión del gen ETV6 en el cromosoma 12 con el gen RUNX1 en el cromosoma 21 se presenta en alrededor del 27 % de los casos de riesgo estándar del NCI y del 10 % de los casos de riesgo alto del NCI.[
1 ,34 ]La fusión ETV6::RUNX1 produce una translocación críptica que se detecta por métodos como la FISH, pero no por las pruebas citogenéticas convencionales; y se presenta de manera más frecuente en niños de 2 a 9 años.[
43 ,44 ] Los niños hispanos con LLA tienen una incidencia más baja de fusiones ETV6::RUNX1 que los niños blancos.[45 ]Por lo general, en los informes se indican tasas de SSC y supervivencia general (SG) favorables para los niños con la fusión ETV6::RUNX1; sin embargo, los siguientes factores modifican la repercusión pronóstica de esta característica genética:[
26 ,46 ,47 ,48 ,49 ,50 ]; [17 ][Nivel de evidencia B4]- Respuesta temprana al tratamiento.
- Categoría de riesgo según el NCI (edad y recuento de GB en el momento del diagnóstico).
- Régimen de tratamiento.
En un estudio sobre el tratamiento de niños con diagnóstico nuevo de LLA, el análisis multivariante de los factores pronósticos indicó que la edad y el recuento leucocitario fueron factores pronósticos independientes, pero no el estado de la fusión ETV6::RUNX1.[
46 ] Sin embargo, en otro ensayo numeroso solo se inscribieron pacientes con LLA-B clasificada como de riesgo bajo, con características clínicas de riesgo bajo como trisomías de 4, 10 y 17 o fusión ETV6::RUNX1, y menos del 0,01 % de ERM al final de la inducción. Los pacientes tuvieron una tasa de remisión completa continua a 5 años del 93,7 % y una tasa de SG a 6 años del 98,2 % para los pacientes con ETV6::RUNX1.[17 ] La presencia de anomalías citogenéticas secundarias, como la deleción de ETV6 (12p) o CDKN2A/B (9p), no parece afectar el desenlace de los pacientes con la fusión ETV6::RUNX1.[50 ,51 ]Las recaídas tardías son más frecuentes en los pacientes con fusiones ETV6::RUNX1 en comparación con otros casos de LLA-B recidivante.[
46 ,52 ] Los pacientes que exhiben la fusión ETV6::RUNX1 y recaen tienen un pronóstico un poco mejor que otros pacientes en recaída,[53 ] el pronóstico es en particular favorable para los pacientes que recaen después de 36 meses del diagnóstico.[54 ] Algunas recaídas en pacientes con fusiones ETV6::RUNX1 quizás indiquen la presencia de una lesión secundaria independiente en un clon preleucémico persistente (la lesión inicial sería la translocación ETV6::RUNX1).[55 ,56 ] - Fusión BCR::ABL1 (t(9;22)(q34.1;q11.2); Ph+).
La fusión BCR::ABL1 conduce a la producción de una proteína de fusión BCR::ABL1 con actividad de tirosina–cinasas (consultar la Figura ).[
1 ] En los pacientes pediátricos con LLA-B, la fusión BCR::ABL1 se produce en alrededor del 2 % de los casos de riesgo estándar del NCI y del 5 % de los casos de riesgo alto del NCI.[1 ] La fusión BCR::ABL1 también es un iniciador leucemógeno de la leucemia mieloide crónica (LMC). El punto de ruptura de BCR más común en la LMC es diferente del punto de ruptura de BCR más común en la LLA. Los puntos de ruptura típicos de la LMC producen una proteína de fusión más grande (llamada p210) que la proteína que producen los puntos de ruptura que se observan con mayor frecuencia en la LLA (llamada p190, una proteína de fusión más pequeña).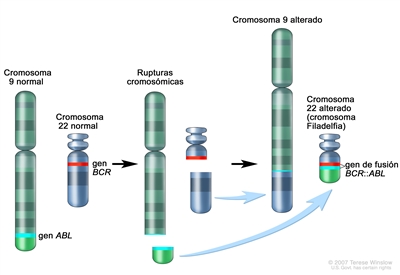
La LLA Ph+ es más común en niños de más edad con LLA-B y recuentos de GB altos; la incidencia de las fusiones BCR::ABL1 aumenta a cerca del 25 % en adultos jóvenes con LLA.
Tradicionalmente, la fusión BCR::ABL1 se relacionó con un pronóstico muy adverso (en especial, en aquellos con un recuento de GB alto en el momento del diagnóstico, o con una respuesta temprana lenta al tratamiento inicial) y su presencia se ha considerado una indicación para el trasplante de células madres hematopoyéticas (TCMH) alogénico durante la primera remisión.[
30 ,57 ,58 ,59 ] Los inhibidores de tirosina–cinasas de BCR::ABL1, como el mesilato de imatinib, son eficaces en los pacientes con LLA con fusión BCR::ABL1.[60 ] En un estudio del Children's Oncology Group (COG), en el que se administró quimioterapia intensiva y mesilato de imatinib simultáneo cada día, se observó una tasa de SSC a 5 años del 70 % (± 12 %), que fue superior a la tasa de SSC de los controles históricos de la era anterior a los inhibidores de tirosina–cinasas (mesilato de imatinib). Este resultado eliminó la recomendación de TCMH para pacientes con una buena respuesta temprana a la quimioterapia con inhibidores de tirosina–cinasas.[61 ,62 ]La International Consensus Classification de la leucemia o linfoma linfoblásticos de 2022 divide la LLA-B con fusión BCR::ABL1 en dos subtipos: casos con compromiso linfoide solo y casos con compromiso de varios linajes.[
63 ] Estos subtipos difieren en el momento del evento de transformación. Una célula progenitora multipotente es la célula de origen de la LLA-B con fusión BCR::ABL1 y compromiso de varios linajes, y una célula progenitora en estadio posterior es la célula de origen de la LLA-B con fusión BCR::ABL1 que solo compromete el linaje linfoide.- La LLA-B con fusión BCR::ABL1 y compromiso linfoide solo es el subtipo predominante. Solamente una minoría de los casos en niños y adultos exhibe compromiso de varios linajes (estimado, 15 %–30 %).[
64 ] - Los casos de LLA-B con fusión BCR::ABL1 y compromiso linfoide solo son similares a los casos con compromiso de varios linajes en aspectos como cuadro clínico inicial e inmunofenotipo. Además, ambos subtipos suelen exhibir la proteína de fusión p190.[
64 ,65 ] - Una manera de diferenciar los pacientes con compromiso linfoide solo de los pacientes con compromiso de varios linajes es mediante la detección de la fusión BCR::ABL1 en células B, T y mieloides no afectadas por LLA.[
65 ] - Otra forma de diferenciar entre los pacientes con compromiso linfoide solo o compromiso de varios linajes es mediante la detección de diferencias cuantitativas en las concentraciones de ERM (por lo general, 1 log) con métodos que cuantifican DNA o RNA con la fusión BCR::ABL1 y métodos que se basan en citometría de flujo, reacción en cadena de la polimerasa (PCR) cuantitativa en tiempo real o secuenciación de última generación (NGS) para la medición de los reordenamientos de inmunoglobulina (IG) o del receptor de células T (TCR) específicos de la leucemia.[
64 ,65 ,66 ]- En pacientes de LLA-B con fusión BCR::ABL1 y compromiso linfoide solo, los cálculos de ERM mediante estos métodos se correlacionan entre sí.
- En pacientes de LLA-B con fusión BCR::ABL1 y compromiso de varios linajes, los cálculos de ERM después del tratamiento que se basan en la detección de DNA o RNA con la fusión BCR::ABL1 a menudo darán resultados más altos que la citometría de flujo o la cuantificación de los reordenamientos de IG o TCR específicos de la leucemia.
- En los pacientes de LLA-B con fusión BCR::ABL1 y compromiso de varios linajes, las concentraciones de trascriptos de BCR::ABL1 y DNA a veces permanecen estables en el tiempo a pesar de continuar el tratamiento con quimioterapia e inhibidores de tirosina–cinasas. En estas circunstancias, la persistencia de DNA o RNA con la fusión BCR::ABL1 posiblemente indique la presencia de un clon preleucémico, no de células leucémicas. Por lo tanto, el término ERM es inapropiado.
- Una conclusión sobre la diferencia en la detección de la ERM mediante métodos basados en medición del DNA o RNA que exhibe la fusión BCR::ABL1 versus la detección de la ERM por métodos de citometría de flujo o reordenamientos de IG o TCR es que ésta última opción es el método que produce el pronóstico más confiable.[
64 ,66 ,67 ] Por ejemplo, la presencia de ERM medida por detección de DNA o RNA con la fusión BCR::ABL1 en ausencia de detección de ERM por reordenamientos de IG o TCR no confiere un pronóstico más precario. - A partir del número escaso de pacientes que se han estudiado hasta el momento, el pronóstico es semejante en adultos y niños con LLA-B con fusión de BCR::ABL1 y compromiso linfoide solo o compromiso de varios linajes.[
64 ,66 ] - Hay informes de casos de pacientes con LLA-B con fusión BCR::ABL1 y compromiso de varios linajes que recaen años después del diagnóstico inicial. Además, su recaída presenta el mismo punto de ruptura de la fusión BCR::ABL1 que la leucemia inicial, pero presentan un reordenamiento diferente de IG o TCR.[
66 ] Estos informes de casos indican que los pacientes de LLA-B con fusión BCR::ABL1 y compromiso de varios linajes están en riesgo de un segundo evento leucemógeno que conlleva a otra leucemia con fusión BCR::ABL1. - No hay evidencia de que un plan de vigilancia específico o el tratamiento prolongado con un inhibidor de tirosina–cinasas produzca un beneficio clínico en pacientes de LLA-B con fusión BCR::ABL1 y compromiso de varios linajes que mantienen la expresión de trascriptos BCR::ABL1 o DNA con esta fusión en el momento de terminar el tratamiento estándar de la leucemia.
- La LLA-B con fusión BCR::ABL1 y compromiso linfoide solo es el subtipo predominante. Solamente una minoría de los casos en niños y adultos exhibe compromiso de varios linajes (estimado, 15 %–30 %).[
- LLA con reordenamiento de KMT2A (t(v;11q23.3)).
Los reordenamientos que afectan el gen KMT2A con más de 100 genes compañeros de translocación producen oncoproteínas de fusión. Los reordenamientos en el gen KMT2A se presentan hasta en el 80 % de los lactantes con LLA. En los pacientes pediátricos de más de un año de edad con LLA-B, cerca del 1 % de los casos de alto riesgo estándar del NCI y del 4 % de los casos de riesgo alto del NCI tienen reordenamientos de KMT2A.[
1 ]Estos reordenamientos se suelen relacionar con aumento del riesgo de fracaso del tratamiento, sobre todo en lactantes.[
68 ,69 ,70 ,71 ] En los niños con LLA, la fusión KMT2A::AFF1 (t(4;11)(q21;q23)) es el reordenamiento más común que afecta el gen KMT2A y se presenta en el 1 % al 2 % de los casos.[69 ,72 ]Los pacientes con fusiones KMT2A::AFF1 por lo general son lactantes con recuentos de GB altos. Estos pacientes son más propensos que otros niños con LLA a presentar enfermedad en el sistema nervioso central (SNC) y una respuesta precaria al tratamiento inicial.[
73 ] Si bien, tanto los lactantes como los adultos con la fusión KMT2A::AFF1 tienen un riesgo alto de fracaso del tratamiento, los niños con esta fusión tienen mejores desenlaces.[68 ,69 ,74 ] Con independencia del tipo de reordenamiento del gen KMT2A, los lactantes con LLA que exhibe reordenamientos del gen KMT2A presentan tasas de supervivencia sin complicaciones mucho más desfavorables que los pacientes pediátricos de más de 1 año con LLA que exhibe reordenamientos de KMT2A.[68 ,69 ,74 ]Mediante secuenciación de genoma completo se determinó que los casos de LLA infantil con reordenamientos del gen KMT2A tienen variantes subclonales frecuentes de NRAS o KRAS y pocas anomalías genómicas adicionales, ninguna de importancia clínica bien definida.[
12 ,75 ] La deleción del gen KMT2A no se ha relacionado con un pronóstico adverso.[76 ]Como dato interesante, la fusión KMT2A::MLLT1 (t(11;19)(q23;p13.3)) se presenta en alrededor del 1 % de los casos de LLA, tanto en la LLA de linaje B temprano como en la LLA-T.[
77 ] El desenlace de los lactantes con la fusión KMT2A::MLLT1 es precario, pero es relativamente favorable en los niños de más edad con LLA-T que presentan esta fusión.[77 ] - Fusión TCF3::PBX1 (t(1;19)(q23;p13.3)) y fusión TCF3::HLF (t(17;19)(q22;p13)).
En los pacientes pediátricos con LLA-B, la fusión del gen TCF3 en el cromosoma 19 con el gen PBX1 en el cromosoma 1 se presenta en alrededor del 4 % de los casos de riesgo estándar del NCI y del 5 % de los casos de riesgo alto del NCI.[
1 ,78 ,79 ] La fusión TCF3::PBX1 se presenta como una translocación equilibrada o desequilibrada, y es la principal anomalía genómica recurrente del inmunofenotipo de LLA pre-B (positiva para inmunoglobulina citoplasmática).[80 ] Los niños negros son más propensos a presentar LLA pre-B con la fusión TCF3::PBX1 que los niños blancos.[81 ]La fusión TCF3::PBX1 se relacionó con un desenlace inferior en el contexto de un tratamiento a base de antimetabolitos,[
82 ] pero la importancia para determinar un pronóstico adverso se invalidó casi por completo cuando se usó un tratamiento multifarmacológico más intensivo.[79 ,83 ] En particular, en un ensayo realizado por el St. Jude Children's Research Hospital (SJCRH) en el que todos los pacientes se trataron sin radiación craneal, aquellos con la fusión TCF3::PBX1 tuvieron un desenlace general comparable al de los niños sin esta translocación, pero presentaron un riesgo más alto de recaída en el SNC y una tasa más baja de recaída en la médula ósea; esto indica que quizás estos pacientes necesiten un tratamiento dirigido al SNC más intensivo.[84 ,85 ]La fusión TCF3::HLF se presenta en menos del 1 % de los casos de LLA infantil. La LLA con la fusión TCF3::HLF se relaciona con coagulación intravascular diseminada e hipercalcemia en el momento del diagnóstico. El desenlace es muy precario en niños con la fusión TCF3::HLF: en una revisión de la literatura se indicó una mortalidad de 20 de 21 casos notificados.[
86 ] Además de la fusión TCF3::HLF, el panorama genómico de este subtipo de LLA se caracterizó por deleciones en genes que participan en el desarrollo de las células B (PAX5, BTG1 y VPREB1) y por variantes de los genes de la vía RAS (NRAS, KRAS y PTPN11).[80 ] - LLA con reordenamiento de DUX4 y deleciones de ERG frecuentes.
En los pacientes pediátricos con LLA-B, casi el 3 % de los casos de riesgo estándar del NCI y del 6 % de los casos de riesgo alto del NCI tienen un reordenamiento que afecta el gen DUX4 y conduce a su sobreexpresión.[
1 ,6 ,7 ] Tener ascendencia de Asia oriental se relacionó con un aumento de la prevalencia de LLA con reordenamiento de DUX4 (favorable).[87 ] El reordenamiento más común produce fusiones IGH::DUX4; y también se observan fusiones ERG::DUX4.[88 ] Los casos con reordenamiento de DUX4 exhiben un perfil de expresión génica característico que al principio se identificó como asociado con deleciones focales en ERG,[88 ,89 ,90 ,91 ] y entre la mitad y más de dos tercios de estos casos tienen deleciones intragénicas focales que afectan el gen ERG pero que no se encuentran en otros subtipos de LLA.[6 ,88 ] Las deleciones de ERG a menudo parecen ser clonales, pero al usar métodos de detección más sensibles, se observa que la mayoría de los casos son policlonales.[88 ] Se observan alteraciones de IKZF1 en el 20 % al 40 % de los casos de LLA con reordenamiento de DUX4.[6 ,7 ]La deleción de ERG conlleva un pronóstico excelente, con tasas de SG superiores al 90 %. El pronóstico sigue siendo favorable incluso cuando hay una deleción de IZKF1.[
89 ,90 ,91 ,92 ] Si bien los pacientes de LLA con reordenamiento de DUX4 tienen un pronóstico general favorable, no se sabe si esto es cierto en los casos con deleción de ERG y en los casos con ERG intacto. En un estudio de 50 pacientes de LLA con reordenamiento de DUX4, los pacientes con deleción de ERG detectada mediante PCR (n = 33) presentaron una tasa de SSC más favorable, de alrededor del 90 %, que los pacientes con ERG intacto (n = 17), con una tasa de SSC de alrededor del 70 %.[90 ] - LLA con reordenamiento de MEF2D.
En los pacientes pediátricos con LLA-B, las fusiones génicas que afectan a MEF2D, un factor de transcripción que se expresa durante el desarrollo de las células B, se observan en alrededor del 0,3 % de los casos de riesgo estándar del NCI y del 3 % de los casos de riesgo alto del NCI.[
1 ,93 ,94 ]Aunque hay múltiples compañeros de fusión, la mayoría de los casos afectan el gen BCL9, en el cromosoma 1q21, al igual que el gen MEF2D.[
93 ,95 ] La deleción intersticial que produce la fusión MEF2D::BCL9 es muy pequeña como para ser detectada por métodos citogenéticos convencionales. Los casos con fusiones del gen MEF2D exhiben un perfil de expresión génica característico, excepto por casos infrecuentes que tienen MEF2D::CSFR1 y un perfil de expresión génica similar a BCR::ABL1.[93 ,96 ]La mediana de edad en el momento del diagnóstico para los casos de LLA con reordenamiento de MEF2D fue de 12 a 14 años en los estudios que incluyeron pacientes adultos y niños.[
93 ,94 ] En los 22 niños de LLA con reordenamiento de MEF2D inscritos en un ensayo clínico de LLA de riesgo alto, la tasa de SSC a 5 años fue del 72 % (error estándar, ±10 %), que fue inferior a la de otros pacientes.[93 ] - LLA con reordenamiento de ZNF384.
En los pacientes pediátricos con LLA-B, los reordenamientos en el gen ZNF384, que codifica un factor de transcripción, se presentan en cerca del 0,3 % de los casos de riesgo estándar del NCI y del 2,7 % de los casos del riesgo alto del NCI.[
1 ,93 ,97 ,98 ]Tener ascendencia de Asia oriental se relacionó con un aumento en la prevalencia de ZNF384.[
87 ] Se han descrito múltiples compañeros de fusión para ZNF384, entre ellos, ARID1B, CREBBP, EP300, SMARCA2, TAF15 y TCF3. Sin importar el compañero de fusión, los casos de LLA con reordenamiento de ZNF384 exhiben un perfil de expresión génica característico.[93 ,97 ,98 ] El reordenamiento de ZNF384 no parece tener importancia pronóstica independiente.[99 ,97 ,98 ] Sin embargo, dentro del subgrupo de pacientes con reordenamientos de ZNF384, los pacientes con fusiones EP300::ZNF384 tienen tasas de recaídas más bajas que los pacientes con fusiones de ZNF384 en las que intervienen otros compañeros de fusión.[99 ] El inmunofenotipo de LLA-B con reordenamiento de ZNF384 se caracteriza por expresión débil de CD10 o ausencia de expresión de este producto, mientras que es común que exprese CD13 o CD33.[97 ,98 ] Se han notificado casos de leucemia aguda de fenotipo mixto (LAFM) (B/mieloide) que exhiben fusiones génicas de ZNF384, [100 ,101 ] y en la evaluación genómica de la LAFM se encontró que las fusiones génicas de ZNF384 estaban presentes en cerca de la mitad de los casos con fenotipo B/mieloide.[102 ] - LLA-B con reordenamiento de NUTM1.
La LLA-B con reordenamiento de NUTM1 se observa con más frecuencia en lactantes, y representa el 3 % al 5 % de todos los casos de LLA-B en ese grupo etario y alrededor del 20 % de los casos de lactantes con LLA-B sin reordenamiento de KMT2A.[
103 ] La frecuencia del reordenamiento de NUTM1 es más baja en los niños después del primer año de vida (<1% % de casos).[1 ,103 ]El gen NUTM1 se encuentra en el cromosoma 15q14, y algunos casos de LLA-B con reordenamientos de NUTM1 presentan anomalías en el cromosoma 15q, pero otros son casos crípticos y no presentan anomalías citogenéticas.[
104 ] La secuenciación del RNA, así como la técnica FISH con sondas de separación, se usan para detectar la presencia de reordenamiento de NUTM1.[103 ]El reordenamiento de NUTM1 está relacionado con un desenlace favorable.[
103 ,105 ] De 35 lactantes con LLA-B con reordenamiento de NUTM1 que se trataron en los protocolos Interfant, todos los pacientes lograron la remisión y no se observaron recaídas.[103 ] En los 32 niños mayores de 12 meses que presentaban LLA-B con reordenamiento de NUTM1, las tasas de SSC a 4 años y de SG fueron del 92 % y del 100 %, respectivamente. - Fusión IGH::IL3 (t(5;14)(q31.1;q32.3)).
Esta entidad se incluyó en la revisión de 2016 de la clasificación de tumores de tejidos hematopoyéticos y linfoides de la Organización Mundial de la Salud (OMS).[
106 ] El hallazgo de la t(5;14)(q31.1;q32.3) en pacientes con LLA e hipereosinofilia en la década de 1980 fue seguido por la identificación de la fusión IGH::IL3 como la causa genética subyacente de esta afección.[107 ,108 ] La unión del locus de IGH a la región promotora del gen IL3 lleva a la desregulación de la expresión de IL3.[109 ] Las anomalías citogenéticas en niños con LLA y eosinofilia son variables, solo un subgrupo se produce a partir de la fusión IGH::IL3.[110 ]El número de casos de LLA con IGH::IL3 descritos en la bibliografía publicada es demasiado pequeño como para evaluar la importancia pronóstica de la fusión IGH::IL3. El diagnóstico de los casos de LLA con IGH::IL3 quizá se retrase porque el clon de LLA en la médula ósea a veces es pequeño y porque es posible que se presente con hipereosinofilia en ausencia de citopenias y blastocitos circulantes.[
106 ] - Amplificación intracromosómica del cromosoma 21 (iAMP21).
La iAMP21 se presenta en alrededor del 5 % de los casos pediátricos de LLA-B del NCI de riesgo estándar y el 7 % de los casos del NCI de riesgo alto.[
1 ] Por lo general la iAMP21 se diagnostica mediante FISH y se define por la presencia de 5 o más señales de RUNX1 por célula (o ≥3 copias adicionales de RUNX1 en un cromosoma anormal único).[106 ] La iAMP21 también se puede identificar mediante análisis de micromatriz cromosómica. Rara vez, la iAMP21 con un patrón genómico atípico (por ejemplo, amplificación de la región genómica, pero con menos de 5 señales RUNX1 o que tiene al menos 5 señales RUNX1 con algunas separadas del cromosoma iAMP21 anormal) se identifica mediante micromatriz, pero no con FISH de RUNX1.[111 ] No se ha descrito la importancia pronóstica de iAMP21 definida solo mediante micromatriz.La iAMP21 se relaciona con mayor edad (mediana, alrededor 10 años), recuento de GB inferior a 50 × 109 /l, un leve predominio en mujeres y una ERM alta al final de la inducción.[
112 ,113 ,114 ] El análisis de las firmas de variantes indican que las amplificaciones génicas en iAMP21 se presentan más tarde en la leucemogénesis, lo cual contrasta con las de la LLA hiperdiploide que pueden presentarse temprano en la vida e incluso in utero.[1 ]El grupo de ensayos clínicos United Kingdom Acute Lymphoblastic Leukaemia (UKALL), inicialmente notificó que la presencia de iAMP21 confirió un pronóstico precario a los pacientes tratados en el ensayo MRC ALL 97/99 (tasa de SSC a 5 años, 29 %).[
18 ] En el ensayo posterior del mismo grupo (UKALL2003 [NCT00222612]), se asignó a los pacientes con iAMP21 a recibir un régimen de quimioterapia más intensivo, y estos pacientes presentaron un desenlace mucho más favorable (tasa de SSC a 5 años, 78 %).[113 ] De manera similar, el COG informó que la iAMP21 se relacionó con un desenlace significativamente inferior en los pacientes de riesgo estándar del NCI (tasa de SSC a 4 años, 73 % para iAMP21 vs. 92 % en otros), pero no en los pacientes con riesgo alto del NCI (tasa de SSC a 4 años, 73 % vs. 80 %).[112 ] En un análisis multivariante, la iAMP21 fue un factor de pronóstico independiente de desenlace precario solo en los pacientes de riesgo estándar del NCI.[112 ] Los resultados de los estudios UKALL2003 y COG indican que en los pacientes que tienen una iAMP21, el tratamiento con regímenes de quimioterapia de riesgo alto anula la repercusión de iAMP21 como factor de pronóstico adverso y evita la necesidad de un TCMH en el momento de la primera remisión.[114 ] - Alteraciones en PAX5.
En análisis de expresión génica se identificaron dos subtipos de LLA diferenciados con alteraciones genómicas en PAX5, denominadas PAX5alt y PAX5 p.P80R (NP_057953.1).[
115 ] Las alteraciones en el subtipo PAX5alt incluyen reordenamientos, variantes de secuencia y amplificaciones intragénicas focales.PAX5alt. En los pacientes pediátricos con LLA-B, se han notificado reordenamientos de PAX5 en alrededor del 3 % de los casos de riesgo estándar del NCI y del 11 % de los casos de riesgo alto del NCI.[
1 ] Se identificaron más de 20 genes compañeros de PAX5,[115 ]. PAX5::ETV6, la alteración genómica primaria en dic(9;12)(p13;p13),[116 ] fue la fusión génica más común.[115 ]Se identificó una amplificación intragénica de PAX5 en cerca del 1 % de los casos de LLA-B, y por lo general se detectó en casos que no tenían alteraciones genómicas que se sabe son iniciadoras de leucemia.[
117 ] Los casos con amplificación de PAX5 son de predominio masculino (66 %), y la mayoría (55 %) se clasifican en un estado de riesgo alto del NCI. En una cohorte de pacientes con amplificación de PAX5 que recibieron el diagnóstico entre 1993 y 2015, la tasa de SSC a 5 años fue del 49 % (intervalo de confianza [IC] 95 %, 36–61 %), y la tasa de SG fue del 67 % (IC 95 %, 54–77 %), lo que indica un pronóstico relativamente más precario para pacientes con este subtipo de LLA-B.PAX5 p.P80R (NP_057953.1). La leucemia que tiene PAX5 con la variante p.P80R exhibe un perfil de expresión génica diferente al de otros casos con alteraciones en PAX5.[
115 ] En los pacientes pediátricos con LLA-B, los casos con PAX5 p.P80R representan alrededor del 0,3 % de los casos de riesgo estándar del NCI y del 1,8 % de los casos de riesgo alto del NCI.[1 ] La LLA-B con PAX5 p.P80R se presenta con más frecuencia en los adolescentes y adultos jóvenes (AAJ) y en los adultos (3,1 % y 4,2 %, respectivamente).[115 ]Para los pacientes pediátricos con la variante p.P80R en PAX5 y con la variante PAX5alt tratados en los ensayos clínicos del COG, el desenlace es intermedio (SSC a 5 años, alrededor del 75 %).[
115 ] Los reordenamientos de PAX5alt también se detectaron en pacientes lactantes con LLA, y los desenlaces notificados fueron similares a los de los niños de LLA con reordenamiento de KMT2A.[105 ] - Similar a BCR::ABL1 (similar a Ph).
Los pacientes con un resultado negativo para BCR::ABL1 que exhiben un perfil de expresión génica semejante al de los pacientes con resultado positivo para BCR::ABL1 se considera que tienen una LLA similar a Ph,[
118 ,119 ,120 ] que ahora se conoce como similar a BCR::ABL1.[19 ] Esto sucede en el 10 % al 20 % de los pacientes de LLA-B infantil; su frecuencia aumenta con la edad y se ha relacionado con una variante o deleción de IKZF1.[1 ,9 ,118 ,119 ,121 ,122 ]En análisis retrospectivos se indicó que los pacientes con LLA similar a BCR::ABL1 tienen un pronóstico precario.[
5 ,118 ] En una serie, la tasa de SSC a 5 años de los niños y adolescentes de riesgo alto según el NCI con LLA similar a BCR::ABL1 fue del 58 % y el 41 %, respectivamente.[5 ] Si bien el subtipo similar a BCR::ABL1 es más frecuente en pacientes de más edad y de riesgo más alto, también se ha identificado en pacientes de riesgo estándar según el NCI. En un estudio del COG, se encontró que el 13,6 % de 1023 pacientes con LLA-B de riesgo estándar según el NCI tenían una LLA similar a BCR::ABL1; estos pacientes exhibieron una tasa de SSC inferior comparada con los pacientes de riesgo estándar con una LLA de otro tipo no similar a BCR::ABL1 (82 % vs. 91 %), aunque no se indicaron diferencias en la tasa de SG (93 % vs. 96 %).[123 ] En un estudio de 40 pacientes con LLA similar a BCR::ABL1, la importancia pronóstica adversa de este subtipo se anuló cuando los pacientes recibieron tratamiento dirigido según el riesgo a partir de las concentraciones de ERM.[124 ]El sello distintivo de la LLA similar a BCR::ABL1 es la activación de la señalización de cinasa; alrededor del 35 % al 50 % exhibe alteraciones genómicas en CRLF2[
1 ,120 ,125 ] y de esos, la mitad exhibe de manera simultánea variantes de JAK.[126 ]Se observó que en muchos de los casos restantes de LLA similar a BCR::ABL1 hay una serie de translocaciones que afectan a genes de fusión de clase ABL codificadores de tirosina–cinasas, como ABL1, ABL2, CSF1R, y PDGFRB.[
5 ,121 ,127 ] En algunos casos, se ha observado que las proteínas de fusión de estas combinaciones de genes son transformadoras y responden a los inhibidores de tirosina–cinasas in vitro e in vivo,[121 ,128 ] lo que indica posibles estrategias terapéuticas para estos pacientes. Las pruebas preclínicas de sensibilidad al medicamento indicaron que es posible que la sensibilidad a diferentes inhibidores de tirosina–cinasas (TKI) varíe según qué gen específico de clase ABL participe en la fusión. En un estudio de sensibilidad a TKI ex vivo, las muestras de pacientes con fusiones de PDGFRB fueron sensibles al imatinib. Sin embargo, estas muestras fueron menos sensibles al dasatinib y al bosutinib que las muestras de pacientes con fusiones de ABL1 (incluso BCR::ABL1).[128 ] En los ensayos clínicos no se han confirmado aún las diferentes respuestas a los distintos TKI según el tipo de fusión de clase ABL.En los pacientes pediátricos con LLA-B, los casos de LLA similar a BCR::ABL1 con alteraciones genómicas que no afectan CRLF2 representan alrededor del 3 % de los casos de riesgo estándar del NCI y del 8 % de los casos de riesgo alto del NCI.[
1 ] En un estudio retrospectivo de 122 pacientes pediátricos (edad 1–18 años) con fusiones de clase ABL (todos recibieron tratamiento sin inhibidores de tirosina–cinasas), la tasa de SSC a 5 años fue del 59 % y la tasa de SG fue del 76 %.[129 ]Alrededor del 9 % de los casos de LLA similar a BCR::ABL1 se producen a partir de reordenamientos que llevan a la sobreexpresión de un receptor de eritropoyetina (EPOR) incompleto.[
130 ] La región del extremo C del receptor que se pierde es la región alterada en la policitemia congénita familiar primaria, y es la que controla la estabilidad de EPOR. La porción de EPOR que queda es suficiente para la activación de JAK-STAT y para conducir a la aparición de la leucemia. Las variantes de un solo nucleótido de los genes de cinasas, distintos a JAK1 y JAK2, son poco comunes en los casos de LLA similar a BCR::ABL1.[9 ]CRLF2. Se han identificado alteraciones genómicas en CRLF2, un gen de un receptor de citocina ubicado en las regiones pseudoautosómicas de los cromosomas sexuales, en el 5 % al 10 % de los casos de LLA-B. Estas variaciones representan alrededor del 50 % de los casos de LLA similar a BCR::ABL1.[
131 ,132 ,133 ] Las anomalías cromosómicas que con mayor frecuencia conducen a la sobreexpresión de CRLF2 incluyen las translocaciones del locus de IGH (cromosoma 14) al CRLF2 y las deleciones intersticiales en las regiones pseudoautosómicas de los cromosomas sexuales, lo que produce una fusión P2RY8::CRLF2.[9 ,125 ,131 ,132 ] Estas dos alteraciones genómicas se relacionan con características clínicas y biológicas diferenciadoras.En los pacientes pediátricos con LLA-B, la LLA-B similar a BCR::ABL1 con alteraciones genómicas que afectan CRLF2 se observa en alrededor del 2 % de los casos de riesgo estándar del NCI y del 5 % de los casos de riesgo alto del NCI.[
1 ]La LLA con variaciones genómicas en CRLF2 presenta una mayor incidencia en niños que tienen ascendencia genética hispana o latina,[
125 ,134 ,135 ] e indígena americana.[87 ] En un estudio de 205 niños con LLA-B de riesgo alto, 18 de 51 (35,3 %) pacientes hispanos presentaron reordenamientos de CRLF2, en comparación con 11 casos de 154 (7,1 %) pacientes en el resto de las etnias manifestadas.[125 ] En un segundo estudio, se observó que la frecuencia de fusiones IGH::CRLF2 estaba aumentada en los niños hispanos en comparación con los niños con LLA-B que no eran hispanos ni latinos (13,2 vs. 3,6 %).[134 ,135 ] En este estudio, el porcentaje de LLA-B con fusiones P2RY8::CRLF2 fue de alrededor del 6 % y no se vio modificado por la etnia.La fusión P2RY8::CRLF2 se observa en el 70 % al 75 % de los pacientes pediátricos con alteraciones genómicas en CRLF2, y se presenta en pacientes jóvenes (mediana de edad, 4 vs. 14 años en los pacientes con IGH::CRLF2).[
136 ,137 ] A menudo la P2RY8::CRLF2 se presenta junto a otras anomalías cromosómicas establecidas (por ejemplo, hiperdiploidía, iAMP21, dic(9;20)), por el contrario IGH::CRLF2 en general es mutuamente excluyente de los subtipos citogenéticos conocidos. Se observan alteraciones genómicas en CRLF2 en cerca del 60 % de los pacientes con LLA y síndrome de Down, y la fusión P2RY8::CRLF2 es más frecuente que la fusión IGH::CRLF2 (cerca del 80–85 % vs. 15–20 %).[132 ,136 ]La presencia de IGH::CRLF2 y P2RY8::CRLF2 a menudo es una alteración temprana en la formación de una LLA-B y exhibe prevalencia clonal.[
138 ] Sin embargo, en algunos casos es una alteración tardía y exhibe prevalencia subclonal.[138 ] En estos casos, la pérdida de la anormalidad genómica de CRLF2 en el momento de una recaída confirma la naturaleza subclonal de esta alteración.[136 ,139 ]Las anormalidades en CRLF2 se asocian directamente con deleciones de IKZF1. Estas deleciones son más frecuentes en casos con fusiones IGH::CRLF2 que en casos con fusiones P2RY8::CRLF2.[
137 ] Los niños hispanos tienen una frecuencia mayor de reordenamientos de CRLF2 con deleciones de IKZF1 que los niños no hispanos.[135 ]Otras alteraciones genómicas recurrentes asociadas con las alteraciones en CRLF2 son las deleciones en los genes vinculados con la diferenciación de las células B (por ejemplo, PAX5, BTG1, EBF1, etc.) y el control del ciclo celular (CDKN2A), así como las alteraciones genómicas que activan la vía de señalización JAK-STAT (por ejemplo, variantes de IL7R y JAK).[
5 ,125 ,126 ,132 ,140 ]Aunque los resultados de varios estudios retrospectivos indican que las anomalías en CRLF2 quizás denoten un pronóstico adverso en los análisis univariantes, la mayoría no indica que esta anomalía sea un factor de predicción independiente del desenlace.[
125 ,131 ,132 ,141 ,142 ] Por ejemplo, en un estudio europeo grande, la expresión elevada de CRLF2 no se relacionó con un desenlace desfavorable en los análisis multivariantes, mientras que las firmas de expresión de deleción de IKZF1 y similar a BCR::ABL1 se relacionaron con desenlaces desfavorables.[122 ] También hay polémica sobre si el análisis de la importancia pronóstica de las anomalías en CRLF2 debe hacerse en relación con la sobreexpresión de CRLF2 o con la presencia de anomalías genómicas en CRLF2.[141 ,142 ] - Deleciones de IKZF1.
Las deleciones de IKZF1, incluso las deleciones del gen completo y de exones específicos, están presentes en alrededor del 15 % de los casos de LLA-B. Con menor frecuencia, el gen IKZF1 se inactiva por variantes de un solo nucleótido deletéreas.[
119 ]Los casos con deleciones de IKZF1 se suelen presentar en niños mayores, que tienen un recuento de GB más alto en el momento del diagnóstico y, por lo tanto, son más frecuentes en los pacientes de riesgo alto según el NCI en comparación con los de riesgo estándar según el NCI.[
3 ,119 ,140 ,143 ,144 ] Una proporción alta de casos positivos para BCR::ABL1 tienen una deleción de IKZF1,[4 ,140 ] y las LLA que surgen en niños con síndrome de Down tienen tasas elevadas de deleciones de IKZF1.[145 ] Las deleciones de IKZF1 también son comunes en los casos con alteraciones genómicas de CRLF2 y en la LLA similar a BCR::ABL1.[89 ,118 ,140 ] Las deleciones de IKZF1 también se presentaron con más frecuencia en los niños hispanos. En un estudio de un solo centro oncológico, las deleciones de IKZF1 se observaron en el 29 % de los niños hispanos, en comparación con el 11 % de los niños no hispanos (P = 0,001).[135 ]En varios informes se ha documentado la relevancia de la deleción de IKZF1 para definir un pronóstico adverso, y, en la mayoría de los estudios con análisis multivariantes, se notificó que esta deleción es un factor independiente de pronóstico precario.[
89 ,118 ,119 ,122 ,140 ,146 ,147 ,148 ,149 ,150 ,151 ,152 ,153 ]; [154 ][Nivel de evidencia B4] Sin embargo, la importancia pronóstica de IKZF1 quizás no sea equivalente en todos los subtipos biológicos de LLA, como se demuestra por la aparente falta de relevancia pronóstica en los pacientes con deleciones de ERG.[89 ,90 ,91 ] De manera parecida, la importancia pronóstica de la deleción de IKZF1 también se redujo en una cohorte de pacientes del COG de LLA con reordenamiento de DUX4 y desregulación transcripcional de ERG, lo que a menudo se presenta por la deleción de ERG.[7 ] El grupo de la Associazione Italiana di Ematologia e Oncologia Pediatrica y el Berlin-Frankfurt-Münster notificó que las deleciones de IKZF1 indicaron un pronóstico adverso solo en los pacientes con LLA-B que exhibían ERM alta al final de la inducción y en quienes se detectaron al mismo tiempo deleciones en CDKN2A, CDKN2B, PAX5 o PAR1 (en ausencia de la deleción de ERG).[155 ] Esta combinación de deleción de IKZF1 acompañada de deleción de otros genes particulares se denomina IKZF1PLUS.[155 ] En un estudio de una sola institución, el perfil de IKZF1PLUS se observó con más frecuencia en los niños hispanos que en los niños no hispanos (20 vs. 5 %, P = 0,001).[135 ]El pronóstico precario relacionado con las alteraciones en IKZF1 empeora con el hallazgo concomitante de la deleción 22q11.22. En un estudio de 1310 pacientes con LLA-B, cerca de la mitad de los pacientes con alteraciones en IKZF1 también tenían una deleción 22q11.22. La tasa de SSC a 5 años fue del 43,3 % para aquellos con las 2 anomalías, en comparación con el 68,5 % de los pacientes con alteraciones en IKZF1 y 22q11.22 natural (P < 0,001).[
156 ]Hay pocos resultados publicados sobre el cambio de tratamiento a partir del estado del gen IKZF1. El grupo Malasia-Singapur publicó los resultados de dos ensayos consecutivos. En el primer ensayo (MS2003), el estado de IKZF1 no se consideró en la estratificación del riesgo, mientras que en el ensayo posterior (MS2010), los pacientes con deleción de IKZF1 se excluyeron del grupo de riesgo estándar. Además, en el ensayo MS2010, más pacientes con deleción de IKZF1 recibieron terapia intensificada. Los pacientes de LLA con deleción de IKZF1 exhibieron mejores desenlaces en el ensayo MS2010 en comparación con los pacientes del ensayo MS2003, pero la interpretación de esta observación se ve limitada por otros cambios en la estratificación del riesgo y las diferencias de tratamiento entre los dos ensayos.[
157 ][Nivel de evidencia B4]En el estudio holandés ALL11, los pacientes con deleciones en IKZF1 prolongaron su terapia de mantenimiento durante 1 año con el objetivo de mejorar el desenlace.[
158 ] En el análisis de referencia se demostró una reducción de casi 3 veces en la tasa de recidiva y una mejora en la tasa de SSC a 2 años (del 74,4 % al 91,2 %), en comparación con los controles históricos. - LLA con reordenamiento de MYC (8q24).
Los reordenamientos del gen MYC son un hallazgo infrecuente pero recurrente en pacientes pediátricos con LLA-B. Se han notificado pacientes con reordenamientos del gen MYC, así como de los genes IGH2, IGK y IGL en 14q32, 2p12 y 22q11.2, respectivamente.[
159 ,160 ,161 ] Los linfoblastos por lo general exhiben un inmunofenotipo de células B precursoras, con una morfología francesa-americana-británica (FAB) L2 o L3, sin expresión de inmunoglobulina de superficie ni cadenas ligeras κ o λ. Se han observado reordenamientos simultáneos del gen MYC junto con otros reordenamientos citogenéticos como IGH::BCL2 o KMT2A.[161 ] Los pacientes mencionados en la bibliografía, recibieron diversas terapias para la LLA o se trataron siguiendo los protocolos de linfoma o leucemia de células B maduras; sin embargo, todavía no está claro cuál es el mejor tratamiento para estos pacientes.[161 ]
- Fusión ETV6::RUNX1 (t(12;21)(p13.2;q22.1)).
Características genómicas de la leucemia linfoblástica aguda en los niños con síndrome de Down
El estudio más grande en el que se analizó el panorama genómico de la leucemia linfoblástica aguda (LLA) que se presenta en niños con síndrome de Down incluyó 295 pacientes que participaron en ensayos clínicos del COG.[
- Casi todos los casos de LLA en niños con síndrome de Down son de LLA-B. La LLA-T es infrecuente.
- Las alteraciones genómicas recurrentes más comunes que se encuentran en casos de LLA no relacionada con el síndrome de Down (por ejemplo, hiperdiploidía elevada y ETV6::RUNX1) se presentan con mucha menor frecuencia en niños con síndrome de Down y LLA. Hay otras alteraciones que se presentan más frecuentemente en niños con síndrome de Down y LLA.
- Entre un 50 % a un 60 % de los niños con síndrome de Down y LLA tienen reordenamientos de CRLF2 que comprometen a IGH o P2RY8; en la mayoría de los casos (85 %) comprometen a P2RY8.
- Cerca de la mitad de los casos con reordenamientos de CRLF2 tienen variantes de JAK2, que no se observan en niños con síndrome de Down y LLA que no tienen reordenamientos de CRLF2.
- Las alteraciones de IKZF1 se presentan en cerca del 30 % de los casos con reordenamientos de CRLF2, pero solo en alrededor del 10 % de los casos con reordenamientos de CRLF2.
- El 25 % de los casos de LLA con reordenamientos de CRLF2 en los pacientes con síndrome de Down se clasifican según la expresión génica como similares a BCR::ABL1, en comparación con el 54 % de los casos de LLA con reordenamientos de CRLF2 y sin síndrome de Down.
- En general, los pacientes con LLA, reordenamientos CRLF2 y síndrome de Down tienen un pronóstico intermedio. Sin embargo, los pacientes con una firma de expresión génica similar a BCR::ABL1 tienen desenlaces más precarios que aquellos sin una firma de expresión génica similar a BCR::ABL1 y reordenamientos de CRLF2 (tasas de SSC, 39,5 % ± 8,1 % vs. 82 % ± 4,4 %; tasas de SG, 70,3 % ± 8,7 % vs. 86,9 % ± 4,8 %).
- La fusión génica IGH::IGF2BP1 se presenta en cerca del 3 % de los pacientes con síndrome de Down. Esta fusión génica es infrecuente en los pacientes con LLA que no tienen síndrome de Down. En un análisis retrospectivo esta fusión se relacionó con un desenlace relativamente favorable (tasa de SSC, 87,5 % ± 11,7 %).
- La LLA-B con alteración C/EBP (C/EBPalt), que se caracteriza por la activación anormal de la familia de genes C/EBP, también se encuentra con mucha frecuencia en los niños con síndrome de Down (10,5 % de los niños con LLA-B y síndrome de Down vs. 0,1 % de los niños con LLA-B sin síndrome de Down).
- Los reordenamientos de CEBPD son la lesión C/EBPalt más común y se presenta en el 7,5 % de los casos de LLA y síndrome de Down. El compañero de fusión en más del 80 % de los reordenamientos de CEBPD es IGH. Otros compañeros de fusión menos comunes son MME, TPM4, 9p13.2 y 6q25.3.
- Un 4 % a un 5 % de los casos de LLA y síndrome de Down se caracterizan por presentar alteraciones en otros miembros de la familia C/EBP, como CEBPA y CEBPE.
- Los casos con C/EBPalt a menudo albergan variantes simultáneas de FLT3, KDM6A y SETD2.
- C/EBPalt se relacionó con tasas elevadas de remisión negativa para la ERM al final de la terapia de inducción (87,1 %) y un desenlace intermedio (tasa de SSC a 10 años, 73,9 % ± 9,9 %; SG a 10 años, 76,7 % ± 12,8 %).
Características citogenéticas y genómicas de la leucemia linfoblástica aguda de células T
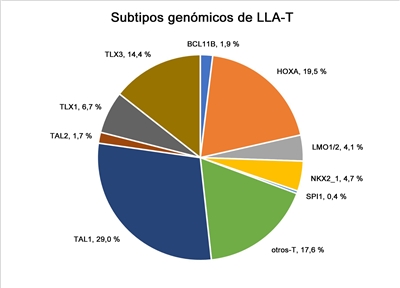
La LLA-T se caracteriza por alteraciones genómicas que producen la activación de programas transcripcionales relacionados con el desarrollo de las células T y por una frecuencia alta de casos (casi el 60 %) con variantes de NOTCH1 o FBXW7 que producen la activación de la vía NOTCH1.[
En la Figura de más abajo, los casos de LLA-T se dividen en 10 subtipos moleculares según la expresión del RNA y la presencia de variantes de los genes. Estos casos provienen de pacientes inscritos en los ensayos clínicos SJCRH y COG.[
- Señalización de la vía Notch.
La señalización de la vía Notch a menudo se activa por variantes de los genes NOTCH1 y FBXW7 en casos de LLA-T, y estos son los genes alterados con mayor frecuencia en los casos de LLA-T infantil.[
162 ,167 ] Las variantes que activan el gen NOTCH1 se presentan en cerca del 50 % al 60 % de los casos de LLA-T; las variantes que inactivan el gen FBXW7 se presentan en cerca del 15 % de los casos. Casi el 60 % de los casos de LLA-T exhiben activación de la vía Notch por variantes de por lo menos uno de estos genes.[168 ,169 ]La importancia pronóstica de las variantes de NOTCH1 y FBXW7 quizás esté modulada por alteraciones genómicas en RAS y PTEN. El French Acute Lymphoblastic Leukaemia Study Group (FRALLE) y el Group for Research on Adult Acute Lymphoblastic Leukemia notificaron que los pacientes con variantes de NOTCH1 o FBXW7 que además tienen tipos naturales de PTEN y RAS forman un grupo de pronóstico favorable (es decir, riesgo bajo), mientras que los pacientes con variantes de PTEN o RAS, sin importar el estadio de NOTCH1 y FBXW7, tienen un riesgo significativamente más alto de fracaso del tratamiento (es decir, grupo de riesgo alto).[
170 ,171 ] En el estudio FRALLE, la tasa de supervivencia sin enfermedad a 5 años fue del 88 % para el grupo de pacientes de riesgo genético bajo y del 60 % para el grupo de pacientes de riesgo genético alto.[170 ] Sin embargo, al usar los mismos criterios para definir el grupo de riesgo genético, no fue posible que el consorcio Dana-Farber Cancer Institute replicara estos resultados. Ellos informaron una tasa de SSC a 5 años del 86 % para los pacientes de riesgo genético bajo y del 79 % para los pacientes de riesgo genético alto, una diferencia que no fue estadísticamente significativa (P = 0,26).[169 ] - Translocaciones cromosómicas.
En la LLA-T se han identificado múltiples translocaciones cromosómicas que llevan a la alteración en la expresión de genes específicos. Estos reordenamientos cromosómicos producen fusiones de genes codificadores de factores de transcripción (por ejemplo, TAL1, TAL2, LMO1, LMO2, LYL1, TLX1, TLX3, NKX2-I, HOXA y MYB) con uno de los locus de los receptores de las células T (o con otros genes), lo que lleva a la alteración en la expresión de los factores de transcripción en las células leucémicas.[
162 ,163 ,172 ,173 ,174 ,175 ,176 ] A menudo, estas translocaciones no son evidentes al examinar el cariotipo estándar, pero se logran confirmar con técnicas de detección más sensibles, como FISH o PCR.[163 ] Las variantes de una región no codificante cerca del gen TAL1 que produce un superpotenciador antes de la secuencia del gen TAL1 representan alteraciones genómicas que no son translocaciones, pero que también activan la transcripción de TAL1 e inducen la LLA-T.[165 ]En la LLA-T también se observan translocaciones que producen proteínas de fusión quiméricas.[
170 ]- Se ha observado una fusión NUP214::ABL1 en el 4 % al 6 % de los casos de LLA-T en adultos y niños, con predomino masculino.[
177 ,178 ,179 ] La fusión es críptica en el análisis citogenético, y se observa en la FISH en los episomas amplificados, o con menor frecuencia, como una región pequeña de tinción homogénea.[179 ] En casos poco frecuentes, la LLA-T exhibe proteínas de fusión de ABL1 con otros genes compañeros (por ejemplo, ETV6, BCR y EML1).[179 ] Los inhibidores de tirosina–cinasas de ABL, como el imatinib o el dasatinib, quizás produzcan beneficios terapéuticos en este subtipo de LLA-T,[177 ,178 ,180 ] pero la experiencia clínica con esta estrategia es muy limitada.[181 ,182 ,183 ] - Se notificaron fusiones génicas que afectan el gen SPI1 (codificador del factor de transcripción PU.1) en el 4 % de los niños japoneses con LLA-T.[
184 ] Los compañeros de fusión fueron STMN1 y TCF7. Los casos de LLA-T con fusiones de SPI1 tienen un pronóstico muy adverso; 6 de 7 personas afectadas murieron en el transcurso de 3 años desde el diagnóstico de una recaída temprana. - El BCL11B es un factor de transcripción con dedos de cinc que cumple una función doble al activar y reprimir la transcripción. Se sabe que desempeña un papel importante en la diferenciación de las células T. En la LLA-T, el gen BCL11B participa en una translocación t(5;14)(q35;q32) donde un potenciador de BCL11B produce la expresión anómala de TLX3 (o NKX2-5).[
185 ] En el proceso de donación de su potenciador, se inactiva un alelo de BCL11B. Sin embargo, el estado haploinsuficiente resultante quizás también cumpla una función en la patogénesis del tumor. La función de BCL11B como gen supresor de tumores se apoya en los hallazgos que indican que cerca del 16 % de los pacientes tienen LLA-T que alberga deleciones o variantes de cambio de sentido.[162 ,186 ] Como se describe en las secciones para leucemia linfoblástica aguda de células T precursoras tempranas y leucemia aguda de fenotipo mixto T/mieloide, en ocasiones BCL11B es leucemógeno por sobrexpresión. - Otras fusiones génicas recurrentes en los pacientes con LLA-T son las que afectan los genes MLLT10, KMT2A, NUP214 y NUP98.[
162 ,166 ]
- Se ha observado una fusión NUP214::ABL1 en el 4 % al 6 % de los casos de LLA-T en adultos y niños, con predomino masculino.[
- Ploidía.
- Las anomalías recurrentes en el número de cromosomas son mucho menos frecuentes en la LLA-T que en la LLA-B. En un estudio se incluyeron 2250 pacientes pediátricos con LLA-T que se trataron en los protocolos de la Associazione Italiana di Ematologia e Oncologia Pediatrica y del Berlin-Frankfurt-Münster. En el estudio se encontró que la casi tetraploidía (índice de DNA, 1,79–2,28 o 81–103 cromosomas), presente en el 1,4 % de los pacientes, se relacionaba con características de la enfermedad y desenlaces favorables.[
187 ]
- Las anomalías recurrentes en el número de cromosomas son mucho menos frecuentes en la LLA-T que en la LLA-B. En un estudio se incluyeron 2250 pacientes pediátricos con LLA-T que se trataron en los protocolos de la Associazione Italiana di Ematologia e Oncologia Pediatrica y del Berlin-Frankfurt-Münster. En el estudio se encontró que la casi tetraploidía (índice de DNA, 1,79–2,28 o 81–103 cromosomas), presente en el 1,4 % de los pacientes, se relacionaba con características de la enfermedad y desenlaces favorables.[
Características citogenéticas y genómicas de la leucemia linfoblástica aguda de células T precursoras tempranas
En la caracterización molecular detallada de la LLA de células T precursoras tempranas (TPT), se observó que esta entidad es muy heterogénea a nivel molecular, y no hay un solo gen afectado por una variante o alteración en el número de copias que se presente en más de un tercio de los casos.[
En algunos estudios se encontró que la ausencia de la deleción bialélica del locus TCR-γ (ABD), detectado por hibridación genómica comparativa o DNA-PCR cuantitativa, se relacionó con un fracaso terapéutico temprano en los pacientes con LLA-T.[
La expresión aleloespecífica, por lo general alta, de BCL11B cumple una función oncogénica en un subgrupo de casos identificados como LLA TPT (7 de 58 casos en un estudio), así como en hasta de un 30 % a un 40 % de la leucemia aguda de fenotipo mixto T/M (LAFM T/M) de linaje ambiguo.[
- Una de esas alteraciones es la t(2;14)(q22;q32), que produce una fusión génica ZEB2::BCL11B dentro del marco de lectura.
- Otras variantes estructurales que conducen a la desregulación de la expresión aleloespecífica de BCL11B incluyen variantes estructurales que unen secuencias reguladoras de genes activos (por ejemplo, ARID1B [cromosoma 6], BENC [cromosoma 7] y CDK6 [cromosoma 7]) secuencia arriba o secuencia abajo del locus de BCL11B lo que conduce a la expresión anómala en un proceso llamado secuestro de potenciador.
- Por último, en alrededor del 20 % de los casos con desregulación de la expresión de BCL11B, no se identifica ninguna translocación. En muchos de esos casos, la amplificación de un potenciador secuencia abajo, la amplificación en tándem del potenciador BCL11B (BETA), conduce a la transcripción impulsada por el promotor BCL11B.
- Hay una alta prevalencia de alteraciones de FLT3 y activación de JAK/STAT en las leucemias agudas causadas por alteraciones genómicas que conducen a la sobrexpresión de BCL11B.[
191 ]
Características citogenéticas y genómicas de la leucemia aguda de fenotipo mixto
El sistema de clasificación de la OMS para las leucemias de linaje ambiguo se resume en el Cuadro .[
| Afección | Definición |
|---|---|
| LAFM = leucemia aguda de fenotipo mixto; SAI = sin otra indicación. | |
| a Adaptación de Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.[ |
|
| Leucemia aguda indiferenciada | Leucemia aguda que no expresa ningún marcador que se considere específico para el linaje linfoide ni el linaje mieloide |
| LAFM conBCR::ABL1(t(9;22)(q34;q11.2)) | Leucemia aguda que cumple con los criterios diagnósticos de LAFM y se identifican blastocitos que también expresan la translocación (9;22) o el reordenamientoBCR::ABL1 |
| LAFM conKMT2A(t(v;11q23)) | Leucemia aguda que cumple con los criterios diagnósticos de la LAFM y se identifican blastocitos que también expresan una translocación que afecta el genKMT2A |
| LAFM, B o mieloide, SAI (LAFM B/M) | Leucemia aguda que cumple con los criterios diagnósticos para asignar un linaje B y un linaje mieloide, y se identifican blastocitos que carecen de anomalías genéticas que afectenBCR::ABL1oKMT2A |
| LAFM, T o mieloide, SAI (LAFM T/M) | Leucemia aguda que cumple con los criterios diagnósticos para asignar un linaje T y un linaje mieloide, y se identifican blastocitos que carecen de anomalías genéticas que afectanBCR::ABL1oKMT2A |
| LAFM, B o mieloide, SAI (tipos poco frecuentes) | Leucemia aguda que cumple con los criterios diagnósticos para asignar un linaje B y un linaje T |
| Otras leucemias de linaje ambiguo | Leucemia o linfoma linfoblásticos de células citolíticas naturales |
| Linaje | Criterios |
|---|---|
| a Adaptación de Arber et al.[ |
|
| b Fuerte se define como más brillante o igual a las células B o T normales de la muestra. | |
| Linaje mieloide | Mieloperoxidasa (citometría de flujo, prueba inmunohistoquímica o citoquímica);o diferenciación monocítica (por lo menos dos de los siguientes aspectos: prueba citoquímica de esterasa inespecífica, CD11c, CD14, CD64 o lisozima) |
| Linaje T | CD3 citoplasmático fuerteb(con anticuerpos contra la cadena ε de CD3);o CD3 de superficie |
| Linaje B | CD19 con expresión fuerteb de por lo menos una de las siguientes moléculas: CD79a, CD22 citoplasmático o CD10;o CD19 débil y por lo menos expresión fuerte de dos de las siguientes moléculas: CD79a, CD22 citoplasmático o CD10 |
El sistema de clasificación de la LAFM incluye dos entidades definidas a partir de la alteración molecular principal: LAFM con translocación BCR::ABL1 y LAFM con reordenamiento de KMT2A. Las alteraciones genómicas asociadas con cada una de las entidades LAFM, B/mieloide, SAI (LAFM B/M) y LAFM, T/mieloide, SAI (LAFM T/M) son diferentes, como se describe a continuación:
- LAFM B/M.
- Entre 115 casos de LAFM en los que se hizo la caracterización genómica, se encontraron 35 (30 %) casos de LAFM B/M. Además hubo 16 casos de LAFM (14 %) con reordenamientos de KMT2A, 15 de ellos exhibían un inmunofenotipo B/mieloide.
- Alrededor de la mitad de los casos de LAFM B/M tenían reordenamientos de ZNF384 con compañeros de fusión recurrentes, como TCF3 y EP300. Estos casos exhibieron perfiles de expresión génica indistinguibles de los casos de LLA-B con reordenamientos de ZNF384.[
102 ] - Cerca de dos tercios de los casos de LAFM B/M presentaban alteraciones en la vía RAS, los genes alterados de manera más frecuente fueron NRAS y PTPN11.[
102 ] - Los genes codificadores de reguladores epigenéticos (por ejemplo, MLLT3, KDM6A, EP300 y CREBBP) están alterados en alrededor de dos tercios de los casos de LAFM B/M.[
102 ]
- LAFM T/M.
- Entre 115 casos de LAFM en los que se hizo la caracterización genómica, se encontraron 49 (43 %) casos de LAFM T/M.[
102 ] Las características genómicas de los casos de LAFM T/M comparten semejanzas con la LLA TPT lo que indica que la LAFM T/M y la LLA TPT son entidades similares a lo largo de la variedad de leucemias inmaduras. - En comparación con la LLA-T, la LAFM T/M presentó una tasa más baja de alteraciones en los factores de transcripción centrales de la LLA-T (TAL1, TAL2, TLX1, TLX3, LMO1, LMO2, NKX2-1, HOXA10 y LYL1) (63 vs. 16 %, respectivamente).[
102 ] También se observó una tasa baja similar en la LLA TPT. - Las variantes de CDKN2A, CDKN2B y NOTCH1, presentes en alrededor de dos tercios de los casos de LLA-T, son mucho menos comunes en la LAFM T/M. Por el contrario, las variantes de WT1 se presentan en alrededor del 40 % de los casos de LAFM T/M, pero en menos del 10 % de los casos de LLA-T.[
102 ] - Un tercio de los casos de LAFM T/M tienen alteraciones genómicas relacionadas con BCL11B que producen la expresión aleloespecífica, por lo general elevada, de BCL11B.[
191 ,192 ]- Una de esas alteraciones es la t(2;14)(q22;q32), que produce una fusión génica ZEB2::BCL11B dentro del marco de lectura que conduce a la desregulación de la expresión de BCL11B.
- Otras variantes estructurales que conducen a la desregulación de la expresión aleloespecífica de BCL11B incluyen variantes estructurales que unen secuencias reguladoras de genes activos (por ejemplo, ARID1B [cromosoma 6], BENC [cromosoma 7] y CDK6 [cromosoma 7]) secuencia arriba o secuencia abajo del locus de BCL11B en un proceso llamado secuestro de potenciador.
- Por último, en alrededor del 20 % de los casos con desregulación de la expresión de BCL11B no se identifica ninguna translocación. En estos casos, la amplificación de un potenciador secuencia abajo, la amplificación en tándem del potenciador BCL11B (BETA), conduce a la transcripción impulsada por el promotor BCL11B.
- Hay una alta prevalencia de alteraciones de FLT3 y activación de JAK/STAT en las leucemias agudas causadas por alteraciones genómicas que conducen a la sobrexpresión de BCL11B.
- Las variantes de las vías RAS y JAK-STAT son comunes en los casos de LAFM T/M y LLA TPT, mientras que las alteraciones en la vía de señalización PI3K son más comunes en la LLA-T.[
102 ] En la LAFM T/M, el gen de vía de señalización alterado de manera más frecuente fue FLT3 (43 % de los casos). Las variantes de FLT3 tienden a ser mutuamente excluyentes de las variantes de la vía RAS. - Los genes codificadores de reguladores epigenéticos (por ejemplo, EZH2 y PHF6) están alterados en alrededor de dos tercios de los casos de LAFM T/M.[
102 ]
- Entre 115 casos de LAFM en los que se hizo la caracterización genómica, se encontraron 49 (43 %) casos de LAFM T/M.[
Polimorfismos génicos en las vías metabólicas de los fármacos
Se ha notificado que varios polimorfismos de los genes involucrados en el metabolismo de fármacos quimioterapéuticos tienen importancia pronóstica en la LLA infantil.[
- TPMT.
Los pacientes con fenotipos de variantes de TPMT (gen que participa en el metabolismo de las tiopurinas, como la mercaptopurina) tienen desenlaces más favorables,[
198 ] aunque estos pacientes también exhiben un riesgo más alto de presentar importantes efectos tóxicos relacionados con el tratamiento, como mielodepresión, infección y segundas neoplasias malignas.[199 ,200 ] Los pacientes con homocigosis para variantes de TPMT relacionadas con actividad enzimática baja solo toleran dosis muy bajas de mercaptopurina (alrededor del 10 % de la dosis estándar) y se tratan con dosis reducidas de este medicamento para evitar toxicidad excesiva. Los pacientes heterocigóticos para la variante de este gen de enzima por lo general toleran la mercaptopurina sin toxicidad grave, pero necesitan reducciones de dosis más frecuentes debido a toxicidad hematopoyética que los pacientes homocigóticos para el alelo normal.[201 ,202 ] - NUDT15.
Las variantes de la línea germinal en NUDT15 que reducen o anulan la actividad de esta enzima también disminuyen la tolerabilidad a las tiopurinas.[
201 ,203 ] Las variantes de NUDT15 son más comunes en los pacientes del este de Asía y en los pacientes hispanos, pero son infrecuentes en los pacientes europeos y africanos. Los pacientes homocigóticos para las variantes de riesgo toleran solo dosis muy bajas de mercaptopurina, mientras que los pacientes heterocigóticos para los alelos de riesgo toleran dosis más bajas que los pacientes homocigóticos para el alelo de tipo natural (reducción promedio de la dosis, 25 %), pero hay una superposición amplia de las dosis toleradas entre los dos grupos.[201 ,204 ] - CEP72.
Los polimorfismos génicos también afectan la expresión de las proteínas que cumplen funciones importantes en los efectos celulares de los antineoplásicos. Por ejemplo, los pacientes homocigóticos para un polimorfismo en la región promotora de CEP72 (una proteína del centrosoma que participa en la formación de microtúbulos) tienen un riesgo aumentado de neurotoxicidad por vincristina.[
205 ] - Polimorfismos de un solo nucleótido.
En el análisis de polimorfismos en el genoma completo, se han identificado polimorfismos de un solo nucleótido específicos que se relacionan con una ERM alta al final de la inducción y riesgo de recaída. Los polimorfismos de la interleucina-15, y los genes asociados con el metabolismo del etopósido y el metotrexato, exhibieron una asociación significativa con la respuesta al tratamiento en dos cohortes numerosas de pacientes con LLA tratados con protocolos del SJCRH y del COG.[
206 ] Las variantes polimórficas que afectan el transportador de folato reducido y el metabolismo del metotrexato se relacionaron con la toxicidad y el desenlace.[207 ,208 ] Aunque estas asociaciones indican que las variaciones individuales en el metabolismo de los fármacos quizás afecten el desenlace, se han realizado pocos estudios para intentar ajustar a partir de dichas variaciones; se desconoce si una modificación personalizada de la dosis a partir de estos hallazgos mejoraría los resultados.
Para obtener información sobre el tratamiento de la LLA infantil, consultar
Leucemia mieloide aguda
Características citogenéticas y moleculares de la leucemia mieloide aguda
En los niños con leucemia mieloide aguda (LMA) se hacen análisis genéticos de los blastocitos leucémicos (mediante métodos citogenéticos convencionales y métodos moleculares) porque las anomalías cromosómicas y moleculares son marcadores diagnósticos y pronósticos importantes.[
A partir de la caracterización molecular integral de la LMA en niños y adultos se demostró que es una enfermedad que exhibe coincidencias y diferencias en todos los grupos de edades.[
- La LMA en la niñez, a diferencia de la que se presenta en la adultez, es por lo general una enfermedad de alteraciones cromosómicas recurrentes. Para obtener una lista de fusiones génicas frecuentes y otras alteraciones genómicas recurrentes, consultar el Cuadro 3.[
2 ,213 ,218 ] Dentro del grupo de edad pediátrica, algunas fusiones génicas se presentan sobre todo en niños menores de 5 años (por ejemplo, las que ocurren en los genes NUP98 y KMT2A, y la fusión génica CBFA2T3::GLIS2), mientras que otras se presentan por lo general en niños de 5 años o más (por ejemplo, las fusiones génicas RUNX1::RUNX1T1, CBFB::MYH11 y PML::RARA). - En general, los pacientes pediátricos con LMA tienen tasas bajas de variantes. La mayoría de los casos exhiben menos de un cambio somático por megabase en las regiones de codificación de proteínas.[
219 ] Esta tasa de variantes es un poco más baja que la observada en pacientes adultos con LMA y mucho más baja que la tasa de variantes de los cánceres que responden a los inhibidores de puntos de control (por ejemplo, el melanoma).[219 ] - El patrón de variantes génicas difiere entre los casos de LMA en niños y adultos. Por ejemplo, las variantes de IDH1, IDH2, TP53, RUNX1 y DNMT3A son más comunes en la LMA de adultos que en la de niños, mientras que las variantes de NRAS y WT1 son mucho más comunes en la LMA del entorno pediátrico.[
218 ,219 ,220 ] - El panorama genómico de los casos pediátricos de LMA puede cambiar desde el momento del diagnóstico hasta la recidiva. Las variantes detectables en el momento del diagnóstico a veces no se encuentran en el momento de la recaída y, a la inversa, aparecen variantes nuevas en el momento de la recaída. Un hallazgo clave en un estudio de 20 casos para los que se disponía de datos de secuenciación en el momento del diagnóstico y de la recaída fue que la frecuencia de una variante alélica en el momento del diagnóstico se correlacionó de manera sólida con persistencia de las variantes en el momento de la recaída.[
221 ] Casi el 90 % de las variantes diagnósticas con una variación alélica mayor de 0,4 persistieron hasta la recaída, en comparación con solo el 28 % en aquellas con frecuencia de variación alélica menor de 0,2 (P < 0,001). Esta observación es congruente con los resultados anteriores en los que se observó que la presencia de una variante del gen FLT3, que resulta de duplicaciones internas en tándem (ITD), predijo un pronóstico precario solo cuando hubo una proporción alélica elevada de FLT3-ITD.
En la 5ª edición (2022) de la clasificación de los tumores hematolinfoides de la Organización Mundial de la Salud (OMS), así como en la clasificación inaugural de tumores pediátricos de la OMS, se hace hincapié en un abordaje de varios niveles para la clasificación de la LMA. Estas clasificaciones consideran numerosos parámetros clínicopatológicos y buscan una base genética para la clasificación de la enfermedad siempre que sea posible.[
Además de las anomalías citogenéticas y moleculares que ayudan al diagnóstico de la LMA, de acuerdo a la OMS, hay otras entidades que, aunque no definen una enfermedad, tienen importancia pronóstica para la LMA en pediatría. Todas las anomalías pronósticas, tanto las caracterizadas por la OMS como las otras anomalías, se han agrupado de acuerdo con su relación con un pronóstico favorable o desfavorable, según lo definen los ensayos clínicos contemporáneos del Children's Oncology Group (COG). Estas entidades se resumen a continuación. Después de la descripción de cada entidad se brinda información sobre las otras características citogenéticas, moleculares y fenotípicas que se relacionan con la LMA en pediatría; sin embargo, es posible que estas características adicionales ya no se usen para la estratificación del riesgo y la asignación del tratamiento.
Si bien la fusión t(15;17) que da origen al producto génico LMP::RARA se considera una lesión que define el riesgo de LMA en pediatría, esta se analiza en Leucemia promielocítica aguda infantil dada su relación con la leucemia promielocítica aguda.
| Categoría diagnóstica | Prevalencia aproximada en la leucemia mieloide aguda en pediatría |
|---|---|
| a Adaptación de Pfister et al.[ |
|
| b Translocación cromosómica críptica. | |
| Leucemia mieloide aguda con t(8;21)(q22;q22);RUNX1::RUNX1T1 | 13–14 % |
| Leucemia mieloide aguda con inv(16)(p13.1q22) o t(16;16)(p13,1;q22);CBFB::MYH11 | 4–9 % |
| Leucemia promielocítica aguda con t(15;17)(q24,1;q21,2);PML::RARA | 6–11 % |
| Leucemia mieloide aguda con reordenamiento deKMT2A | 25 % |
| Leucemia mieloide aguda con t(6;9)(p23;q34,1);DEK::NUP214 | 1,7 % |
| Leucemia mieloide aguda con inv(3)(q21q26)/t(3;3)(q21;q26);GATA2,RPN1::MECOM | <1 % |
| Leucemia mieloide aguda con fusión deETV6 | 0,8 % |
| Leucemia mieloide aguda con t(8;16)(p11,2;p13,3);KAT6A::CREBBP | 0,5 % |
| Leucemia mieloide aguda con t(1;22)(p13,3;q13,1);RBM15::MRTFA (MKL1) | 0,8 % |
| Leucemia mieloide aguda conCBFA2T3::GLIS2(inv(16)(p13q24))b | 3 % |
| Leucemia mieloide aguda con fusión deNUP98b | 10 % |
| Leucemia mieloide aguda con t(16;21)(p11;q22);FUS::ERG | 0,3–0,5 % |
| Leucemia mieloide aguda con variante deNPM1 | 8 % |
| Leucemia mieloide aguda con variantes en el dominio bZIP deCEBPA | 5 % |
A continuación se presenta una descripción breve de las anomalías citogenéticas y moleculares recurrentes específicas. Las anomalías se enumeran según aquellas en uso clínico que identifican a los pacientes con un pronóstico favorable o desfavorable, seguidas de otras anomalías. Cuando se considera relevante, se incluye la nomenclatura de la 5ª edición de la clasificación de la OMS para cada entidad.
Anomalías relacionadas con un pronóstico favorable
Las anomalías citogenéticas o moleculares relacionadas con un pronóstico favorable según los subtipos de leucemia mieloide aguda son las siguientes:
- Leucemia mieloide aguda con factor de unión nuclear (CBF). Incluye casos con las fusiones génicas RUNX1::RUNX1T1 y CBFB::MYH11.
- Leucemia mieloide aguda con fusiones génicas RUNX1::RUNX1T1 (t(8;21)(q22;q22,1)). En las leucemias con t(8;21), el gen RUNX1 del cromosoma 21 se fusiona con el gen RUNX1T1 del cromosoma 8. La translocación t(8;21) se relaciona con el subtipo FAB M2 y con los sarcomas granulocíticos. Los adultos que tienen LMA con t(8;21) presentan un pronóstico más favorable que los adultos que tienen otros tipos de LMA.[
209 ] La translocación t(8;21) se observa en cerca del 12 % de los niños con LMA [210 ,211 ,224 ] y se relaciona con un desenlace más favorable que la LMA caracterizada por cariotipos normales o complejos.[209 ,225 ,226 ,227 ] En conjunto, la translocación se relaciona con tasas de supervivencia general (SG) a 5 años del 74 % al 90 %.[210 ,211 ,224 ] - Leucemia mieloide aguda con fusiones génicas CBFB::MYH11 (inv(16)(p13,1;q22) o t(16;16)(p13,1;q22)). En las leucemias con inv(16), el gen CBFB de la banda cromosómica 16q22 se fusiona con el gen MYH11 de la banda cromosómica 16p13. La translocación inv(16) se relaciona con el subtipo FAB M4Eo y confiere un pronóstico favorable a adultos y niños con niños con LMA.[
209 ,225 ,226 ,227 ] La inv(16) se presenta en el 7 % al 9 % de los niños con LMA; la tasa de SG a 5 años de estos pacientes es de casi el 85 %.[210 ,211 ]Los casos de fusiones CBFB::MYH11 o RUNX1::RUNX1T1 tienen variantes secundarias características; las variantes secundarias tipo CBFB::MYH11 se restringen sobre todo a genes que activan la señalización de receptores tirosina–cinasa (NRAS, FLT3 y KIT).[
228 ,229 ] Se ha estudiado la importancia pronóstica de las variantes activadoras en KIT en adultos con LMA CBF y los resultados fueron contradictorios. En un metanálisis se encontró que las variantes de KIT aumentan el riesgo de recaída sin afectar la SG en adultos con LMA y fusiones RUNX1::RUNX1T1.[230 ] La importancia pronóstica de las variantes de KIT en los casos pediátricos de la LMA CBF sigue estando poco clara. En algunos estudios, no se encontró un efecto de las variantes de KIT sobre el desenlace,[231 ,232 ,233 ] aunque, en ciertos casos, el tratamiento utilizado fue heterogéneo, lo que podría dificultar el análisis. En otros estudios se notificó un riesgo más alto de fracaso del tratamiento cuando se presentan variantes de KIT.[234 ,235 ,236 ,237 ,238 ,239 ] En un análisis de un subconjunto de pacientes pediátricos que se trataron con una quimioterapia de base uniforme en el estudio COG AAML0531, se demostró que el subconjunto de pacientes con variantes del exón 17 de KIT tuvo desenlaces inferiores en comparación con los pacientes con LMA CBF que no presentaron la variante. Sin embargo, el tratamiento con gemtuzumab ozogamicina anuló este efecto pronóstico negativo.[238 ] Si bien hubo una tendencia hacia desenlaces inferiores en los pacientes con LMA CBF y anomalías en el exón 8 de KIT, este hallazgo no fue estadísticamente significativo. En un segundo estudio de 46 pacientes tratados de manera uniforme, se encontró que las variantes del exón 17 de KIT solo tuvieron importancia pronóstica en la LMA con fusiones RUNX1::RUNX1T1, pero no con fusiones CBFB::MYH11.[239 ]Si bien se observan variantes de KIT en ambos subconjuntos de LMA CBF, otras variantes secundarias tienden a agruparse con una de las dos fusiones. Por ejemplo, los pacientes con fusiones RUNX1::RUNX1T1 también tienen variantes frecuentes en los genes que regulan la conformación de la cromatina (por ejemplo, ASXL1 y ASXL2) (40 % de los casos) y en los genes que codifican componentes del complejo de la cohesina (20 % de los casos). Las variantes de ASXL1 y ASXL2, así como las variantes de los componentes del complejo de la cohesina, son poco frecuentes en los casos de leucemia y fusiones CBFB::MYH11.[
228 ,229 ] A pesar de esta correlación, en un estudio de 204 adultos con LMA y fusiones RUNX1::RUNX1T1, se encontró que las variantes de ASXL2 (presentes en el 17 % de los casos) y las variantes de ASXL1 o ASXL2 (presentes en el 25 % de los casos) carecían de importancia pronóstica.[240 ] Se notificaron resultados similares, aunque con números más pequeños, en niños con las mismas anomalías.[241 ]
- Leucemia mieloide aguda con fusiones génicas RUNX1::RUNX1T1 (t(8;21)(q22;q22,1)). En las leucemias con t(8;21), el gen RUNX1 del cromosoma 21 se fusiona con el gen RUNX1T1 del cromosoma 8. La translocación t(8;21) se relaciona con el subtipo FAB M2 y con los sarcomas granulocíticos. Los adultos que tienen LMA con t(8;21) presentan un pronóstico más favorable que los adultos que tienen otros tipos de LMA.[
- Leucemia mieloide aguda con variante de NPM1. La NPM1 es una proteína que se ha relacionado con el transporte y el ensamblaje proteico en los ribosomas; además es una chaperona molecular que previene la agregación proteica en el nucléolo. Se pueden utilizar métodos inmunohistoquímicos para identificar con exactitud a los pacientes que tienen variantes de NPM1 mediante la demostración de la localización citoplasmática de NPM. Las variantes de la proteína NPM1 que reducen su ubicación nuclear se relacionan de manera primaria con un subconjunto de LMA de cariotipo normal, que no expresa CD34, y presenta mejor pronóstico cuando no hay variantes de FLT3 por ITD en adultos y adultos jóvenes.[
242 ,243 ,244 ,245 ,246 ,247 ]Los estudios de LMA en niños indican una menor tasa de variantes de NPM1 en comparación con los casos de adultos con características citogenéticas normales. Las variantes de NPM1 afectan a casi el 8 % de los pacientes pediátricos con LMA y son infrecuentes en niños menores de 2 años.[
214 ,215 ,248 ,249 ] Las variantes de NPM1 se relacionan con un pronóstico favorable en pacientes con LMA caracterizada por un cariotipo normal.[214 ,215 ,249 ] Se publicaron informes contradictorios sobre la importancia pronóstica en la población pediátrica de una variante de NPM1 cuando también se presenta una variante de FLT3 por ITD. En un estudio se informó que una variante de NPM1 no anula por completo el pronóstico precario que acarrea la variante de FLT3 por ITD;[214 ,250 ] sin embargo, en otros estudios se observó que no hay efecto de la variante de FLT3 por ITD sobre el pronóstico favorable relacionado con la variante de NPM1.[215 ,219 ,249 ]En un análisis exhaustivo de ensayos en serie del COG, los desenlaces de los pacientes con una variante de NPM1 que presentaban de manera simultánea variantes de FLT3 por ITD fueron favorables y comparables a los de los pacientes con una variante de NPM1 que no tenían de manera simultánea variantes de FLT3 por ITD. Las tasas de supervivencia sin complicaciones (SSC) y de SG oscilaron entre el 70 % y el 75 % en ambos grupos.[
251 ] Un número significativo de pacientes analizados tenían una variante de NPM1 y recibieron un TCMH en ensayos clínicos anteriores, lo que llevó a la especulación sobre que los desenlaces podrían ser comparables debido al efecto favorable del TCMH en pacientes con LMA con variantes de NPM1 y variantes de FLT3 por ITD. En el ensayo COG AAML1831 (NCT04293562) se determinará si los pacientes con variantes de NPM1 y variantes de FLT3 por ITD que no tienen enfermedad residual medible tras la inducción 1 pueden evitar un TCMH y seguir teniendo desenlaces excelentes comparables a los de los pacientes con variantes de NPM1 que no tienen anomalías FLT3 por ITD. - Leucemia mieloide aguda con variantes de CEBPA. Las variantes del gen CEBPA se presentan en un subconjunto de niños y adultos que tienen LMA con características citogenéticas normales.[
252 ,253 ] En los adultos menores de 60 años, cerca del 15 % de los casos de LMA con características citogenéticas normales tienen variantes de CEBPA.[246 ] Los desenlaces para los adultos que tienen una LMA con variantes de CEBPA son relativamente favorables y similares a los de los pacientes con leucemias CBF.[246 ,254 ] En los estudios iniciales en adultos con LMA se demostró que la variante doble en CEBPA, pero no la variante de un solo alelo, se relacionaba de forma independiente con un pronóstico favorable,[255 ,256 ,257 ,258 ,259 ,260 ] lo que llevó a que, en la revisión de 2016 de la OMS, se incluyeran las variantes bialélicas como una característica para definir la enfermedad.[106 ] Sin embargo, en un estudio de más de 4700 adultos con LMA se observó que los pacientes con variante de un solo alelo de CEBPA en el dominio C-terminal bZIP tienen características clínicas y desenlaces favorables similares a los de los pacientes de LMA con variante de ambos alelos.[260 ]Hay variantes de CEBPA en alrededor del 5 % de los niños con LMA y se encuentran casi siempre en el subtipo de LMA con características citogenéticas normales, tipo FAB M1 o M2.
- Los pacientes con variantes de ambos alelos de CEBPA o variantes de bZIP uno de los alelos de CEBPA tienen una mediana de edad de presentación de 12 a 13 años y perfiles de expresión génica muy parecidos.[
253 ] - Cerca del 80 % de los pacientes pediátricos tienen ambos alelos alterados (es decir, casos que tienen al mismo tiempo una variante del dominio TAD de CEBPA y una variante del dominio bZIP de CEBPA), lo que predice una supervivencia significativamente mejor, similar al efecto observado en los estudios de adultos.[
253 ,261 ] - En un estudio de cerca de 3000 niños con LMA, se observó que los pacientes con variantes de ambos alelos de CEBPA y aquellos que presentaban solo una variante del dominio bZip tenían un pronóstico favorable, en comparación con los pacientes con el tipo natural de CEBPA.[
253 ]
Teniendo en cuenta estos hallazgos en los casos pediátricos de LMA con variante de CEBPA, la presencia de una variante de bZIP sola confiere un pronóstico favorable. Sin embargo, es importante destacar que hay un pequeño subconjunto de pacientes con LMA y variante de CEBPA que tienen desenlaces menos favorables. En particular, las variantes de CSF3R se presentan en el 10 % al 15 % de los pacientes con LMA y variante de CEBPA. Las variantes de CSF3R se relacionan con un aumento del riesgo de recaída, pero sin consecuencias en la SG.[
253 ,262 ] En la actualidad, la presencia de esta variante secundaria no produce estratificación a un tratamiento más intensificado en pacientes pediátricos con LMA.Aunque no es común, un pequeño porcentaje de niños con LMA y variante de CEBPA tienen una variante germinal subyacente. En pacientes con diagnóstico reciente de una LMA con variante de ambos alelos de CEBPA, se debe considerar el análisis de la línea germinal además de las preguntas habituales sobre la historia familiar, porque se ha notificado que del 5 % al 10 % de estos pacientes tiene una anomalía germinal en CEBPA que les confiere un mayor riesgo de neoplasias malignas.[
252 ,263 ] Para obtener más información en inglés, consultarCEBPA-Associated Familial Acute Myeloid Leukemia . - Los pacientes con variantes de ambos alelos de CEBPA o variantes de bZIP uno de los alelos de CEBPA tienen una mediana de edad de presentación de 12 a 13 años y perfiles de expresión génica muy parecidos.[
Anomalía citogenética relacionada con un pronóstico variable: Reordenamientos del genKMT2A(MLL)
La 5ª edición (2022) de la clasificación de tumores hematolinfoides de la OMS incluye una categoría diagnóstica de LMA con reordenamientos de KMT2A. No se enumeran los genes específicos que conforman estas translocaciones porque hay más de 80 genes que forman parejas de fusión con KMT2A.[
- Los reordenamientos del gen KMT2A se presentan en alrededor del 20 % de los niños con LMA.[
210 ,211 ] Estos casos, incluso la mayoría de las LMA secundarias a la exposición a epipodofilotoxinas,[264 ] por lo general se relacionan con diferenciación monocítica (FAB M4 y M5). También se notifican reordenamientos de KMT2A en cerca del 10 % de los pacientes con FAB M7 (AMKL).[265 ,266 ] - En el entorno pediátrico, la mediana de edad de los casos con reordenamiento 11q23/KMT2A es de cerca de 2 años; la mayoría de los subgrupos de translocaciones tienen una mediana de edad de menos de 5 años en el momento del cuadro clínico inicial.[
267 ] Sin embargo, se observa una mediana de edad significativamente mayor en el momento de la presentación de casos pediátricos con t(6;11)(q27;q23) (12 años) y t(11;17)(q23;q21) (9 años).[267 ] - Por lo general, se notifica que el desenlace en los pacientes con diagnóstico nuevo de LMA con reordenamientos del gen KMT2A es similar, o un poco más precario, que el observado en otros pacientes con LMA.[
209 ,210 ,267 ,268 ,269 ] Como el gen KMT2A puede formar translocaciones con muchos genes de fusión diferentes, el gen compañero específico de fusión quizás influya en el pronóstico. Este hallazgo se demostró en un estudio retrospectivo internacional de gran tamaño en el que se evaluaron los desenlaces de 756 niños con LMA y reordenamiento de 11q23 o KMT2A, así como en un segundo análisis del COG en el que se estudiaron los desenlaces de la alteración en KMT2A en el contexto del ensayo AAML0531.[267 ,269 ] - La translocación más común, que representa casi el 50 % de los casos con reordenamiento de KMT2A en la población pediátrica con LMA, es t(9;11)(P22;q23), en la que el gen KMT2A se fusiona con el gen MLLT3.[
267 ,269 ] Los grupos de ensayos clínicos individuales han descrito de manera variable un pronóstico más favorable para estos pacientes. Sin embargo, ni en el estudio retrospectivo internacional ni en el estudio del COG se confirmó el pronóstico favorable para este subgrupo.[209 ,210 ,267 ,269 ] Esta fusión, que se relaciona con un pronóstico intermedio, en la actualidad no se clasifica como de riesgo alto (al menos dentro del COG), excepto cuando la enfermedad residual mínima (ERM) persiste al final de la inducción 1. - Los subgrupos de LMA con reordenamiento de KMT2A que se relacionan con desenlaces precarios son los siguientes:
- Los casos con la translocación t(10;11) son un grupo con riesgo alto de recaída en la médula ósea y el SNC.[
209 ,211 ] Algunos casos con esta translocación tienen una fusión del gen KMT2A con el gen MLLT10 en 10p12, mientras que otros tienen una fusión de KMT2A con ABI1 en 10p11.2. En un estudio retrospectivo internacional, se encontró que estos casos, que se presentan a una mediana de edad de alrededor de 1 a 3 años, tienen una tasa de SSC a 5 años del 17 % al 30 %.[267 ,269 ] - Los pacientes con t(6;11)(q27;q23) (KMT2A::AFDN) tienen desenlaces precarios, con tasas de SSC a 5 años del 11 % al 15 %.[
269 ] - Los pacientes con t(4;11)(q21;q23) (KMT2A::AFF1) a menudo presentan hiperleucocitosis y desenlaces precarios, con tasas de SSC a 5 años del 0 % al 29 %.[
267 ,269 ] - Los pacientes con t(11;19)(q23;p13,3) (KMT2A::MLLT1) tienen desenlaces precarios, con una tasa de SSC a 5 años del 14 %.[
269 ] - A partir de los datos anteriores, el grupo de estudio International Berlin-Frankfurt-Münster (iBFM) analizó los datos sobre los desenlaces en pacientes con reordenamientos de KMT2A inscritos en estudios del BFM, el COG y de otros grupos cooperativos europeos.[
270 ] De acuerdo con las publicaciones anteriores,[270 ] en este estudio se clasificó a los pacientes con 6q27 (KMT2A::AFDN, es decir, MLLT4), 4q21 (KMT2A::AFF1, es decir, MLL::MLLT2), 10p12.3 (KMT2A::MLLT10) 10p12.1 (KMT2A::ABI1) y 19p13.3 (KMT2A::MLLT1, es decir, MLL::ENL) como de riesgo alto, mientras que todos los demás se clasificaron en otro grupo de riesgo.[267 ,269 ,270 ] Según esta clasificación, las tasas de SSC a 5 años en los pacientes con LMA y reordenamientos de KMT2A que no confieren un riesgo alto son del 54 %, en comparación con el 30,3 % en los pacientes con enfermedad de riesgo alto.[269 ] La evaluación de la ERM después de la inducción 2 confiere una importancia pronóstica adicional en el análisis del iBFM. Sin embargo, la ERM al final de la inducción 1 no predijo la recaída en el contexto del análisis del COG.[269 ] - De acuerdo con esta distinción, las fusiones de KMT2A que no confieren un riesgo alto pasan a ser de riesgo alto en la mayoría de los grupos cooperativos si se observa ERM después del tratamiento de inducción.[
269 ] - Al examinar los desenlaces de los pacientes con reordenamientos de KMT2A, en general así como en el contexto de fusiones de riesgo alto y de otro tipo de riesgo, el tratamiento con gemtuzumab ozogamicina anuló el efecto pronóstico negativo de la variante. En particular, la tasa de SSC en los pacientes con LMA y reordenamientos de KMT2A fue superior con gemtuzumab ozogamicina que sin este tratamiento (48 vs. 29 %; P = 0,003) y equivalente a los desenlaces observados en los pacientes sin reordenamientos de KMT2A.[
269 ]
- Los casos con la translocación t(10;11) son un grupo con riesgo alto de recaída en la médula ósea y el SNC.[
Anomalías citogenéticas y moleculares relacionadas con un pronóstico desfavorable
A continuación se describen las anomalías genéticas relacionadas con un pronóstico desfavorable. Algunas de estas son alteraciones que definen el tipo de leucemia mieloide aguda y que se mantienen durante todo el curso de la enfermedad del paciente. Otras entidades que se describen a continuación son alteraciones secundarias (por ejemplo, alteraciones en FLT3). Aunque estas alteraciones secundarias no causan la enfermedad por sí solas, son capaces de promover la proliferación y supervivencia celular de las leucemias que se originan por alteraciones genéticas primarias.
- Leucemia mieloide aguda con anomalías de GATA2 o MECOM (inv(3)(q21,3;q26,2)/t(3;3)(q21,3;q26,2) o t(3;21)(26,2;q22)). El gen MECOM en el cromosoma 3q26 codifica 2 proteínas que regulan la transcripción: la EVI1 y la MDS1::EVI1. Las anomalías inv(3) y t(3;3) conducen a una sobreexpresión de EVI1 y a una reducción de la expresión de GATA2.[
271 ,272 ] Estas anomalías se relacionan con un pronóstico precario en adultos con LMA, [209 ,273 ,274 ] pero son infrecuentes en niños (<1 % de los casos pediátricos de LMA).[210 ,226 ,275 ]Así mismo, se pueden detectar anomalías que afectan MECOM en algunos casos de LMA con otras anomalías de 3q (por ejemplo, t(3;21)(26,2;q22)). La fusión RUNX1::MECOM también se relaciona con un pronóstico precario.[
276 ,277 ] - Leucemia mieloide aguda con fusiones génicas NPM1::MLF1 (t(3;5)(q25;q34)). Esta fusión produce una proteína quimérica que incluye prácticamente todo el gen MLF1. Por lo general, este gen no tiene una función en la hematopoyesis normal, pero en este contexto, se especula que produce la expresión ectópica de la proteína. Aunque es muy infrecuente en pediatría (menos de 0,5 % de los casos, y la mayoría se presenta en la adolescencia),[
278 ] por lo general se relaciona con un pronóstico precario.[279 ] - Leucemia mieloide aguda con fusiones génicas DEK::NUP214 (t(6;9)(p23;q34,1)). Esta fusión conduce a la formación de la proteína de fusión DEK::NUP214 asociada a la leucemia.[
280 ,281 ] Este subgrupo de LMA se relacionó con un pronóstico precario en adultos con LMA,[280 ,282 ,283 ] y se presenta con poca frecuencia en niños (menos del 1 % de los casos de LMA). La mediana de edad de los niños con LMA y fusiones DEK::NUP214 es de 10 a 11 años, y alrededor del 40 % de los pacientes pediátricos tienen FLT3-ITD.[284 ,285 ]La LMA t(6;9) se relaciona con un riesgo alto de fracaso del tratamiento en los niños, en particular en aquellos que no se someten a un TCMH alogénico.[
210 ,281 ,284 ,285 ] - Leucemia mieloide aguda con fusiones génicas KAT6A::CREBBP (t(8;16)(p11,2;p13,3)) (si tiene 90 días o más en el momento del diagnóstico). La translocación t(8;16) fusiona el gen KAT6A del cromosoma 8p11 con el gen CREBBP del cromosoma 16p13. Se relaciona con desenlaces precarios en adultos, pero su importancia pronóstica en pediatría es menos clara.[
219 ,286 ] Aunque esta translocación se presenta con poca frecuencia en niños, en un estudio de LMA que llevó a cabo el iBFM con 62 niños, esta translocación se relacionó con una edad más temprana en el momento del diagnóstico (mediana, 1,2 años), fenotipo FAB M4/M5, eritrofagocitosis, leucemia cutis, y coagulación intravascular diseminada.[287 ] Una proporción importante de lactantes diagnosticados con LMA t(8;16) en el primer mes de vida presenta remisión espontánea, aunque la recidiva de la LMA puede ocurrir meses o años después.[287 ,288 ,289 ,290 ] Estas observaciones indican que se podría considerar una estrategia de observación cautelosa para los casos de LMA con t(8;16) diagnosticada en el período neonatal si se puede garantizar una vigilancia estrecha a largo plazo.[287 ] Para los niños mayores, el pronóstico es menos favorable y la recomendación típica es proceder a un TCMH una vez que se alcanza la remisión. - Leucemia mieloide aguda con fusiones génicas FUS::ERG (t(16;21)(p11;q22)). En las leucemias con t(16;21)(p11;q22), el gen FUS se une al gen ERG y produce un subtipo particular de LMA con un perfil de expresión génica que se agrupa por separado de otros subgrupos citogenéticos.[
291 ] Esta fusión es infrecuente en pediatría y representa del 0,3 % al 0,5 % de los casos pediátricos de LMA. En una cohorte de 31 pacientes con LMA y fusiones FUS::ERG, los desenlaces fueron precarios; la tasa de SSC a 4 años fue del 7 % y la incidencia acumulada de recaída fue del 74 %.[291 ] - Leucemia mieloide aguda con fusiones génicas CBFA2T3::GLIS2. Esta fusión es el resultado de una inversión críptica del cromosoma 16 (inv(16)(p13,3;q24,3)).[
292 ,293 ,294 ,295 ,296 ] Por lo común, se presenta en la LMCA sin síndrome de Down; explica entre el 16 % y el 27 % de las LMCA en edad pediátrica y se manifiesta a una mediana de edad de 1 año.[266 ,294 ,297 ,298 ,299 ] Las células leucémicas con fusiones CBFA2T3::GLIS2 tienen un inmunofenotipo característico (que en el inicio se notificó como fenotipo RAM),[300 ,301 ] con CD56 elevado, expresión débil o negativa de CD45 y CD38, y ausencia de expresión de HLA-DR. Esta fusión es una lesión de riesgo muy alto que se relaciona con desenlaces clínicos precarios.[266 ,292 ,296 ,297 ,298 ,299 ]En un estudio de aproximadamente 2000 niños con LMA, se identificó la fusión CBFA2T3::GLIS2 en 39 casos (1,9 %), con una mediana de edad en el momento de la presentación de 1,5 años. Todos los casos observados en niños fueron en menores de 3 años.[
302 ] Cerca de la mitad de los casos exhibían morfología megacarioblástica M7 y el 29 % de los pacientes eran negros o afroamericanos (excedió la frecuencia del 12,8 % en pacientes sin la fusión). Se encontró que los niños con la fusión tenían ERM positiva después de la inducción 1 en un 80 % de los casos. En un análisis de los resultados de los ensayos en serie del COG de 37 pacientes identificados, la SG a los 5 años desde el momento de inscripción en el estudio fue del 22,0 % en los pacientes con fusiones CBFA2T3::GLIS2 versus el 63,0 % en los pacientes que no presentaban la fusión (n = 1724). Se demostraron desenlaces más precarios cuando se comparó un subconjunto de pacientes con LMCA y CBFA2T3::GLIS2 y un subconjunto de pacientes con LMCA sin la anomalía. En el análisis de los ensayos del COG AAML0531 y AAML1031, se observaron tasas de SG del 43 % (± 37 %) y del 10 % (± 19 %), respectivamente, en niños con LMCA y esta fusión.[299 ] Dado que las leucemias con la fusión CBFA2T3::GLIS2 expresan concentraciones altas de FOLR1 en la superficie celular, un antígeno de superficie que se puede usar como diana terapéutica en abordajes con fármacos de inmunoterapia, se tiene previsto estudiar la función de estos fármacos en esta población de riesgo alto.[303 ,304 ] - Leucemia mieloide aguda con fusiones génicas de NUP98. Se notificó que NUP98 forma fusiones génicas leucemógenas con más de 20 genes compañeros de fusión diferentes. Una proporción significativa de los casos se relaciona con la LMCA sin síndrome de Down, aunque cerca del 50 % se observan fuera de ese subtipo morfológico.[
305 ,306 ] Las dos fusiones génicas más comunes en los casos pediátricos de LMA son NUP98::NSD1 y NUP98::KDM5A. En un informe, la primera fusión se observó en alrededor del 15 % de los casos pediátricos de LMA con características citogenéticas normales y la segunda fusión se observó en alrededor del 10 % de los casos pediátricos de LMCA (consultar más adelante).[266 ,294 ,299 ,307 ] Los casos de LMA con cualquiera de las dos fusiones génicas de NUP98 presentan una expresión alta de los genes HOXA y HOXB, lo que indica un fenotipo de células madre.[281 ,294 ] Algunas de las fusiones menos comunes afectan genes HOX.[306 ]La fusión génica NUP98::NSD1, que a menudo es críptica en el análisis citogenético, es el resultado de la fusión de NUP98 (cromosoma 11p15) con NSD1 (cromosoma 5q35).[
281 ,307 ,308 ,309 ] Esta alteración se presenta en aproximadamente el 4 % al 7 % de los casos pediátricos de LMA.[106 ,281 ,307 ,310 ,311 ] Es la fusión de NUP98 más frecuente. Este fenotipo de enfermedad presenta las siguientes características:- La frecuencia más alta de fusiones NUP98::NSD1 se observa en niños de 5 a 9 años (cerca del 8 %); la frecuencia más baja se detecta en los niños más pequeños (cerca del 2 % en niños menores de 2 años).
- Los pacientes con fusiones NUP98::NSD1 presentan un recuento alto de glóbulos blancos (GB) (mediana, 147 × 109 /l en un estudio).[
307 ,308 ] La mayoría de los pacientes con LMA y fusiones NUP98::NSD1 no tienen anomalías citogenéticas.[281 ,307 ] Hay un ligero predominio de pacientes varones con esta fusión (64,5 vs. 32,2 %).[306 ] - Un porcentaje alto de pacientes con fusiones NUP98::NSD1 (74–90 %) exhiben también LMA con FLT3-ITD.[
307 ,308 ,310 ] - En un estudio de una serie del COG, 108 niños con fusiones NUP98::NSD1 mostraron tasas más bajas de remisión completa (RC) (38 %, P < 0,001) y tasas más altas de ERM (73 %, P < 0,001), en comparación con una cohorte de pacientes sin fusiones de NUP98. Los pacientes con fusiones NUP98::NSD1 también presentaron tasas inferiores de SSC (17 vs. 47 %; P < 0,001) y de SG (36 vs. 64 %; P < 0,001), en comparación con la cohorte de referencia.[
306 ] En otro estudio en el que se incluyeron niños (n = 38) y adultos (n = 7) con LMA y fusiones NUP98::NSD1, la presencia de la fusión NUP98::NSD1 y FLT3-ITD predijo de manera independiente un pronóstico precario. Los pacientes con ambas lesiones tuvieron tasas bajas de RC (alrededor del 30 %) y de SSC a 3 años (alrededor del 15 %).[308 ] - En un estudio de niños con LMA resistente al tratamiento, la anomalía de NUP98 estuvo sobrerrepresentada en comparación con una cohorte que sí alcanzó la remisión (21 % [6 de 28 pacientes] vs. <4 %).[
312 ]
Una translocación críptica en el análisis citogenético, t(11;12)(p15;p13), produce la fusión génica NUP98::KDM5A.[
313 ] Cerca del 2 % de todos los pacientes pediátricos con LMA tienen fusiones NUP98::KDM5A, y estos casos tienden a presentarse a una edad temprana (mediana de edad, 3 años).[314 ] Las características clínicas adicionales son las siguientes:- Los casos con fusiones NUP98::KDM5A suelen ser de LMCA (34 %), seguidas de aquellos con características histológicas FAB M5 (21 %) y FAB M6 (17 %).[
314 ] Las fusiones NUP98::KDM5A se observan en alrededor del 10 % de los casos de LMA en la niñez,[266 ,297 ] y los pacientes con esta fusión suelen presentar recuentos de GB más bajos que los pacientes con fusiones NUP98::NSD1. - Otras anomalías genéticas relacionadas con la LMA en pediatría, como las variantes de FLT3, son infrecuentes en los pacientes con fusiones NUP98::KDM5A.[
314 ] - En una serie, el pronóstico para los niños con fusiones NUP98::KDM5A es inferior al de otros niños con LMA (tasa de SSC a 5 años, 29,6 % ± 14,6 %; tasa de SG, 34,1 % ± 16,1 %).[
314 ] En otro estudio en el que se incluyeron 32 pacientes con fusiones NUP98::KDM5A, se observaron tasas de RC similares a las de la población de referencia, pero tasas inferiores de SG (30 %, P < 0,001) y de SSC (25 %; P = 0,01).[306 ]
- Leucemia mieloide aguda con reordenamientos 12p13.2 (ETV6 y cualquier gen compañero de fusión). La familia de genes ETS codifica los factores de transcripción responsables del crecimiento y desarrollo celular. El gen ETV6 codifica un factor de transcripción que cumple la función de gen supresor de tumores y es el compañero de reordenamiento más frecuente de la familia ETS en la LMA en pediatría. La translocación críptica t(7;12)(Q36;p13) codifica ETV6::MNX1, la combinación más frecuente de los reordenamientos de ETV6, que se presenta en cerca del 1 % de los casos pediátricos de LMA (más frecuente en lactantes). Se relaciona con desenlaces clínicos precarios,[
315 ] y también tiene una fuerte asociación con la trisomía 19.[315 ] Es posible que la translocación sea críptica en un análisis de cariotipo convencional y, en ocasiones, solo se confirma mediante hibridación fluorescente in situ (FISH).[316 ,317 ] Esta alteración se presenta de manera casi exclusiva en niños menores de 2 años, y la mediana de edad de diagnóstico de 6 meses.[315 ] Además, se relaciona con un riesgo alto de fracaso del tratamiento.[210 ,211 ,249 ,316 ,318 ,319 ] En una revisión bibliográfica de 17 casos, se observó una tasa de SSC a 3 años del 24 % y una tasa de SG del 42 %.[219 ,315 ,320 ] - Leucemia mieloide aguda con deleción de 12p que incluye 12p13.2 (pérdida de ETV6). Las deleciones de ETV6 son muy infrecuentes en la LMA del entorno pediátrico. En una serie pediátrica, 4 de 259 pacientes (1,5 %) presentaron deleción de ETV6.[
320 ,321 ] Esta deleción se observa con más frecuencia en pacientes adultos con anomalías en el cromosoma 7 y en pacientes con variantes de TP53.[322 ] Sin embargo, en una segunda serie pediátrica, se notificó una correlación con la LMA CBF.[321 ] Según esta última serie, las tasas de riesgo de recaída en los pacientes con deleciones en ETV6 y sin estas fueron del 63 % y del 45 %, respectivamente (P = 0,3), con tasas de supervivencia sin enfermedad (SSE) correspondientes del 32 % y del 53 %, respectivamente (P = 0,2). Sin embargo, hubo una prevalencia alta de LMA CBF en pacientes con deleciones de ETV6. En el contexto de la LMA CBF, la deleción se relacionó con desenlaces adversos. Los pacientes con LMA CBF, con deleciones de ETV6 y sin estas, tuvieron tasas de SSC del 0 % y del 63 %, respectivamente (P = 0,002). Entre los pacientes con LMA CBF que lograron una RC inicial, aquellos con deleción de ETV6 tuvieron un riesgo de recaída del 88 %, en comparación con el 38 % en aquellos sin deleción (P = 0,08). Las tasas de SSE correspondientes fueron del 0 % en los pacientes con deleción de ETV6, en comparación con el 61 % en aquellos sin deleción (P = 0,009).[321 ] - Anomalías en los cromosomas 5 y 7. Las anomalías cromosómicas relacionadas con un pronóstico precario en adultos con LMA son las que afectan el cromosoma 5 (del(5q)) y el cromosoma 7 (monosomía 7).[
209 ,273 ,323 ] Estos subgrupos citogenéticos representan entre el 2 % y el 4 % de los casos pediátricos de LMA, respectivamente, y también se relacionan con un pronóstico precario en los niños.[210 ,273 ,323 ,324 ,325 ,326 ] Las anomalías en los cromosomas 5 y 7 carecen de importancia pronóstica para los pacientes de LMA con síndrome de Down de 4 años de edad y menos.[327 ]Cada vez hay más datos que demuestran que la presencia de monosomía 7 se relaciona con un riesgo mayor de que el paciente tenga variantes patogénicas germinales de GATA2, SAMD9 o SAMD9L. También se han descrito casos relacionados con un trastorno plaquetario familiar subyacente con alteración de RUNX1, con un trastorno de las características biológicas de los telómeros y con variantes patogénicas germinales de ERCC6L2.[
328 ] Deben considerarse pruebas de la línea germinal cuando se detecta la enfermedad de la monosomía 7.En el pasado, se consideró que los pacientes con del(7q) también tenían un riesgo alto de fracaso del tratamiento; además, los datos de los adultos con LMA apoyan un pronóstico precario tanto para la del(7q) como para la monosomía 7.[
212 ] Sin embargo, los desenlaces en niños con del(7q), pero sin monosomía 7, son comparables a los de otros niños con LMA.[211 ,325 ] La presencia de del(7q) no anula la importancia pronóstica de las características citogenéticas favorables (por ejemplo, inv(16) y t(8;21)).[209 ,325 ] - Leucemia mieloide aguda con reordenamientos 10p12.3 (MLLT10::cualquier gen compañero de fusión). Con frecuencia, MLLT10 forma fusiones con otros genes además de KMT2A, y estas fusiones también se relacionan con un pronóstico precario. En una revisión retrospectiva de 2226 niños inscritos en ensayos en serie del COG, se identificaron 23 niños con fusiones diferentes a KMT2A::MLLT10. Casi la mitad de los pacientes (13 de 23) tenían fusiones MLLT10::PICALM y la tasa de SSC en este grupo heterogéneo fue del 12,7 %.[
329 ] Otro estudio se centró en el efecto pronóstico de la fusión PICALM::MLLT10, que produce hematopoyesis anómala y pérdida de la regulación génica mediada por cromatina. Dentro de este subconjunto específico, los 20 pacientes pediátricos con fusiones MLLT10::PICALM tuvieron un pronóstico precario. La tasa de SSC a 5 años fue del 22 % y la tasa de SG fue del 26 %.[330 ] - Variantes de FLT3. La presencia de una variante FLT3-ITD está relacionada con un pronóstico precario en adultos con LMA,[
331 ] sobre todo cuando ambos alelos están alterados o hay una proporción alta del alelo mutado con respecto al alelo normal.[332 ] Las variantes FLT3-ITD también acarrean un pronóstico precario en los niños con LMA.[217 ,250 ,333 ,334 ,335 ] La frecuencia de las variantes FLT3-ITD en los niños es más baja que la de adultos, en particular en los niños menores de 10 años, en los que la variante se observa en el 5 % al 10 % de los casos (en comparación con casi el 30 % en los adultos).[334 ,335 ]La frecuencia de FLT3 ITD está elevada en ciertos subtipos genómicos de LMA en pediatría, incluyendo casos con el gen de fusión NUP98::NSD1, entre el 80 % y 90 % de ellos presentan de manera simultánea FLT3 ITD.[
307 ,308 ]La importancia pronóstica de FLT3-ITD se modifica por la presencia de otras alteraciones genómicas recurrentes.[
307 ,308 ] En los pacientes con FLT3-ITD, la presencia de variantes de WT1 o fusiones NUP98::NSD1 se relaciona con desenlaces más precarios (tasas de SSC inferiores al 25 %) que los de pacientes con variantes FLT3-ITD sin estas alteraciones.[219 ] Por el contrario, es posible que una fusión críptica DEK::NUP214 concurrente sea más favorable, en particular con la adición de un inhibidor de FLT3 a la quimioterapia de primera línea estándar. Cuando FLT3-ITD se presenta junto con variantes de NPM1, el desenlace es relativamente favorable y similar al de los casos pediátricos de LMA sin FLT3-ITD.[219 ] Este último subconjunto es el único escenario en el que la presencia de la variante FLT3-ITD no necesariamente hace que el riesgo de un paciente se vuelva alto, según los desenlaces favorables observados con las variantes que ocurren en forma simultánea.[219 ]En niños y adultos con LMA también se identificaron variantes activadoras de un solo nucleótido de FLT3, aunque la importancia clínica de estas variantes no está bien definida. Algunas de estas variantes de un solo nucleótido son específicas de pacientes pediátricos.[
219 ] - Fenotipo RAM. El fenotipo RAM se caracteriza por expresión de CD56 de alta intensidad, expresión de CD45 y CD38 de atenuada a negativa y ausencia de expresión de HLA-DR. Estos pacientes suelen ser más jóvenes, con una mediana de edad de 1,6 años en las series notificadas inicialmente. Este fenotipo se encuentra con más frecuencia en pacientes de LMCA no relacionada con el síndrome de Down.[
336 ] Desde el punto de vista clínico, los pacientes con fenotipo RAM tienen desenlaces inferiores. En la serie inicial, los pacientes de la cohorte RAM tuvieron una tasa de SSC a 3 años del 16 %, en comparación con el 51 % en los pacientes de la cohorte que no presentaba fenotipo RAM (P < 0,001). Los pacientes de la cohorte RAM también tuvieron una supervivencia inferior en comparación con los pacientes con expresión alta de CD56, que carecían de otras características fenotípicas de RAM. La SG también fue inferior en comparación con los pacientes sin el fenotipo RAM (26 vs. 69 %, P = 0,001). En un subanálisis, la SG de los pacientes de la cohorte RAM también fue mucho más precaria que la de los pacientes de la cohorte positiva para CD56 (sin fenotipo RAM) (26 vs. 66 %, P < 0,001) y de la cohorte negativa para CD56 (26 vs. 70 %, P < 001).[336 ] Muchos pacientes con fenotipo RAM, pero no todos, tienen indicios de una fusión CBFA2T3::GLIS2 que, en sí misma, confiere un riesgo muy alto. En una serie publicada, se encontró que alrededor del 60 % de los pacientes con fenotipo RAM en el momento del diagnóstico tenían esta fusión críptica que también confiere enfermedad de riesgo más alto.[336 ]
Anomalías citogenéticas o moleculares adicionales que quizás tengan importancia pronóstica
En esta sección se incluyen anomalías citogenéticas o moleculares que se observan en el momento del diagnóstico y que no afectan la estratificación del riesgo de la leucemia mieloide aguda (LMA), pero que pueden tener importancia pronóstica.
- Leucemia mieloide aguda con fusiones génicas RUNX1::CBFA2T3 (t(16;21)(q24;q22)). En las leucemias con t(16;21)(q24;q22), el gen RUNX1 se fusiona con el gen CBFA2T3 y el perfil de expresión génica está estrechamente relacionado con el de los casos de LMA con t(8;21) y fusiones RUNX1::RUNX1T1.[
291 ] Los pacientes presentan la enfermedad a una mediana de edad de 7 años. Este cáncer es poco frecuente, representa del 0,1 % al 0,3 % de los casos pediátricos de LMA. De 23 pacientes con fusiones RUNX1::CBFA2T3, 5 presentaron LMA secundaria, y 2 de ellos tenían diagnóstico primario de sarcoma de Ewing. Los desenlaces fueron favorables en la cohorte de 23 pacientes; la tasa de SSC a 4 años fue del 77 % y la incidencia acumulada de recaída fue del 0 %.[291 ] - Variantes de RAS. Aunque se identificaron variantes de RAS en un 20 % a un 25 % de los pacientes con LMA, no se ha demostrado de forma clara la importancia pronóstica de estas variantes.[
249 ,337 ] Las variantes de NRAS se observan con más frecuencia que las variantes de KRAS en los casos pediátricos de LMA.[249 ,338 ] Las variantes de RAS se presentan con una frecuencia similar en todos los subtipos de alteración de tipo II, a excepción de la LPA, en la que rara vez se observan variantes de RAS.[249 ] - Leucemia mieloide aguda con fusiones génicas RBM15::MRTFA. La translocación t(1;22)(p13;q13) que produce las fusiones RBM15::MRTFA (también conocidas como RBM15::MKL1) es infrecuente (<1 % de la LMA en pediatría) y se limita a la LMCA.[
210 ,298 ,339 ,340 ,341 ,342 ] En estudios se observó que la t(1;22)(p13;q13) se encuentra en el 10 % al 20 % de los niños con LMCA en quienes se pueden evaluar las características citogenéticas o de genética molecular.[265 ,266 ,297 ,299 ] La mayoría de los casos de LMCA con t(1;22) se presentan en lactantes con una mediana de edad en el momento del cuadro clínico inicial (4 a 7 meses) menor que la de otros niños con LMCA.[265 ,294 ,299 ,343 ] También se han notificado casos en los que se detectan los transcritos de la fusión RBM15::MKL1 en ausencia de t(1;22) porque estos pacientes jóvenes por lo general tienen una médula ósea hipoplásica.[340 ]En un estudio retrospectivo de colaboración internacional de 51 casos con t(1;22), se informó que los pacientes con esta anomalía tuvieron una tasa de SSC a 5 años del 54,5 % y una tasa de SG del 58,2 %, similar a las tasas de otros niños con LMCA.[
265 ] En otro análisis retrospectivo internacional de 153 casos de LMCA sin síndrome de Down para los que se contaba con muestras para análisis molecular, la tasa de SSC a 4 años en los pacientes con t(1;22) fue del 59 % y la tasa de SG fue del 70 %; estas fueron significativamente mejores que las de los pacientes con LMCA que tenían otras anomalías genéticas específicas (fusiones CBFA2T3::GUS2, fusiones NUP98::KDM5A, reordenamientos de KMT2A y monosomía 7).[297 ] Se observaron desenlaces similares en los ensayos de fase III del COG AAML0531 y AAML1031 (tasas de SG a 5 años, 86 % ± 26 % [n = 7] y 54 % ± 14 % [n = 14] en el AAML0531 y el AAML1031, respectivamente).[299 ] - Reordenamientos de HOX. En un informe, los casos que tenían una fusión génica que afectaba el complejo génico HOX representaron un 15 % de las LMCA infantiles.[
266 ] En el informe se observó que estos pacientes tienen un pronóstico relativamente favorable, aunque el pequeño número de casos estudiados restringe la confianza de esta evaluación. - Variantes de GATA1. Las variantes interruptoras en GATA1 en la LMCA sin síndrome de Down se presentan en niños pequeños (mediana de edad, 1–2 años) y se relacionan con la amplificación del gen RCAN1 en el cromosoma 21.[
266 ] Estos pacientes representan cerca del 10 % de las LMCA sin síndrome de Down y tienen un pronóstico favorable cuando no se presentan, de manera simultánea, genes de fusión con pronóstico desfavorable; aunque el número de pacientes estudiados fue bajo (n = 8).[266 ] - Hipodiploidía. La hipodiploidía se define como un número modal de cromosomas igual o inferior a 45. Esto ocurre con poca frecuencia en pacientes pediátricos con LMA. En un análisis retrospectivo de cohortes, el grupo del iBFM que estudia la LMA tuvo como objetivo caracterizar la hipodiploidía en pacientes pediátricos con LMA. En el estudio se excluyeron varios grupos de pacientes, incluso aquellos con LPA, síndrome de Down o pérdida del cromosoma 7.[
344 ] Se notificaron las siguientes observaciones:- Se observó hipodiploidía en el 1,3 % de los niños con LMA. Cerca del 80 % de los pacientes tenían un número modal de cromosomas de 45, y el 20 % restante tenía un número modal de cromosomas de 43 o 44.
- La mayoría de los pacientes (>80 %) con un número modal de cromosomas de 43 o 44 también cumplían con los criterios de cariotipo complejo. En este estudio, el cariotipo complejo se definió como la presencia de por lo menos 3 alteraciones cromosómicas diferentes, ya fueran estructurales o defectos en el número de cromosomas, además de la ausencia de las anomalías recurrentes definidas por la OMS.
- Los pacientes con un número modal de cromosomas de 43 o 44 presentaron tasas de SSC y SG inferiores, en comparación con los pacientes con 45 cromosomas (tasa de SSC, 21 vs. 37 %; P = 07; tasa de SG, 33 vs. 56 %; P = 0,1).
- Duplicación en tándem de UBTF. El gen UBTF se encuentra en el cromosoma 17q21.31 y codifica una proteína nucleolar que interactúa con el DNA ribosómico para intervenir en la transcripción del RNA ribosómico de la RNA polimerasa 1.[
345 ]- La duplicación en tándem de UBTF (UBTF-TD) es mutuamente excluyente de otras alteraciones genómicas oncoiniciadoras de la leucemia. Al igual que otros oncoiniciadores leucémicos, persiste en el momento de la recaída.
- Se observan alteraciones genómicas de UBTF que causan variantes somáticas heterocigotas que producen duplicación en tándem del exón 13 de UBTF en aproximadamente el 4 % de los casos pediátricos de LMA.
- La LMA con UBTF-TD en la población pediátrica se presenta sobre todo durante la adolescencia (mediana de edad, 12–14 años). También se observa en adultos menores de 60 años, pero es infrecuente en la LMA de pacientes adultos mayores.
- La FLT3-ITD es común en los casos de LMA con UBTF-TD. Cerca de dos tercios de los casos tienen FLT3-ITD. Además, alrededor del 40 % de los casos de LMA con UBTF-TD tienen variantes de WT1.
- En el ensayo clínico AAML1031, las tasas de SSC y SG en los pacientes con UBTF-TD fueron del 30 % y del 44 %, respectivamente. Estos valores fueron más bajos que los de los pacientes inscritos en el AAML1031 que no tenían UBTF-TD (45 % y 64 %, respectivamente). El desenlace de los pacientes con UBTF-TD fue similar al de los pacientes con reordenamientos de KMT2A.
- En el ensayo AAML1031, la presencia de UBTF-TD junto con FLT3-ITD o variantes de WT1 se relacionó con un pronóstico inferior en comparación con el de los pacientes con UBTF-TD solo.
- Leucemia mieloide aguda con inserciones CBFB::GDXY. El gen CBFB codifica la proteína CBFB que forma parte del complejo multiproteico del factor de transcripción de unión nuclear, que regula un programa de expresión génica fundamental para la hematopoyesis. El gen CBFB se fusiona de forma recurrente con MYH11 en la LMA con inv(16)/t(16;16).[
346 ]- Se identificaron inserciones en el exón 3 de CBFB en cerca del 0,4 % de los casos pediátricos de LMA en el momento del diagnóstico. Todas las inserciones descritas producen la sustitución del ácido aspártico en la posición 87 (D87) por glicina, ácido aspártico, serina y tirosina (GDSY) o glicina, ácido aspártico, treonina y tirosina (GDTY).
- Las inserciones CBFB::GDXY se relacionan con un perfil de expresión génica que se superpone con la LMA que expresa CBFB::MYH11, a excepción de un aumento de la expresión de genes de células madre como MEIS1 y los genes de grupo HOXA.
- Las inserciones CBFB::GDXY con frecuencia se producen junto con las variantes de BCOR1 y en el dominio tirosina–cinasa (TKD) de FLT3, pero en ausencia de las variantes de KIT, que se encuentran con frecuencia en la LMA con CBFB::MYH11.
- Las inserciones CBFB::GDXY son más comunes en adolescentes y adultos jóvenes.
- El efecto de las inserciones CBFB::GDXY en los desenlaces de los pacientes no está claro debido a la escasez de datos. Sin embargo, el análisis preliminar indica que es posible que estos pacientes no tengan el mismo desenlace favorable que los pacientes con fusiones CBFB::MYH11.
- Variantes de RUNX1. La LMA con variante de RUNX1 fue una entidad provisional en la clasificación de la OMS de 2016. En la 5ª edición de la clasificación de la OMS, se incluye en la categoría de LMA con otras alteraciones genéticas definidas.[
223 ] Este subtipo de LMA es más común en adultos que en niños. En los adultos, la variante de RUNX1 se relaciona con un riesgo alto de fracaso del tratamiento. En un metanálisis de los desenlaces de pacientes adultos con variantes de RUNX1 también se demostró la enfermedad de riesgo alto, aunque esta significación estadística no se observó en el contexto de características citogenéticas de riesgo intermedio.[347 ]En un estudio de niños con LMA, se observaron variantes de RUNX1 en 11 de 503 pacientes (cerca del 2 %). De los 11 pacientes, 6 con LMA y variante de RUNX1 no alcanzaron la remisión y la tasa de SSC a 5 años fue del 9 %, lo que indica que la variante de RUNX1 confiere un pronóstico precario tanto en niños como en adultos.[
348 ] Sin embargo, en un segundo estudio de 488 niños con LMA, en el que 23 de ellos tenían variantes de RUNX1, no se observó un efecto significativo de las variantes de RUNX1 sobre la respuesta o el desenlace. Además, en el análisis se identificó que los pacientes pediátricos con variantes de RUNX1 eran con mayor frecuencia varones, adolescentes, y que tenían una incidencia más alta de FLT3-ITD y otras variantes. Sin embargo, en los análisis univariantes y multivariantes de cada uno de estos grupos, no se encontraron diferencias en la supervivencia según la presencia de variantes de RUNX1.[349 ] Las variantes de RUNX1 producen un trastorno plaquetario familiar con neoplasia maligna mieloide relacionada (FPD-MM).[223 ] - Variantes de WT1. La WT1 es una proteína con dedos de cinc que regula la transcripción génica y que está alterada en alrededor del 10 % de los casos de LMA con características citogenéticas normales en adultos.[
350 ,351 ,352 ,353 ] En algunos estudios,[350 ,351 ,353 ] pero no en todos,[352 ] se encontró que la variante de WT1 fue un factor de predicción independiente de SSE, SSC y SG más precarias en pacientes adultos.En los niños con LMA, se observan variantes de WT1 en cerca del 10 % de los casos.[
354 ,355 ] Los casos con variantes de WT1 ocurren con mucha frecuencia en niños con características citogenéticas normales y FLT3-ITD, pero son menos comunes en niños menores de 3 años.[354 ,355 ] Los casos de LMA con fusiones NUP98::NSD1 tienen abundantes variantes FLT3-ITD y variantes de WT1.[307 ] En los análisis univariantes, las variantes de WT1 predicen un desenlace más precario en los pacientes pediátricos, pero la importancia pronóstica independiente del estado de la variante de WT1 no está clara debido a su fuerte relación con FLT3-ITD y su relación con la fusión NUP98::NSD1.[307 ,354 ,355 ] En el estudio más grande sobre variantes de WT1 en niños con LMA, se observó que estos niños que tenían variantes de WT1 pero no exhibían FLT3-ITD presentaron desenlaces similares a los de los niños que no tenían variantes de WT1, mientras que aquellos que tenían al mismo tiempo una variante de WT1 y FLT3-ITD presentaron tasas de supervivencia inferiores al 20 %.[354 ]En un estudio de niños con LMA resistente al tratamiento, la variante de WT1 se encontraba sobrerrepresentada, en comparación con una cohorte que alcanzó la remisión (54 % [15 de 28 pacientes] vs. 15 %).[
312 ] - Variantes de DNMT3A. Se identificaron variantes del gen DNMT3A en cerca del 20 % de los pacientes adultos con LMA. Estas variantes son infrecuentes en los pacientes con características citogenéticas favorables, pero se presentan en un tercio de los adultos con características citogenéticas de riesgo intermedio.[
356 ] Las variantes de este gen se relacionan de forma independiente con un desenlace precario.[356 ,357 ,358 ] Las variantes de DNMT3A están prácticamente ausentes en los niños.[359 ] - Variantes de IDH1 e IDH2. Las variantes de IDH1 e IDH2, que codifican la isocitrato–deshidrogenasa, se presentan en alrededor del 20 % de los adultos con LMA [
220 ,360 ,361 ,362 ,363 ,364 ] y son muy frecuentes en pacientes con variantes de NPM1.[361 ,362 ,365 ] Las variantes específicas que se presentan en IDH1 e IDH2 crean una actividad enzimática nueva que promueve la conversión del α-cetoglutarato en 2-hidroxiglutarato.[366 ,367 ] Esta actividad nueva induce un fenotipo de hipermetilación del DNA similar al que se observa en los casos de LMA con variantes de pérdida de la función en TET2.[365 ]Las variantes de IDH1 e IDH2 son infrecuentes en la LMA del entorno pediátrico; se presentan en un 0 % a un 4 % de los casos.[
220 ,359 ,368 ,369 ,370 ,371 ,372 ] No hay indicios de que las variantes de IDH1 e IDH2 acarreen un efecto pronóstico negativo en niños con LMA.[220 ,368 ] - Variantes de CSF3R. El gen CSF3R codifica el receptor del factor estimulante de colonias de granulocitos (G-CSF), y se observan variantes activadoras de CSF3R en un 2 % a un 3 % de los casos pediátricos de LMA.[
373 ] Estas variantes aumentan la señalización mediada por el receptor G-CSF. Se observan sobre todo en la LMA con variantes de CEBPA o anomalías de CBF (fusiones RUNX1::RUNX1T1 y CBFB::MYH11).[373 ] En un estudio de 2150 pacientes pediátricos con LMA, se encontró que 35 pacientes (1,6 %) tenían variantes de CSF3R; 30 de estos casos (89 %) tenían fusiones RUNX1::RUNX1T1 (n = 18) o variantes de CEBPA (n = 12).[262 ] El riesgo de recaída fue significativamente más alto en los pacientes con variantes simultáneas en CSF3R y CEBPA, en comparación con los pacientes con fusiones RUNX1::RUNX1T1 y variantes de CSF3R.[262 ] Si bien las tasas de recaídas en los pacientes de LMA con variantes simultáneas en CSF3R y CEBPA son más elevadas, la supervivencia general no se afecta de manera desfavorable, lo que indica una tasa de rescate alta con la terapia de reinducción y el TCMH.[253 ]También se observan variantes activadoras de CSF3R en los pacientes con neutropenia congénita grave. Estas variantes no causan la neutropenia congénita grave; más bien, surgen como variantes somáticas y pueden reflejar un paso inicial en la vía que lleva a la LMA.[
374 ] En un estudio de pacientes con neutropenia congénita grave, el 34 % de los pacientes que no tenían una neoplasia maligna mieloide exhibieron variantes de CSF3R detectables en neutrófilos de sangre periférica y células mononucleares, mientras que el 78 % de los pacientes con neoplasia maligna mieloide exhibieron variantes de CSF3R.[374 ] En un estudio de 31 pacientes con neutropenia congénita grave que presentaron LMA o síndrome mielodisplásico, se observaron variantes de CSF3R en cerca del 80 % de los pacientes. En el estudio también se observó una frecuencia alta de variantes de RUNX1 (cerca del 60 %), lo que indica que la presencia de variantes simultáneas en CSF3R y RUNX1 cumple una función en la formación de la leucemia en el contexto de neutropenia congénita grave.[375 ]
Para obtener información sobre el tratamiento de la LMA infantil, consultar
Leucemia promielocítica aguda
Proteínas de fusión RARA
La anomalía cromosómica característica relacionada con la leucemia promielocítica aguda (LPA) es t(15;17)(q22;q21). Esta translocación afecta un punto de ruptura que incluye el receptor del ácido retinoico y conduce a la producción de la proteína de fusión PML::RARA.[
El diagnóstico de los pacientes, en quienes se sospecha una LPA, se confirma cuando se detecta la proteína de fusión PML::RARA (por ejemplo, mediante hibridación fluorescente in situ [FISH], reacción en cadena de la polimerasa con retrotranscripción [PCR-RT] o análisis citogenéticos convencionales). La PCR-RT cuantitativa permite identificar las tres variantes frecuentes de los transcritos, y se usa para vigilar la respuesta al tratamiento y detectar de manera precoz una recidiva molecular.[
Las variantes moleculares poco frecuentes de la LPA producen proteínas de fusión por la unión de genes específicos (por ejemplo, PLZF, NPM, STAT5B y NuMA) con RARA.[
- Variante con el gen de fusión PLZF::RARA. La variante con la fusión PLZF::RARA, caracterizada por la t(11;17)(q23;q21), representa alrededor del 0,8 % de la LPA, que además expresa CD56 de superficie y tiene gránulos muy finos, en comparación con la LPA que presenta la t(15;17).[
384 ,385 ,386 ] La LPA con el gen de fusión PLZF::RARA se ha relacionado con un pronóstico precario y, por lo general, no responde a la tretinoína ni al trióxido de arsénico.[383 ,384 ,385 ,386 ] - Variantes con los genes de fusión NPM::RARA o NuMA::RARA. Es posible que las variantes poco frecuentes de LPA que tienen las translocaciones NPM::RARA (t(5;17)(q35;q21)) o NuMA::RARA (t(11;17)(q13;q21)) respondan bien a la tretinoína.[
383 ,387 ,388 ,389 ,390 ] - Variante con el gen de fusión PML::RARA. En escasas ocasiones, se notificaron casos de pacientes con LPA negativa para la fusión PML::RARA. Una de estas LPA es el subtipo producido por el torque teno minivirus (TTMV).[
345 ,391 ,392 ] Esta es una entidad descrita de manera reciente en la que el genoma del TTMV se integra en el intrón 2 del gen RARA, lo que resulta en una fusión TTMV::RARA. Las características clínicas y morfológicas de este subtipo de LPA son similares a las de la LPA positiva para la fusión PML::RARA.
Variantes deFLT3
Se observan variantes de FLT3 (duplicación interna en tándem o variantes en el dominio de tirosina–cinasas) en un 40 % a un 50 % de los casos de LPA. La presencia de variantes de FLT3 se correlacionó con un recuento más alto de glóbulos blancos y con el subtipo de la variante microgranular (M3v).[
Para obtener información sobre el tratamiento de la leucemia promielocítica aguda infantil, consultar
Leucemia mieloide crónica
Características genómicas de la leucemia mieloide crónica
La anomalía citogenética necesaria para el diagnóstico de la leucemia mieloide crónica (LMC) es el cromosoma Filadelfia (Ph), una translocación de los cromosomas 9 y 22 (t(9;22)) que produce la proteína de fusión BCR::ABL1.[
Se han encontrado otras anomalías cromosómicas en estudios de adultos con LMC desde que se comenzaron a usar los inhibidores de tirosina–cinasas. En estos estudios se han descrito varias variantes de pronóstico adverso, incluso aquellas que se identificaron como de riesgo alto en la fase crónica.[
Para obtener información en inglés sobre el tratamiento de la leucemia mieloide crónica, consultar
Leucemia mielomonocítica juvenil
Características moleculares de la leucemia mielomonocítica juvenil
El panorama genómico de la leucemia mielomonocítica juvenil (LMMJ) se caracteriza por variantes de 1 de los 5 genes de la vía RAS: NF1, NRAS, KRAS, PTPN11 y CBL.[
La tasa de variantes de las células leucémicas en la JMML es muy baja, pero se observan variantes adicionales de genes diferentes a los 5 genes de la vía RAS descritos antes.[
En un informe en el que se describe el panorama genómico de la LMMJ se encontró que 16 de 150 pacientes (11 %) carecían de variantes canónicas de la vía RAS. De esos 16 pacientes, 3 presentaban fusiones en el marco de lectura que afectaban receptores tirosina–cinasas (fusiones génicas DCTN1::ALK, RANBP2::ALK y TBL1XR1::ROS1). Todos estos pacientes presentaban monosomía 7 y tenían 56 meses o más. Un paciente con fusión del gen ALK se trató con crizotinib y quimioterapia convencional, logró una remisión molecular completa, luego se sometió a un trasplante alogénico de médula ósea.[
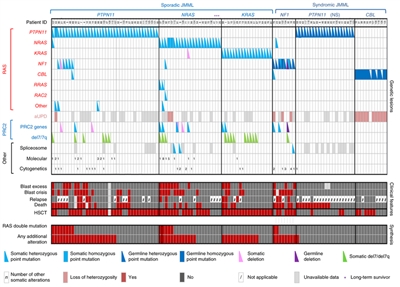
Factores pronósticos genómicos y moleculares
Varios factores genómicos afectan el pronóstico de los pacientes con LMMJ; entre ellos, los siguientes:
- Número de variantes fuera de la vía RAS. Un factor de pronóstico de los niños con LMMJ es el número de variantes diferentes de las variantes de la vía RAS que son definitorias de enfermedad.[
404 ,405 ]- En un estudio se observó que, en el momento del diagnóstico, se encontraron entre 0 a 1 alteraciones somáticas (variante patógena o monosomía 7) en 64 pacientes (65,3 %), mientras que se encontraron 2 o más alteraciones en 34 pacientes (34,7 %).[
405 ] En el análisis multivariante, el número de variantes (2 o más vs. 0 a 1) conservó la significación como factor de pronóstico de supervivencia sin complicaciones (SSC) y supervivencia general (SG) más precarias. Una mayor proporción de pacientes con diagnóstico de 2 o más alteraciones tenían más edad y eran varones; estos pacientes también exhibieron una tasa más alta de monosomía 7 o variantes somáticas de NF1.[405 ] - En otro estudio se observó que alrededor del 60 % de los pacientes tenían 1 o más variantes adicionales a la variante de la vía RAS que son definitorias de la enfermedad. Estos pacientes tenían una SG inferior en comparación con los pacientes que no tenían variantes adicionales (tasa de SG a 3 años, 61 vs 85 %, respectivamente).[
404 ] - En un tercer estudio se observó una tendencia hacia una SG inferior para los pacientes con 2 variantes o más en comparación con los pacientes con 1 o ninguna variante.[
406 ]
- En un estudio se observó que, en el momento del diagnóstico, se encontraron entre 0 a 1 alteraciones somáticas (variante patógena o monosomía 7) en 64 pacientes (65,3 %), mientras que se encontraron 2 o más alteraciones en 34 pacientes (34,7 %).[
- Variantes dobles de la vía RAS. A pesar de que las variantes de los 5 genes canónicos de la vía RAS relacionados con la LMMJ (NF1, NRAS, KRAS, PTPN11, y CBL) son por lo general mutuamente excluyentes, el 4 % al 17 % de los casos presentan variantes de 2 de estos genes de la vía RAS.[
404 ,405 ] Este hallazgo se relacionó con un pronóstico más precario.[404 ,405 ]- En un informe se identificaron 2 variantes de la vía RAS en el 11 % de los pacientes de LMMJ, y estos pacientes tuvieron una tasa de SSC significativamente inferior (14 %) en comparación con la de los pacientes que tenían una sola variante de la vía RAS (62 %). Los pacientes con el síndrome de Noonan se excluyeron del análisis.[
405 ] - Se informaron resultados similares en un segundo estudio. En este estudio se observó que los pacientes con variantes dobles de la vía RAS (15 de 96 pacientes) tenían tasas de supervivencia más bajas que los pacientes sin variantes adicionales o con variantes adicionales a la variante de la vía RAS.[
404 ]
- En un informe se identificaron 2 variantes de la vía RAS en el 11 % de los pacientes de LMMJ, y estos pacientes tuvieron una tasa de SSC significativamente inferior (14 %) en comparación con la de los pacientes que tenían una sola variante de la vía RAS (62 %). Los pacientes con el síndrome de Noonan se excluyeron del análisis.[
- Perfil de metilación del DNA.
- En un estudio se aplicó un perfil de metilación del DNA a una cohorte de descubrimiento de 39 pacientes de LMMJ y a una cohorte de validación de 40 pacientes. En ambas cohortes se observaron subgrupos de LMMJ característicos con grados de metilación alto, medio o bajo. Los pacientes con los grados de metilación más bajos tuvieron las tasas de supervivencia más altas, y en la cohorte de metilación baja todos menos 1 de los 15 pacientes presentaron resolución espontánea. El estado de metilación alta se relacionó con tasas más bajas de SSC.[
408 ] - En otro estudio se aplicó un perfil de metilación del DNA a una cohorte de 106 pacientes de LMMJ. En el estudio se observó un subgrupo de pacientes con un perfil de hipermetilación y un subgrupo de pacientes con un perfil de hipometilación. Los pacientes del grupo de hipermetilación tuvieron una SG significativamente más baja que la de los pacientes del grupo de hipometilación (tasa de SG a 5 años, 46 vs. 73 %, respectivamente). Los pacientes en el grupo de hipermetilación también tuvieron una tasa de supervivencia sin trasplante a 5 años significativamente más precaria que la de los pacientes del grupo de hipometilación (2,2 %, IC 95 %, 0,2–10,1 vs. 41,2 %; IC 95 %, 27,1–54,8 %). El estado de hipermetilación se relacionó con 2 o más variantes, concentraciones más altas de hemoglobina fetal, mayor edad y recuento de plaquetas más bajo en el momento del diagnóstico. Todos los pacientes con síndrome de Noonan estaban en el grupo de hipometilación.[
406 ] - En un estudio se examinaron 33 pacientes de LMMJ que tenían variantes de CBL. En el estudio se identificaron 31 pacientes con metilación baja y 2 pacientes con metilación intermedia. Los 2 niños con metilación intermedia recayeron después de someterse a TCMH. Debido a que el tratamiento, que incluyó la observación sola, varió entre los 31 pacientes con metilación baja, no se pudo valorar por completo el efecto del perfil de metilación en las decisiones terapéuticas y los desenlaces. Sin embargo, el estado de metilación no fue pronóstico de resolución espontánea.[
409 ]
- En un estudio se aplicó un perfil de metilación del DNA a una cohorte de descubrimiento de 39 pacientes de LMMJ y a una cohorte de validación de 40 pacientes. En ambas cohortes se observaron subgrupos de LMMJ característicos con grados de metilación alto, medio o bajo. Los pacientes con los grados de metilación más bajos tuvieron las tasas de supervivencia más altas, y en la cohorte de metilación baja todos menos 1 de los 15 pacientes presentaron resolución espontánea. El estado de metilación alta se relacionó con tasas más bajas de SSC.[
- Sobreexpresión de LIN28B. La sobreexpresión de LIN28B se presenta en casi la mitad de los niños con LMMJ e identifica un subgrupo de LMMJ distintivo desde el punto de vista biológico. La LIN28B es una proteína de unión al RNA que regula la renovación de células madre.[
410 ]- La sobreexpresión de LIN28B se correlacionó de forma directa con las concentraciones sanguíneas altas de hemoglobina fetal y la edad (ambos relacionados con un pronóstico más precario), y se correlacionó de forma inversa con monosomía 7 (también relacionada con un pronóstico más precario). Aunque la sobreexpresión de LIN28B permite identificar un subconjunto de pacientes con aumento de riesgo de fracaso del tratamiento, se encontró que no era un factor de pronóstico independiente cuando se consideran otros factores como la edad o la monosomía 7.[
410 ] - En otro estudio también se observó un subgrupo de pacientes de LMMJ con aumento de la expresión de LIN28B. En el estudio se identificó a LIN28B como el gen cuya expresión se relacionó de manera más contundente con el estado de hipermetilación.[
406 ]
- La sobreexpresión de LIN28B se correlacionó de forma directa con las concentraciones sanguíneas altas de hemoglobina fetal y la edad (ambos relacionados con un pronóstico más precario), y se correlacionó de forma inversa con monosomía 7 (también relacionada con un pronóstico más precario). Aunque la sobreexpresión de LIN28B permite identificar un subconjunto de pacientes con aumento de riesgo de fracaso del tratamiento, se encontró que no era un factor de pronóstico independiente cuando se consideran otros factores como la edad o la monosomía 7.[
Para obtener información sobre el tratamiento de la LMMJ, consultar PDQ
Neoplasias mielodisplásicas
Características moleculares de las neoplasias mielodisplásicas
Las neoplasias mielodisplásicas, tradicionalmente conocidas como síndromes mielodisplásicos (SMD), que se presentan durante la niñez se caracterizan por un conjunto específico de alteraciones genéticas, a diferencia de lo que ocurre con las neoplasias mielodisplásicas que se presentan durante la adultez. En adultos, las neoplasias mielodisplásicas a menudo surgen a partir de una hematopoyesis clonal y se caracterizan por la presencia de variantes de TET2, DNMT3A, DDX41 y TP53. Por el contrario, las variantes de estos genes son poco frecuentes en las neoplasias mielodisplásicas que se presentan durante la niñez, aunque en subgrupos de niños con esta patología se observan variantes de los genes GATA2, SAMD9, SAMD9L, ETV6, SETBP1, ASXL1 y de la vía RAS/MAPK.[
En un informe del panorama genómico de las neoplasias mielodisplásicas infantiles se describieron los resultados de la secuenciación del exoma completo de 32 pacientes pediátricos con neoplasias mielodisplásicas primarias infantiles y de la secuenciación dirigida de otros 14 casos.[
- Se observaron variantes de los genes de la vía RAS/MAPK en el 43 % de los casos de neoplasias mielodisplásicas primarias; las más comunes afectaban los genes PTPN11 y NRAS, pero también se observaron variantes de otros genes de la vía RAS/MAPK (por ejemplo, BRAF [diferentes a la variante BRAF V600E], CBL y KRAS). Las variantes de la vía RAS/MAPK fueron más comunes en pacientes con aumento de blastocitos (65 %) que en aquellos con número bajo de blastocitos (17 %).
- Se observaron variantes patogénicas germinales en SAMD9 (n = 4) o en SAMD9L (n = 4) en el 17 % de los pacientes con neoplasias mielodisplásicas primarias, donde 7 de las 8 variantes ocurrieron en pacientes con número bajo de blastocitos. En todos estos casos se observó una pérdida de material en el cromosoma 7. Alrededor del 40 % de los pacientes con deleciones de parte o de la totalidad del cromosoma 7 presentaron variantes germinales en SAMD9 o en SAMD9L.
- Se observaron variantes patogénicas de GATA2 en 3 casos (7 %), y en todos ellos se confirmó o se sospechó que eran germinales.
- Las alteraciones en el número de copias más comunes fueron las deleciones en el cromosoma 7, que se observaron en el 41 % de los casos. La pérdida parcial o total del cromosoma 7 fue más frecuente en los casos con alteraciones en los genes SAMD9 y SAMD9L (100 %), y en los pacientes con aumento de blastocitos que presentaban variante de la vía RAS/MAPK (71 %).
- En más de 1 de los 46 casos, se presentó alteración en otros genes (SETBP1, ETV6 y TP53).
En un segundo informe se describió la utilización de un perfil de secuenciación dirigida de 105 genes en 50 pacientes pediátricos con neoplasias mielodisplásicas (pacientes con número bajo de blastocitos = 31 y pacientes con aumento de blastocitos = 19) entre los que se encontraron abundantes casos con monosomía 7 (48 %).[
- Se observaron variantes patogénicas germinales de GATA2 en el 30 % de los pacientes y variantes patogénicas germinales de RUNX1 en el 6 % de los pacientes.
- Se observaron variantes somáticas en el 34 % de los pacientes y fueron más comunes en pacientes con aumento de blastocitos que en pacientes con número bajo de blastocitos (68 vs. 13 %).
- El gen que presentó alteración con más frecuencia fue SETBP1 (18 %). Los genes que presentaron alteración con menor frecuencia fueron ASXL1, RUNX1 y los genes de la vía RAS/MAPK (PTPN11, NRAS, KRAS, NF1). Se encontraron variantes de los genes de la vía RAS/MAPK en el 12 % de los casos.
Los pacientes con variantes patogénicas germinales de GATA2, además de neoplasias mielodisplásicas, exhiben un amplio abanico de defectos hematopoyéticos e inmunológicos así como manifestaciones no hematopoyéticas.[
Se estudiaron las variantes patogénicas germinales de GATA2 de 426 pacientes pediátricos con neoplasias mielodisplásicas primarias y 82 casos con neoplasias mielodisplásicas secundarias que participaron en estudios consecutivos del European Working Group of MDS in Childhood (EWOG-MDS).[
- En el 7 % de los pacientes pediátricos con neoplasias mielodisplásicas primarias se identificaron variantes patogénicas germinales de GATA2. Si bien la mediana de edad de los pacientes con variantes de GATA2 fue de 12,3 años en la población pediátrica del EWOG-MDS, la mayor parte de los casos de neoplasias mieloides relacionadas con la línea germinal en GATA2 se presentan durante la edad adulta.[
415 ] - Las variantes de GATA2 fueron más comunes en los pacientes con aumento de blastocitos (15 %) que en los pacientes con número bajo de blastocitos (4 %).
- Entre los pacientes con variantes de GATA2, el 46 % presentaba aumento de blastocitos y el 70 % exhibió monosomía 7.
- Se identificaron neoplasias mielodisplásicas o leucemia mieloide aguda (LMA) familiar en 12 de los 53 pacientes con variantes de GATA2 para los que se disponía de una historia familiar detallada.
- Los fenotipos no hematológicos de la deficiencia de GATA2 se presentaron en el 51 % de los pacientes con neoplasias mielodisplásicas y variantes de GATA2, e incluyeron hipoacusia (9 %), linfedema o hidrocele (23 %) e inmunodeficiencia (39 %).
Las variantes patogénicas germinales en SAMD9 y SAMD9L se relacionan con casos de neoplasias mielodisplásicas con pérdida adicional, total o parcial, del cromosoma 7.[
En 2016, se identificó el gen SAMD9 como la causa del síndrome MIRAGE (mielodisplasia, infección, restricción del crecimiento, hipoplasia suprarrenal, fenotipos genitales y enteropatía) que se relaciona con las neoplasias mielodisplásicas de aparición temprana con monosomía 7.[
- Las variantes causales de SAMD9 y SAMD9L son variantes de ganancia de función que intensifican la actividad supresora del crecimiento de SAMD9 o SAMD9L.[
418 ,420 ] - Los genes SAMD9 y SAMD9L están en el cromosoma 7q21.2. Los casos de neoplasias mielodisplásicas en pacientes con variantes de SAMD9 o SAMD9L a menudo exhiben monosomía 7 y el otro cromosoma 7 tiene SAMD9 y SAMD9L naturales. Esto lleva a que se pierda el efecto intensificador del gen alterado en la actividad supresora del crecimiento.
- Los pacientes con fenotipo normal, variantes de SAMD9 o SAMD9L y monosomía 7, a veces, presentan neoplasias mielodisplásicas o LMA, o por el contrario, pierden la monosomía 7 y su hematopoyesis se normaliza.[
420 ] El primer caso se relaciona con la aparición de variantes de los genes vinculados con las neoplasias mielodisplásicas o la LMA (por ejemplo, ETV6 o SETBP1). El segundo caso se relaciona con modificaciones genéticas (por ejemplo, variantes inversas o pérdida de la heterocigosidad sin cambio en el número de copias con retención del alelo natural) que llevan a la normalización de la actividad de SAMD9 y SAMD9L. Estas observaciones indican que el seguimiento de pacientes con monosomía 7 relacionada con SAMD9 o SAMD9L mediante la secuenciación clínica de las variantes somáticas adquiridas en genes vinculados con la formación de la LMA, permitiría identificar a personas con riesgo alto de transformación leucémica. Dichos pacientes quizás obtengan mayor beneficio de un trasplante de células madre hematopoyéticas.[420 ]
La presencia de una monosomía 7 aislada es la anomalía citogenética más común, si bien no parece augurar un pronóstico adverso en comparación con su presencia en la LMA manifiesta. No obstante, la presencia de monosomía 7 en combinación con otras anomalías citogenéticas se relaciona con un pronóstico precario.[
Para obtener información sobre el tratamiento de los SMD, consultar
Mielopoyesis anormal transitoria
Características genómicas de la mielopoyesis anormal transitoria
En presencia de trisomía 21, los blastocitos de la mielopoyesis anormal transitoria (MAT), a menudo exhiben características de diferenciación megacariocítica y variantes específicas que afectan el gen GATA1.[
Las variantes de GATA1 se encuentran en la mayoría de los niños con síndrome de Down (si no en todos) que tienen MAT o leucemia megacarioblástica aguda (LMCA).[
En un análisis de 2024 se examinaron 143 muestras de MAT para determinar la presencia de variantes somáticas en las células anormales. Además de la anomalía típica de GATA1, las únicas anomalías adicionales que se encontraron en el estudio fueron variantes raras de STAG2.[
Alrededor del 20 % de los lactantes con MAT y síndrome de Down presentan LMA en algún momento de su vida. La mayoría de estos casos se diagnostican dentro de los primeros 3 años de vida.[
Para obtener información sobre el tratamiento de la mielopoyesis anormal transitoria, consultar
Referencias:
- Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet 54 (9): 1376-1389, 2022.
- Liu Y, Klein J, Bajpai R, et al.: Etiology of oncogenic fusions in 5,190 childhood cancers and its clinical and therapeutic implication. Nat Commun 14 (1): 1739, 2023.
- Mullighan CG, Goorha S, Radtke I, et al.: Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446 (7137): 758-64, 2007.
- Mullighan CG, Miller CB, Radtke I, et al.: BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453 (7191): 110-4, 2008.
- Roberts KG, Li Y, Payne-Turner D, et al.: Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 371 (11): 1005-15, 2014.
- Lilljebjörn H, Henningsson R, Hyrenius-Wittsten A, et al.: Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun 7: 11790, 2016.
- Zhang J, McCastlain K, Yoshihara H, et al.: Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet 48 (12): 1481-1489, 2016.
- Holmfeldt L, Wei L, Diaz-Flores E, et al.: The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45 (3): 242-52, 2013.
- Loh ML, Zhang J, Harvey RC, et al.: Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children's Oncology Group TARGET Project. Blood 121 (3): 485-8, 2013.
- Bercovich D, Ganmore I, Scott LM, et al.: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484-92, 2008.
- Li Z, Chang TC, Junco JJ, et al.: Genomic landscape of Down syndrome-associated acute lymphoblastic leukemia. Blood 142 (2): 172-184, 2023.
- Andersson AK, Ma J, Wang J, et al.: The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet 47 (4): 330-7, 2015.
- Ma X, Edmonson M, Yergeau D, et al.: Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun 6: 6604, 2015.
- Meyer JA, Wang J, Hogan LE, et al.: Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 45 (3): 290-4, 2013.
- Li B, Li H, Bai Y, et al.: Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med 21 (6): 563-71, 2015.
- Mullighan CG, Zhang J, Kasper LH, et al.: CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471 (7337): 235-9, 2011.
- Mattano LA, Devidas M, Maloney KW, et al.: Favorable Trisomies and ETV6-RUNX1 Predict Cure in Low-Risk B-Cell Acute Lymphoblastic Leukemia: Results From Children's Oncology Group Trial AALL0331. J Clin Oncol 39 (14): 1540-1552, 2021.
- Moorman AV, Ensor HM, Richards SM, et al.: Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol 11 (5): 429-38, 2010.
- Alaggio R, Amador C, Anagnostopoulos I, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 36 (7): 1720-1748, 2022.
- Paulsson K, Johansson B: High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 48 (8): 637-60, 2009.
- Aricò M, Valsecchi MG, Rizzari C, et al.: Long-term results of the AIEOP-ALL-95 Trial for Childhood Acute Lymphoblastic Leukemia: insight on the prognostic value of DNA index in the framework of Berlin-Frankfurt-Muenster based chemotherapy. J Clin Oncol 26 (2): 283-9, 2008.
- Dastugue N, Suciu S, Plat G, et al.: Hyperdiploidy with 58-66 chromosomes in childhood B-acute lymphoblastic leukemia is highly curable: 58951 CLG-EORTC results. Blood 121 (13): 2415-23, 2013.
- Synold TW, Relling MV, Boyett JM, et al.: Blast cell methotrexate-polyglutamate accumulation in vivo differs by lineage, ploidy, and methotrexate dose in acute lymphoblastic leukemia. J Clin Invest 94 (5): 1996-2001, 1994.
- Moorman AV, Richards SM, Martineau M, et al.: Outcome heterogeneity in childhood high-hyperdiploid acute lymphoblastic leukemia. Blood 102 (8): 2756-62, 2003.
- Chilton L, Buck G, Harrison CJ, et al.: High hyperdiploidy among adolescents and adults with acute lymphoblastic leukaemia (ALL): cytogenetic features, clinical characteristics and outcome. Leukemia 28 (7): 1511-8, 2014.
- Purvis K, Zhou Y, Karol SE, et al.: Outcomes in patients with ETV6::RUNX1 or high-hyperdiploid B-ALL treated in the St. Jude Total Therapy XV/XVI studies. Blood 145 (2): 190-201, 2025.
- Sutcliffe MJ, Shuster JJ, Sather HN, et al.: High concordance from independent studies by the Children's Cancer Group (CCG) and Pediatric Oncology Group (POG) associating favorable prognosis with combined trisomies 4, 10, and 17 in children with NCI Standard-Risk B-precursor Acute Lymphoblastic Leukemia: a Children's Oncology Group (COG) initiative. Leukemia 19 (5): 734-40, 2005.
- Harris MB, Shuster JJ, Carroll A, et al.: Trisomy of leukemic cell chromosomes 4 and 10 identifies children with B-progenitor cell acute lymphoblastic leukemia with a very low risk of treatment failure: a Pediatric Oncology Group study. Blood 79 (12): 3316-24, 1992.
- Enshaei A, Vora A, Harrison CJ, et al.: Defining low-risk high hyperdiploidy in patients with paediatric acute lymphoblastic leukaemia: a retrospective analysis of data from the UKALL97/99 and UKALL2003 clinical trials. Lancet Haematol 8 (11): e828-e839, 2021.
- Heerema NA, Harbott J, Galimberti S, et al.: Secondary cytogenetic aberrations in childhood Philadelphia chromosome positive acute lymphoblastic leukemia are nonrandom and may be associated with outcome. Leukemia 18 (4): 693-702, 2004.
- Carroll AJ, Shago M, Mikhail FM, et al.: Masked hypodiploidy: Hypodiploid acute lymphoblastic leukemia (ALL) mimicking hyperdiploid ALL in children: A report from the Children's Oncology Group. Cancer Genet 238: 62-68, 2019.
- Nachman JB, Heerema NA, Sather H, et al.: Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood 110 (4): 1112-5, 2007.
- Raimondi SC, Zhou Y, Shurtleff SA, et al.: Near-triploidy and near-tetraploidy in childhood acute lymphoblastic leukemia: association with B-lineage blast cells carrying the ETV6-RUNX1 fusion, T-lineage immunophenotype, and favorable outcome. Cancer Genet Cytogenet 169 (1): 50-7, 2006.
- Attarbaschi A, Mann G, König M, et al.: Incidence and relevance of secondary chromosome abnormalities in childhood TEL/AML1+ acute lymphoblastic leukemia: an interphase FISH analysis. Leukemia 18 (10): 1611-6, 2004.
- Lemez P, Attarbaschi A, Béné MC, et al.: Childhood near-tetraploid acute lymphoblastic leukemia: an EGIL study on 36 cases. Eur J Haematol 85 (4): 300-8, 2010.
- Paulsson K, Lilljebjörn H, Biloglav A, et al.: The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet 47 (6): 672-6, 2015.
- Harrison CJ, Moorman AV, Broadfield ZJ, et al.: Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol 125 (5): 552-9, 2004.
- Mullighan CG, Jeha S, Pei D, et al.: Outcome of children with hypodiploid ALL treated with risk-directed therapy based on MRD levels. Blood 126 (26): 2896-9, 2015.
- Pui CH, Rebora P, Schrappe M, et al.: Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Retrospective Multinational Study. J Clin Oncol 37 (10): 770-779, 2019.
- McNeer JL, Devidas M, Dai Y, et al.: Hematopoietic Stem-Cell Transplantation Does Not Improve the Poor Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group. J Clin Oncol 37 (10): 780-789, 2019.
- Irving J, Matheson E, Minto L, et al.: Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 124 (23): 3420-30, 2014.
- Qian M, Cao X, Devidas M, et al.: TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J Clin Oncol 36 (6): 591-599, 2018.
- Rubnitz JE, Wichlan D, Devidas M, et al.: Prospective analysis of TEL gene rearrangements in childhood acute lymphoblastic leukemia: a Children's Oncology Group study. J Clin Oncol 26 (13): 2186-91, 2008.
- Kanerva J, Saarinen-Pihkala UM, Niini T, et al.: Favorable outcome in 20-year follow-up of children with very-low-risk ALL and minimal standard therapy, with special reference to TEL-AML1 fusion. Pediatr Blood Cancer 42 (1): 30-5, 2004.
- Aldrich MC, Zhang L, Wiemels JL, et al.: Cytogenetics of Hispanic and White children with acute lymphoblastic leukemia in California. Cancer Epidemiol Biomarkers Prev 15 (3): 578-81, 2006.
- Loh ML, Goldwasser MA, Silverman LB, et al.: Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood 107 (11): 4508-13, 2006.
- Borowitz MJ, Devidas M, Hunger SP, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood 111 (12): 5477-85, 2008.
- Madzo J, Zuna J, Muzíková K, et al.: Slower molecular response to treatment predicts poor outcome in patients with TEL/AML1 positive acute lymphoblastic leukemia: prospective real-time quantitative reverse transcriptase-polymerase chain reaction study. Cancer 97 (1): 105-13, 2003.
- Bhojwani D, Pei D, Sandlund JT, et al.: ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy. Leukemia 26 (2): 265-70, 2012.
- Enshaei A, Schwab CJ, Konn ZJ, et al.: Long-term follow-up of ETV6-RUNX1 ALL reveals that NCI risk, rather than secondary genetic abnormalities, is the key risk factor. Leukemia 27 (11): 2256-9, 2013.
- Barbany G, Andersen MK, Autio K, et al.: Additional aberrations of the ETV6 and RUNX1 genes have no prognostic impact in 229 t(12;21)(p13;q22)-positive B-cell precursor acute lymphoblastic leukaemias treated according to the NOPHO-ALL-2000 protocol. Leuk Res 36 (7): 936-8, 2012.
- Forestier E, Heyman M, Andersen MK, et al.: Outcome of ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia in the NOPHO-ALL-1992 protocol: frequent late relapses but good overall survival. Br J Haematol 140 (6): 665-72, 2008.
- Seeger K, Stackelberg AV, Taube T, et al.: Relapse of TEL-AML1--positive acute lymphoblastic leukemia in childhood: a matched-pair analysis. J Clin Oncol 19 (13): 3188-93, 2001.
- Gandemer V, Chevret S, Petit A, et al.: Excellent prognosis of late relapses of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia: lessons from the FRALLE 93 protocol. Haematologica 97 (11): 1743-50, 2012.
- Zuna J, Ford AM, Peham M, et al.: TEL deletion analysis supports a novel view of relapse in childhood acute lymphoblastic leukemia. Clin Cancer Res 10 (16): 5355-60, 2004.
- van Delft FW, Horsley S, Colman S, et al.: Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 117 (23): 6247-54, 2011.
- Aricò M, Schrappe M, Hunger SP, et al.: Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol 28 (31): 4755-61, 2010.
- Schrappe M, Aricò M, Harbott J, et al.: Philadelphia chromosome-positive (Ph+) childhood acute lymphoblastic leukemia: good initial steroid response allows early prediction of a favorable treatment outcome. Blood 92 (8): 2730-41, 1998.
- Ribeiro RC, Broniscer A, Rivera GK, et al.: Philadelphia chromosome-positive acute lymphoblastic leukemia in children: durable responses to chemotherapy associated with low initial white blood cell counts. Leukemia 11 (9): 1493-6, 1997.
- Biondi A, Schrappe M, De Lorenzo P, et al.: Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol 13 (9): 936-45, 2012.
- Schultz KR, Bowman WP, Aledo A, et al.: Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 27 (31): 5175-81, 2009.
- Schultz KR, Carroll A, Heerema NA, et al.: Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's Oncology Group study AALL0031. Leukemia 28 (7): 1467-71, 2014.
- Duffield AS, Mullighan CG, Borowitz MJ: International Consensus Classification of acute lymphoblastic leukemia/lymphoma. Virchows Arch 482 (1): 11-26, 2023.
- Short NJ, Jabbour E, Macaron W, et al.: Ultrasensitive NGS MRD assessment in Ph+ ALL: Prognostic impact and correlation with RT-PCR for BCR::ABL1. Am J Hematol 98 (8): 1196-1203, 2023.
- Hovorkova L, Zaliova M, Venn NC, et al.: Monitoring of childhood ALL using BCR-ABL1 genomic breakpoints identifies a subgroup with CML-like biology. Blood 129 (20): 2771-2781, 2017.
- Zuna J, Hovorkova L, Krotka J, et al.: Minimal residual disease in BCR::ABL1-positive acute lymphoblastic leukemia: different significance in typical ALL and in CML-like disease. Leukemia 36 (12): 2793-2801, 2022.
- Hunger SP, Tran TH, Saha V, et al.: Dasatinib with intensive chemotherapy in de novo paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (CA180-372/COG AALL1122): a single-arm, multicentre, phase 2 trial. Lancet Haematol 10 (7): e510-e520, 2023.
- Pui CH, Chessells JM, Camitta B, et al.: Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia 17 (4): 700-6, 2003.
- Johansson B, Moorman AV, Haas OA, et al.: Hematologic malignancies with t(4;11)(q21;q23)--a cytogenetic, morphologic, immunophenotypic and clinical study of 183 cases. European 11q23 Workshop participants. Leukemia 12 (5): 779-87, 1998.
- Raimondi SC, Peiper SC, Kitchingman GR, et al.: Childhood acute lymphoblastic leukemia with chromosomal breakpoints at 11q23. Blood 73 (6): 1627-34, 1989.
- Harrison CJ, Moorman AV, Barber KE, et al.: Interphase molecular cytogenetic screening for chromosomal abnormalities of prognostic significance in childhood acute lymphoblastic leukaemia: a UK Cancer Cytogenetics Group Study. Br J Haematol 129 (4): 520-30, 2005.
- Pui CH, Pei D, Campana D, et al.: A revised definition for cure of childhood acute lymphoblastic leukemia. Leukemia 28 (12): 2336-43, 2014.
- Pieters R, Schrappe M, De Lorenzo P, et al.: A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370 (9583): 240-50, 2007.
- Attarbaschi A, Möricke A, Harrison CJ, et al.: Outcomes of Childhood Noninfant Acute Lymphoblastic Leukemia With 11q23/KMT2A Rearrangements in a Modern Therapy Era: A Retrospective International Study. J Clin Oncol 41 (7): 1404-1422, 2023.
- Isobe T, Takagi M, Sato-Otsubo A, et al.: Multi-omics analysis defines highly refractory RAS burdened immature subgroup of infant acute lymphoblastic leukemia. Nat Commun 13 (1): 4501, 2022.
- Pui CH, Gaynon PS, Boyett JM, et al.: Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet 359 (9321): 1909-15, 2002.
- Rubnitz JE, Camitta BM, Mahmoud H, et al.: Childhood acute lymphoblastic leukemia with the MLL-ENL fusion and t(11;19)(q23;p13.3) translocation. J Clin Oncol 17 (1): 191-6, 1999.
- Hunger SP: Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood 87 (4): 1211-24, 1996.
- Uckun FM, Sensel MG, Sather HN, et al.: Clinical significance of translocation t(1;19) in childhood acute lymphoblastic leukemia in the context of contemporary therapies: a report from the Children's Cancer Group. J Clin Oncol 16 (2): 527-35, 1998.
- Fischer U, Forster M, Rinaldi A, et al.: Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet 47 (9): 1020-9, 2015.
- Pui CH, Sandlund JT, Pei D, et al.: Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA 290 (15): 2001-7, 2003.
- Crist WM, Carroll AJ, Shuster JJ, et al.: Poor prognosis of children with pre-B acute lymphoblastic leukemia is associated with the t(1;19)(q23;p13): a Pediatric Oncology Group study. Blood 76 (1): 117-22, 1990.
- Andersen MK, Autio K, Barbany G, et al.: Paediatric B-cell precursor acute lymphoblastic leukaemia with t(1;19)(q23;p13): clinical and cytogenetic characteristics of 47 cases from the Nordic countries treated according to NOPHO protocols. Br J Haematol 155 (2): 235-43, 2011.
- Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.
- Jeha S, Pei D, Raimondi SC, et al.: Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia 23 (8): 1406-9, 2009.
- Minson KA, Prasad P, Vear S, et al.: t(17;19) in Children with Acute Lymphocytic Leukemia: A Report of 3 Cases and a Review of the Literature. Case Rep Hematol 2013: 563291, 2013.
- Lee SHR, Antillon-Klussmann F, Pei D, et al.: Association of Genetic Ancestry With the Molecular Subtypes and Prognosis of Childhood Acute Lymphoblastic Leukemia. JAMA Oncol 8 (3): 354-363, 2022.
- Zaliova M, Potuckova E, Hovorkova L, et al.: ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on screening method sensitivity. Haematologica 104 (7): 1407-1416, 2019.
- Harvey RC, Mullighan CG, Wang X, et al.: Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 116 (23): 4874-84, 2010.
- Zaliova M, Zimmermannova O, Dörge P, et al.: ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia 28 (1): 182-5, 2014.
- Clappier E, Auclerc MF, Rapion J, et al.: An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia 28 (1): 70-7, 2014.
- Li Z, Lee SHR, Chin WHN, et al.: Distinct clinical characteristics of DUX4- and PAX5-altered childhood B-lymphoblastic leukemia. Blood Adv 5 (23): 5226-5238, 2021.
- Gu Z, Churchman M, Roberts K, et al.: Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun 7: 13331, 2016.
- Liu YF, Wang BY, Zhang WN, et al.: Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 8: 173-83, 2016.
- Suzuki K, Okuno Y, Kawashima N, et al.: MEF2D-BCL9 Fusion Gene Is Associated With High-Risk Acute B-Cell Precursor Lymphoblastic Leukemia in Adolescents. J Clin Oncol 34 (28): 3451-9, 2016.
- Lilljebjörn H, Ågerstam H, Orsmark-Pietras C, et al.: RNA-seq identifies clinically relevant fusion genes in leukemia including a novel MEF2D/CSF1R fusion responsive to imatinib. Leukemia 28 (4): 977-9, 2014.
- Hirabayashi S, Ohki K, Nakabayashi K, et al.: ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 102 (1): 118-129, 2017.
- Qian M, Zhang H, Kham SK, et al.: Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res 27 (2): 185-195, 2017.
- Hirabayashi S, Butler ER, Ohki K, et al.: Clinical characteristics and outcomes of B-ALL with ZNF384 rearrangements: a retrospective analysis by the Ponte di Legno Childhood ALL Working Group. Leukemia 35 (11): 3272-3277, 2021.
- Shago M, Abla O, Hitzler J, et al.: Frequency and outcome of pediatric acute lymphoblastic leukemia with ZNF384 gene rearrangements including a novel translocation resulting in an ARID1B/ZNF384 gene fusion. Pediatr Blood Cancer 63 (11): 1915-21, 2016.
- Yao L, Cen J, Pan J, et al.: TAF15-ZNF384 fusion gene in childhood mixed phenotype acute leukemia. Cancer Genet 211: 1-4, 2017.
- Alexander TB, Gu Z, Iacobucci I, et al.: The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 562 (7727): 373-379, 2018.
- Boer JM, Valsecchi MG, Hormann FM, et al.: Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia 35 (10): 2978-2982, 2021.
- De Lorenzo P, Moorman AV, Pieters R, et al.: Cytogenetics and outcome of infants with acute lymphoblastic leukemia and absence of MLL rearrangements. Leukemia 28 (2): 428-30, 2014.
- Fazio G, Bardini M, De Lorenzo P, et al.: Recurrent genetic fusions redefine MLL germ line acute lymphoblastic leukemia in infants. Blood 137 (14): 1980-1984, 2021.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Hogan TF, Koss W, Murgo AJ, et al.: Acute lymphoblastic leukemia with chromosomal 5;14 translocation and hypereosinophilia: case report and literature review. J Clin Oncol 5 (3): 382-90, 1987.
- Grimaldi JC, Meeker TC: The t(5;14) chromosomal translocation in a case of acute lymphocytic leukemia joins the interleukin-3 gene to the immunoglobulin heavy chain gene. Blood 73 (8): 2081-5, 1989.
- Meeker TC, Hardy D, Willman C, et al.: Activation of the interleukin-3 gene by chromosome translocation in acute lymphocytic leukemia with eosinophilia. Blood 76 (2): 285-9, 1990.
- Sutton R, Lonergan M, Tapp H, et al.: Two cases of hypereosinophilia and high-risk acute lymphoblastic leukemia. Leukemia 22 (7): 1463-5, 2008.
- Koleilat A, Smadbeck JB, Zepeda-Mendoza CJ, et al.: Characterization of unusual iAMP21 B-lymphoblastic leukemia (iAMP21-ALL) from the Mayo Clinic and Children's Oncology Group. Genes Chromosomes Cancer 61 (12): 710-719, 2022.
- Heerema NA, Carroll AJ, Devidas M, et al.: Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk children's oncology group studies: a report from the children's oncology group. J Clin Oncol 31 (27): 3397-402, 2013.
- Moorman AV, Robinson H, Schwab C, et al.: Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: a comparison of the MRC ALL97/99 and UKALL2003 trials. J Clin Oncol 31 (27): 3389-96, 2013.
- Harrison CJ, Moorman AV, Schwab C, et al.: An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia 28 (5): 1015-21, 2014.
- Gu Z, Churchman ML, Roberts KG, et al.: PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet 51 (2): 296-307, 2019.
- Strehl S, König M, Dworzak MN, et al.: PAX5/ETV6 fusion defines cytogenetic entity dic(9;12)(p13;p13). Leukemia 17 (6): 1121-3, 2003.
- Schwab C, Nebral K, Chilton L, et al.: Intragenic amplification of PAX5: a novel subgroup in B-cell precursor acute lymphoblastic leukemia? Blood Adv 1 (19): 1473-7, 2017.
- Den Boer ML, van Slegtenhorst M, De Menezes RX, et al.: A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 10 (2): 125-34, 2009.
- Mullighan CG, Su X, Zhang J, et al.: Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 360 (5): 470-80, 2009.
- Reshmi SC, Harvey RC, Roberts KG, et al.: Targetable kinase gene fusions in high-risk B-ALL: a study from the Children's Oncology Group. Blood 129 (25): 3352-3361, 2017.
- Roberts KG, Morin RD, Zhang J, et al.: Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 22 (2): 153-66, 2012.
- van der Veer A, Waanders E, Pieters R, et al.: Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood 122 (15): 2622-9, 2013.
- Roberts KG, Reshmi SC, Harvey RC, et al.: Genomic and outcome analyses of Ph-like ALL in NCI standard-risk patients: a report from the Children's Oncology Group. Blood 132 (8): 815-824, 2018.
- Roberts KG, Pei D, Campana D, et al.: Outcomes of children with BCR-ABL1–like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol 32 (27): 3012-20, 2014.
- Harvey RC, Mullighan CG, Chen IM, et al.: Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 115 (26): 5312-21, 2010.
- Mullighan CG, Collins-Underwood JR, Phillips LA, et al.: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243-6, 2009.
- Schwab C, Roberts K, Boer JM, et al.: SSBP2-CSF1R is a recurrent fusion in B-lineage acute lymphoblastic leukemia with diverse genetic presentation and variable outcome. Blood 137 (13): 1835-1838, 2021.
- van Outersterp I, Tasian SK, Reichert CEJ, et al.: Tyrosine kinase inhibitor response of ABL-class acute lymphoblastic leukemia: the role of kinase type and SH3 domain. Blood 143 (21): 2178-2189, 2024.
- den Boer ML, Cario G, Moorman AV, et al.: Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: a multicentre, retrospective, cohort study. Lancet Haematol 8 (1): e55-e66, 2021.
- Iacobucci I, Li Y, Roberts KG, et al.: Truncating Erythropoietin Receptor Rearrangements in Acute Lymphoblastic Leukemia. Cancer Cell 29 (2): 186-200, 2016.
- Cario G, Zimmermann M, Romey R, et al.: Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood 115 (26): 5393-7, 2010.
- Ensor HM, Schwab C, Russell LJ, et al.: Demographic, clinical, and outcome features of children with acute lymphoblastic leukemia and CRLF2 deregulation: results from the MRC ALL97 clinical trial. Blood 117 (7): 2129-36, 2011.
- Schmäh J, Fedders B, Panzer-Grümayer R, et al.: Molecular characterization of acute lymphoblastic leukemia with high CRLF2 gene expression in childhood. Pediatr Blood Cancer 64 (10): , 2017.
- Raca G, Abdel-Azim H, Yue F, et al.: Increased Incidence of IKZF1 deletions and IGH-CRLF2 translocations in B-ALL of Hispanic/Latino children-a novel health disparity. Leukemia 35 (8): 2399-2402, 2021.
- Kovach AE, Wengyn M, Vu MH, et al.: IKZF1PLUS alterations contribute to outcome disparities in Hispanic/Latino children with B-lymphoblastic leukemia. Pediatr Blood Cancer 71 (7): e30996, 2024.
- Vesely C, Frech C, Eckert C, et al.: Genomic and transcriptional landscape of P2RY8-CRLF2-positive childhood acute lymphoblastic leukemia. Leukemia 31 (7): 1491-1501, 2017.
- Russell LJ, Jones L, Enshaei A, et al.: Characterisation of the genomic landscape of CRLF2-rearranged acute lymphoblastic leukemia. Genes Chromosomes Cancer 56 (5): 363-372, 2017.
- Potter N, Jones L, Blair H, et al.: Single-cell analysis identifies CRLF2 rearrangements as both early and late events in Down syndrome and non-Down syndrome acute lymphoblastic leukaemia. Leukemia 33 (4): 893-904, 2019.
- Morak M, Attarbaschi A, Fischer S, et al.: Small sizes and indolent evolutionary dynamics challenge the potential role of P2RY8-CRLF2-harboring clones as main relapse-driving force in childhood ALL. Blood 120 (26): 5134-42, 2012.
- Schwab CJ, Chilton L, Morrison H, et al.: Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica 98 (7): 1081-8, 2013.
- Chen IM, Harvey RC, Mullighan CG, et al.: Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 119 (15): 3512-22, 2012.
- Palmi C, Vendramini E, Silvestri D, et al.: Poor prognosis for P2RY8-CRLF2 fusion but not for CRLF2 over-expression in children with intermediate risk B-cell precursor acute lymphoblastic leukemia. Leukemia 26 (10): 2245-53, 2012.
- Clappier E, Grardel N, Bakkus M, et al.: IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children's Leukemia Group study 58951. Leukemia 29 (11): 2154-61, 2015.
- Srinivasan S, Ramanathan S, Kumar S, et al.: Prevalence and prognostic significance of IKZF1 deletion in paediatric acute lymphoblastic leukemia: A systematic review and meta-analysis. Ann Hematol 102 (8): 2165-2179, 2023.
- Buitenkamp TD, Pieters R, Gallimore NE, et al.: Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia 26 (10): 2204-11, 2012.
- Krentz S, Hof J, Mendioroz A, et al.: Prognostic value of genetic alterations in children with first bone marrow relapse of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia 27 (2): 295-304, 2013.
- Feng J, Tang Y: Prognostic significance of IKZF1 alteration status in pediatric B-lineage acute lymphoblastic leukemia: a meta-analysis. Leuk Lymphoma 54 (4): 889-91, 2013.
- Dörge P, Meissner B, Zimmermann M, et al.: IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica 98 (3): 428-32, 2013.
- Olsson L, Castor A, Behrendtz M, et al.: Deletions of IKZF1 and SPRED1 are associated with poor prognosis in a population-based series of pediatric B-cell precursor acute lymphoblastic leukemia diagnosed between 1992 and 2011. Leukemia 28 (2): 302-10, 2014.
- Boer JM, van der Veer A, Rizopoulos D, et al.: Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia 30 (1): 32-8, 2016.
- Tran TH, Harris MH, Nguyen JV, et al.: Prognostic impact of kinase-activating fusions and IKZF1 deletions in pediatric high-risk B-lineage acute lymphoblastic leukemia. Blood Adv 2 (5): 529-533, 2018.
- Vrooman LM, Blonquist TM, Harris MH, et al.: Refining risk classification in childhood B acute lymphoblastic leukemia: results of DFCI ALL Consortium Protocol 05-001. Blood Adv 2 (12): 1449-1458, 2018.
- Öfverholm I, Rezayee F, Heyman M, et al.: The prognostic impact of IKZF1 deletions and UKALL genetic classifiers in paediatric B-cell precursor acute lymphoblastic leukaemia treated according to NOPHO 2008 protocols. Br J Haematol 202 (2): 384-392, 2023.
- van der Veer A, Zaliova M, Mottadelli F, et al.: IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood 123 (11): 1691-8, 2014.
- Stanulla M, Dagdan E, Zaliova M, et al.: IKZF1plus Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J Clin Oncol 36 (12): 1240-1249, 2018.
- Mangum DS, Meyer JA, Mason CC, et al.: Association of Combined Focal 22q11.22 Deletion and IKZF1 Alterations With Outcomes in Childhood Acute Lymphoblastic Leukemia. JAMA Oncol 7 (10): 1521-1528, 2021.
- Yeoh AEJ, Lu Y, Chin WHN, et al.: Intensifying Treatment of Childhood B-Lymphoblastic Leukemia With IKZF1 Deletion Reduces Relapse and Improves Overall Survival: Results of Malaysia-Singapore ALL 2010 Study. J Clin Oncol 36 (26): 2726-2735, 2018.
- Pieters R, de Groot-Kruseman H, Fiocco M, et al.: Improved Outcome for ALL by Prolonging Therapy for IKZF1 Deletion and Decreasing Therapy for Other Risk Groups. J Clin Oncol 41 (25): 4130-4142, 2023.
- Herbrueggen H, Mueller S, Rohde J, et al.: Treatment and outcome of IG-MYC+ neoplasms with precursor B-cell phenotype in childhood and adolescence. Leukemia 34 (3): 942-946, 2020.
- Sakaguchi K, Imamura T, Ishimaru S, et al.: Nationwide study of pediatric B-cell precursor acute lymphoblastic leukemia with chromosome 8q24/MYC rearrangement in Japan. Pediatr Blood Cancer 67 (7): e28341, 2020.
- Bomken S, Enshaei A, Schwalbe EC, et al.: Molecular characterization and clinical outcome of B-cell precursor acute lymphoblastic leukemia with IG-MYC rearrangement. Haematologica 108 (3): 717-731, 2023.
- Liu Y, Easton J, Shao Y, et al.: The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 49 (8): 1211-1218, 2017.
- Armstrong SA, Look AT: Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol 23 (26): 6306-15, 2005.
- Karrman K, Forestier E, Heyman M, et al.: Clinical and cytogenetic features of a population-based consecutive series of 285 pediatric T-cell acute lymphoblastic leukemias: rare T-cell receptor gene rearrangements are associated with poor outcome. Genes Chromosomes Cancer 48 (9): 795-805, 2009.
- Mansour MR, Abraham BJ, Anders L, et al.: Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346 (6215): 1373-7, 2014.
- Steimlé T, Dourthe ME, Alcantara M, et al.: Clinico-biological features of T-cell acute lymphoblastic leukemia with fusion proteins. Blood Cancer J 12 (1): 14, 2022.
- Weng AP, Ferrando AA, Lee W, et al.: Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306 (5694): 269-71, 2004.
- Gallo Llorente L, Luther H, Schneppenheim R, et al.: Identification of novel NOTCH1 mutations: increasing our knowledge of the NOTCH signaling pathway. Pediatr Blood Cancer 61 (5): 788-96, 2014.
- Burns MA, Place AE, Stevenson KE, et al.: Identification of prognostic factors in childhood T-cell acute lymphoblastic leukemia: Results from DFCI ALL Consortium Protocols 05-001 and 11-001. Pediatr Blood Cancer 68 (1): e28719, 2021.
- Petit A, Trinquand A, Chevret S, et al.: Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood 131 (3): 289-300, 2018.
- Trinquand A, Tanguy-Schmidt A, Ben Abdelali R, et al.: Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a Group for Research in Adult Acute Lymphoblastic Leukemia study. J Clin Oncol 31 (34): 4333-42, 2013.
- Bergeron J, Clappier E, Radford I, et al.: Prognostic and oncogenic relevance of TLX1/HOX11 expression level in T-ALLs. Blood 110 (7): 2324-30, 2007.
- van Grotel M, Meijerink JP, Beverloo HB, et al.: The outcome of molecular-cytogenetic subgroups in pediatric T-cell acute lymphoblastic leukemia: a retrospective study of patients treated according to DCOG or COALL protocols. Haematologica 91 (9): 1212-21, 2006.
- Cavé H, Suciu S, Preudhomme C, et al.: Clinical significance of HOX11L2 expression linked to t(5;14)(q35;q32), of HOX11 expression, and of SIL-TAL fusion in childhood T-cell malignancies: results of EORTC studies 58881 and 58951. Blood 103 (2): 442-50, 2004.
- Baak U, Gökbuget N, Orawa H, et al.: Thymic adult T-cell acute lymphoblastic leukemia stratified in standard- and high-risk group by aberrant HOX11L2 expression: experience of the German multicenter ALL study group. Leukemia 22 (6): 1154-60, 2008.
- Ferrando AA, Neuberg DS, Dodge RK, et al.: Prognostic importance of TLX1 (HOX11) oncogene expression in adults with T-cell acute lymphoblastic leukaemia. Lancet 363 (9408): 535-6, 2004.
- Burmeister T, Gökbuget N, Reinhardt R, et al.: NUP214-ABL1 in adult T-ALL: the GMALL study group experience. Blood 108 (10): 3556-9, 2006.
- Graux C, Stevens-Kroef M, Lafage M, et al.: Heterogeneous patterns of amplification of the NUP214-ABL1 fusion gene in T-cell acute lymphoblastic leukemia. Leukemia 23 (1): 125-33, 2009.
- Hagemeijer A, Graux C: ABL1 rearrangements in T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer 49 (4): 299-308, 2010.
- Quintás-Cardama A, Tong W, Manshouri T, et al.: Activity of tyrosine kinase inhibitors against human NUP214-ABL1-positive T cell malignancies. Leukemia 22 (6): 1117-24, 2008.
- Clarke S, O'Reilly J, Romeo G, et al.: NUP214-ABL1 positive T-cell acute lymphoblastic leukemia patient shows an initial favorable response to imatinib therapy post relapse. Leuk Res 35 (7): e131-3, 2011.
- Deenik W, Beverloo HB, van der Poel-van de Luytgaarde SC, et al.: Rapid complete cytogenetic remission after upfront dasatinib monotherapy in a patient with a NUP214-ABL1-positive T-cell acute lymphoblastic leukemia. Leukemia 23 (3): 627-9, 2009.
- Crombet O, Lastrapes K, Zieske A, et al.: Complete morphologic and molecular remission after introduction of dasatinib in the treatment of a pediatric patient with t-cell acute lymphoblastic leukemia and ABL1 amplification. Pediatr Blood Cancer 59 (2): 333-4, 2012.
- Seki M, Kimura S, Isobe T, et al.: Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat Genet 49 (8): 1274-1281, 2017.
- Nagel S, Scherr M, Kel A, et al.: Activation of TLX3 and NKX2-5 in t(5;14)(q35;q32) T-cell acute lymphoblastic leukemia by remote 3'-BCL11B enhancers and coregulation by PU.1 and HMGA1. Cancer Res 67 (4): 1461-71, 2007.
- Gutierrez A, Kentsis A, Sanda T, et al.: The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood 118 (15): 4169-73, 2011.
- Ceppi F, Gotti G, Möricke A, et al.: Near-tetraploid T-cell acute lymphoblastic leukaemia in childhood: Results of the AIEOP-BFM ALL studies. Eur J Cancer 175: 120-124, 2022.
- Zhang J, Ding L, Holmfeldt L, et al.: The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481 (7380): 157-63, 2012.
- Gutierrez A, Dahlberg SE, Neuberg DS, et al.: Absence of biallelic TCRgamma deletion predicts early treatment failure in pediatric T-cell acute lymphoblastic leukemia. J Clin Oncol 28 (24): 3816-23, 2010.
- Yang YL, Hsiao CC, Chen HY, et al.: Absence of biallelic TCRγ deletion predicts induction failure and poorer outcomes in childhood T-cell acute lymphoblastic leukemia. Pediatr Blood Cancer 58 (6): 846-51, 2012.
- Montefiori LE, Bendig S, Gu Z, et al.: Enhancer Hijacking Drives Oncogenic BCL11B Expression in Lineage-Ambiguous Stem Cell Leukemia. Cancer Discov 11 (11): 2846-2867, 2021.
- Di Giacomo D, La Starza R, Gorello P, et al.: 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T-lymphoid and myeloid immature acute leukemia. Blood 138 (9): 773-784, 2021.
- Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.
- Borowitz MJ, Béné MC, Harris NL, et al.: Acute leukaemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. International Agency for Research on Cancer, 2017, pp 179-87.
- Davies SM, Bhatia S, Ross JA, et al.: Glutathione S-transferase genotypes, genetic susceptibility, and outcome of therapy in childhood acute lymphoblastic leukemia. Blood 100 (1): 67-71, 2002.
- Krajinovic M, Costea I, Chiasson S: Polymorphism of the thymidylate synthase gene and outcome of acute lymphoblastic leukaemia. Lancet 359 (9311): 1033-4, 2002.
- Krajinovic M, Lemieux-Blanchard E, Chiasson S, et al.: Role of polymorphisms in MTHFR and MTHFD1 genes in the outcome of childhood acute lymphoblastic leukemia. Pharmacogenomics J 4 (1): 66-72, 2004.
- Schmiegelow K, Forestier E, Kristinsson J, et al.: Thiopurine methyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. Leukemia 23 (3): 557-64, 2009.
- Relling MV, Hancock ML, Boyett JM, et al.: Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 93 (9): 2817-23, 1999.
- Stanulla M, Schaeffeler E, Flohr T, et al.: Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA 293 (12): 1485-9, 2005.
- Yang JJ, Landier W, Yang W, et al.: Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol 33 (11): 1235-42, 2015.
- Relling MV, Hancock ML, Rivera GK, et al.: Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 91 (23): 2001-8, 1999.
- Moriyama T, Nishii R, Perez-Andreu V, et al.: NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet 48 (4): 367-73, 2016.
- Tanaka Y, Kato M, Hasegawa D, et al.: Susceptibility to 6-MP toxicity conferred by a NUDT15 variant in Japanese children with acute lymphoblastic leukaemia. Br J Haematol 171 (1): 109-15, 2015.
- Diouf B, Crews KR, Lew G, et al.: Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA 313 (8): 815-23, 2015.
- Yang JJ, Cheng C, Yang W, et al.: Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA 301 (4): 393-403, 2009.
- Gregers J, Christensen IJ, Dalhoff K, et al.: The association of reduced folate carrier 80G>A polymorphism to outcome in childhood acute lymphoblastic leukemia interacts with chromosome 21 copy number. Blood 115 (23): 4671-7, 2010.
- Radtke S, Zolk O, Renner B, et al.: Germline genetic variations in methotrexate candidate genes are associated with pharmacokinetics, toxicity, and outcome in childhood acute lymphoblastic leukemia. Blood 121 (26): 5145-53, 2013.
- Grimwade D, Walker H, Oliver F, et al.: The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood 92 (7): 2322-33, 1998.
- Harrison CJ, Hills RK, Moorman AV, et al.: Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J Clin Oncol 28 (16): 2674-81, 2010.
- von Neuhoff C, Reinhardt D, Sander A, et al.: Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J Clin Oncol 28 (16): 2682-9, 2010.
- Grimwade D, Hills RK, Moorman AV, et al.: Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 116 (3): 354-65, 2010.
- Creutzig U, van den Heuvel-Eibrink MM, Gibson B, et al.: Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 120 (16): 3187-205, 2012.
- Brown P, McIntyre E, Rau R, et al.: The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood 110 (3): 979-85, 2007.
- Hollink IH, Zwaan CM, Zimmermann M, et al.: Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia 23 (2): 262-70, 2009.
- Ho PA, Alonzo TA, Gerbing RB, et al.: Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): a report from the Children's Oncology Group. Blood 113 (26): 6558-66, 2009.
- Meshinchi S, Alonzo TA, Stirewalt DL, et al.: Clinical implications of FLT3 mutations in pediatric AML. Blood 108 (12): 3654-61, 2006.
- Tarlock K, Meshinchi S: Pediatric acute myeloid leukemia: biology and therapeutic implications of genomic variants. Pediatr Clin North Am 62 (1): 75-93, 2015.
- Bolouri H, Farrar JE, Triche T, et al.: The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med 24 (1): 103-112, 2018.
- Zarnegar-Lumley S, Alonzo TA, Gerbing RB, et al.: Characteristics and prognostic impact of IDH mutations in AML: a COG, SWOG, and ECOG analysis. Blood Adv 7 (19): 5941-5953, 2023.
- Farrar JE, Schuback HL, Ries RE, et al.: Genomic Profiling of Pediatric Acute Myeloid Leukemia Reveals a Changing Mutational Landscape from Disease Diagnosis to Relapse. Cancer Res 76 (8): 2197-205, 2016.
- Pfister SM, Reyes-Múgica M, Chan JKC, et al.: A Summary of the Inaugural WHO Classification of Pediatric Tumors: Transitioning from the Optical into the Molecular Era. Cancer Discov 12 (2): 331-355, 2022.
- Khoury JD, Solary E, Abla O, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 36 (7): 1703-1719, 2022.
- Klein K, Kaspers G, Harrison CJ, et al.: Clinical Impact of Additional Cytogenetic Aberrations, cKIT and RAS Mutations, and Treatment Elements in Pediatric t(8;21)-AML: Results From an International Retrospective Study by the International Berlin-Frankfurt-Münster Study Group. J Clin Oncol 33 (36): 4247-58, 2015.
- Creutzig U, Zimmermann M, Ritter J, et al.: Definition of a standard-risk group in children with AML. Br J Haematol 104 (3): 630-9, 1999.
- Raimondi SC, Chang MN, Ravindranath Y, et al.: Chromosomal abnormalities in 478 children with acute myeloid leukemia: clinical characteristics and treatment outcome in a cooperative pediatric oncology group study-POG 8821. Blood 94 (11): 3707-16, 1999.
- Lie SO, Abrahamsson J, Clausen N, et al.: Treatment stratification based on initial in vivo response in acute myeloid leukaemia in children without Down's syndrome: results of NOPHO-AML trials. Br J Haematol 122 (2): 217-25, 2003.
- Duployez N, Marceau-Renaut A, Boissel N, et al.: Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood 127 (20): 2451-9, 2016.
- Faber ZJ, Chen X, Gedman AL, et al.: The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet 48 (12): 1551-1556, 2016.
- Chen W, Xie H, Wang H, et al.: Prognostic Significance of KIT Mutations in Core-Binding Factor Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis. PLoS One 11 (1): e0146614, 2016.
- Shih LY, Liang DC, Huang CF, et al.: Cooperating mutations of receptor tyrosine kinases and Ras genes in childhood core-binding factor acute myeloid leukemia and a comparative analysis on paired diagnosis and relapse samples. Leukemia 22 (2): 303-7, 2008.
- Goemans BF, Zwaan CM, Miller M, et al.: Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia 19 (9): 1536-42, 2005.
- Pollard JA, Alonzo TA, Gerbing RB, et al.: Prevalence and prognostic significance of KIT mutations in pediatric patients with core binding factor AML enrolled on serial pediatric cooperative trials for de novo AML. Blood 115 (12): 2372-9, 2010.
- Shimada A, Taki T, Tabuchi K, et al.: KIT mutations, and not FLT3 internal tandem duplication, are strongly associated with a poor prognosis in pediatric acute myeloid leukemia with t(8;21): a study of the Japanese Childhood AML Cooperative Study Group. Blood 107 (5): 1806-9, 2006.
- Manara E, Bisio V, Masetti R, et al.: Core-binding factor acute myeloid leukemia in pediatric patients enrolled in the AIEOP AML 2002/01 trial: screening and prognostic impact of c-KIT mutations. Leukemia 28 (5): 1132-4, 2014.
- Chen X, Dou H, Wang X, et al.: KIT mutations correlate with adverse survival in children with core-binding factor acute myeloid leukemia. Leuk Lymphoma 59 (4): 829-836, 2018.
- Tokumasu M, Murata C, Shimada A, et al.: Adverse prognostic impact of KIT mutations in childhood CBF-AML: the results of the Japanese Pediatric Leukemia/Lymphoma Study Group AML-05 trial. Leukemia 29 (12): 2438-41, 2015.
- Tarlock K, Alonzo TA, Wang YC, et al.: Functional Properties of KIT Mutations Are Associated with Differential Clinical Outcomes and Response to Targeted Therapeutics in CBF Acute Myeloid Leukemia. Clin Cancer Res 25 (16): 5038-5048, 2019.
- Srinivasan S, Dhamne C, Patkar N, et al.: KIT exon 17 mutations are predictive of inferior outcome in pediatric acute myeloid leukemia with RUNX1::RUNX1T1. Pediatr Blood Cancer 71 (2): e30791, 2024.
- Jahn N, Agrawal M, Bullinger L, et al.: Incidence and prognostic impact of ASXL2 mutations in adult acute myeloid leukemia patients with t(8;21)(q22;q22): a study of the German-Austrian AML Study Group. Leukemia 31 (4): 1012-1015, 2017.
- Yamato G, Shiba N, Yoshida K, et al.: ASXL2 mutations are frequently found in pediatric AML patients with t(8;21)/ RUNX1-RUNX1T1 and associated with a better prognosis. Genes Chromosomes Cancer 56 (5): 382-393, 2017.
- Döhner K, Schlenk RF, Habdank M, et al.: Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood 106 (12): 3740-6, 2005.
- Verhaak RG, Goudswaard CS, van Putten W, et al.: Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood 106 (12): 3747-54, 2005.
- Schnittger S, Schoch C, Kern W, et al.: Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 106 (12): 3733-9, 2005.
- Falini B, Mecucci C, Tiacci E, et al.: Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 352 (3): 254-66, 2005.
- Schlenk RF, Döhner K, Krauter J, et al.: Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 358 (18): 1909-18, 2008.
- Gale RE, Green C, Allen C, et al.: The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 111 (5): 2776-84, 2008.
- Cazzaniga G, Dell'Oro MG, Mecucci C, et al.: Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood 106 (4): 1419-22, 2005.
- Balgobind BV, Hollink IH, Arentsen-Peters ST, et al.: Integrative analysis of type-I and type-II aberrations underscores the genetic heterogeneity of pediatric acute myeloid leukemia. Haematologica 96 (10): 1478-87, 2011.
- Staffas A, Kanduri M, Hovland R, et al.: Presence of FLT3-ITD and high BAALC expression are independent prognostic markers in childhood acute myeloid leukemia. Blood 118 (22): 5905-13, 2011.
- Tarlock K, Gerbing RB, Ries RE, et al.: Prognostic impact of cooccurring mutations in FLT3-ITD pediatric acute myeloid leukemia. Blood Adv 8 (9): 2094-2103, 2024.
- Tawana K, Wang J, Renneville A, et al.: Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood 126 (10): 1214-23, 2015.
- Tarlock K, Lamble AJ, Wang YC, et al.: CEBPA-bZip mutations are associated with favorable prognosis in de novo AML: a report from the Children's Oncology Group. Blood 138 (13): 1137-1147, 2021.
- Marcucci G, Maharry K, Radmacher MD, et al.: Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B Study. J Clin Oncol 26 (31): 5078-87, 2008.
- Wouters BJ, Löwenberg B, Erpelinck-Verschueren CA, et al.: Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 113 (13): 3088-91, 2009.
- Dufour A, Schneider F, Metzeler KH, et al.: Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol 28 (4): 570-7, 2010.
- Taskesen E, Bullinger L, Corbacioglu A, et al.: Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood 117 (8): 2469-75, 2011.
- Fasan A, Haferlach C, Alpermann T, et al.: The role of different genetic subtypes of CEBPA mutated AML. Leukemia 28 (4): 794-803, 2014.
- Wakita S, Sakaguchi M, Oh I, et al.: Prognostic impact of CEBPA bZIP domain mutation in acute myeloid leukemia. Blood Adv 6 (1): 238-247, 2022.
- Taube F, Georgi JA, Kramer M, et al.: CEBPA mutations in 4708 patients with acute myeloid leukemia: differential impact of bZIP and TAD mutations on outcome. Blood 139 (1): 87-103, 2022.
- Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, et al.: Characterization of CEBPA mutations and promoter hypermethylation in pediatric acute myeloid leukemia. Haematologica 96 (3): 384-92, 2011.
- Tarlock K, Alonzo T, Wang YC, et al.: Prognostic impact of CSF3R mutations in favorable risk childhood acute myeloid leukemia. Blood 135 (18): 1603-1606, 2020.
- Tawana K, Rio-Machin A, Preudhomme C, et al.: Familial CEBPA-mutated acute myeloid leukemia. Semin Hematol 54 (2): 87-93, 2017.
- Pui CH, Relling MV, Rivera GK, et al.: Epipodophyllotoxin-related acute myeloid leukemia: a study of 35 cases. Leukemia 9 (12): 1990-6, 1995.
- Inaba H, Zhou Y, Abla O, et al.: Heterogeneous cytogenetic subgroups and outcomes in childhood acute megakaryoblastic leukemia: a retrospective international study. Blood 126 (13): 1575-84, 2015.
- de Rooij JD, Branstetter C, Ma J, et al.: Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet 49 (3): 451-456, 2017.
- Balgobind BV, Raimondi SC, Harbott J, et al.: Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood 114 (12): 2489-96, 2009.
- Swansbury GJ, Slater R, Bain BJ, et al.: Hematological malignancies with t(9;11)(p21-22;q23)--a laboratory and clinical study of 125 cases. European 11q23 Workshop participants. Leukemia 12 (5): 792-800, 1998.
- Pollard JA, Guest E, Alonzo TA, et al.: Gemtuzumab Ozogamicin Improves Event-Free Survival and Reduces Relapse in Pediatric KMT2A-Rearranged AML: Results From the Phase III Children's Oncology Group Trial AAML0531. J Clin Oncol 39 (28): 3149-3160, 2021.
- van Weelderen RE, Klein K, Harrison CJ, et al.: Measurable Residual Disease and Fusion Partner Independently Predict Survival and Relapse Risk in Childhood KMT2A-Rearranged Acute Myeloid Leukemia: A Study by the International Berlin-Frankfurt-Münster Study Group. J Clin Oncol 41 (16): 2963-2974, 2023.
- Gröschel S, Sanders MA, Hoogenboezem R, et al.: A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157 (2): 369-81, 2014.
- Yamazaki H, Suzuki M, Otsuki A, et al.: A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 25 (4): 415-27, 2014.
- Mrózek K, Heerema NA, Bloomfield CD: Cytogenetics in acute leukemia. Blood Rev 18 (2): 115-36, 2004.
- Lugthart S, Gröschel S, Beverloo HB, et al.: Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J Clin Oncol 28 (24): 3890-8, 2010.
- Balgobind BV, Lugthart S, Hollink IH, et al.: EVI1 overexpression in distinct subtypes of pediatric acute myeloid leukemia. Leukemia 24 (5): 942-9, 2010.
- Elsherif M, Hammad M, Hafez H, et al.: MECOM gene overexpression in pediatric patients with acute myeloid leukemia. Acta Oncol 61 (4): 516-522, 2022.
- Baldazzi C, Luatti S, Zuffa E, et al.: Complex chromosomal rearrangements leading to MECOM overexpression are recurrent in myeloid malignancies with various 3q abnormalities. Genes Chromosomes Cancer 55 (4): 375-88, 2016.
- Lim G, Choi JR, Kim MJ, et al.: Detection of t(3;5) and NPM1/MLF1 rearrangement in an elderly patient with acute myeloid leukemia: clinical and laboratory study with review of the literature. Cancer Genet Cytogenet 199 (2): 101-9, 2010.
- Dumézy F, Renneville A, Mayeur-Rousse C, et al.: Acute myeloid leukemia with translocation t(3;5): new molecular insights. Haematologica 98 (4): e52-4, 2013.
- Ageberg M, Drott K, Olofsson T, et al.: Identification of a novel and myeloid specific role of the leukemia-associated fusion protein DEK-NUP214 leading to increased protein synthesis. Genes Chromosomes Cancer 47 (4): 276-87, 2008.
- Shiba N, Ichikawa H, Taki T, et al.: NUP98-NSD1 gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia. Genes Chromosomes Cancer 52 (7): 683-93, 2013.
- Slovak ML, Gundacker H, Bloomfield CD, et al.: A retrospective study of 69 patients with t(6;9)(p23;q34) AML emphasizes the need for a prospective, multicenter initiative for rare 'poor prognosis' myeloid malignancies. Leukemia 20 (7): 1295-7, 2006.
- Alsabeh R, Brynes RK, Slovak ML, et al.: Acute myeloid leukemia with t(6;9) (p23;q34): association with myelodysplasia, basophilia, and initial CD34 negative immunophenotype. Am J Clin Pathol 107 (4): 430-7, 1997.
- Sandahl JD, Coenen EA, Forestier E, et al.: t(6;9)(p22;q34)/DEK-NUP214-rearranged pediatric myeloid leukemia: an international study of 62 patients. Haematologica 99 (5): 865-72, 2014.
- Tarlock K, Alonzo TA, Moraleda PP, et al.: Acute myeloid leukaemia (AML) with t(6;9)(p23;q34) is associated with poor outcome in childhood AML regardless of FLT3-ITD status: a report from the Children's Oncology Group. Br J Haematol 166 (2): 254-9, 2014.
- Lamble AJ, Hagiwara K, Gerbing RB, et al.: CREBBP alterations are associated with a poor prognosis in de novo AML. Blood 141 (17): 2156-2159, 2023.
- Coenen EA, Zwaan CM, Reinhardt D, et al.: Pediatric acute myeloid leukemia with t(8;16)(p11;p13), a distinct clinical and biological entity: a collaborative study by the International-Berlin-Frankfurt-Munster AML-study group. Blood 122 (15): 2704-13, 2013.
- Wong KF, Yuen HL, Siu LL, et al.: t(8;16)(p11;p13) predisposes to a transient but potentially recurring neonatal leukemia. Hum Pathol 39 (11): 1702-7, 2008.
- Wu X, Sulavik D, Roulston D, et al.: Spontaneous remission of congenital acute myeloid leukemia with t(8;16)(p11;13). Pediatr Blood Cancer 56 (2): 331-2, 2011.
- Terui K, Sato T, Sasaki S, et al.: Two novel variants of MOZ-CBP fusion transcripts in spontaneously remitted infant leukemia with t(1;16;8)(p13;p13;p11), a new variant of t(8;16)(p11;p13). Haematologica 93 (10): 1591-3, 2008.
- Noort S, Zimmermann M, Reinhardt D, et al.: Prognostic impact of t(16;21)(p11;q22) and t(16;21)(q24;q22) in pediatric AML: a retrospective study by the I-BFM Study Group. Blood 132 (15): 1584-1592, 2018.
- Gruber TA, Larson Gedman A, Zhang J, et al.: An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell 22 (5): 683-97, 2012.
- Thiollier C, Lopez CK, Gerby B, et al.: Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J Exp Med 209 (11): 2017-31, 2012.
- de Rooij JD, Hollink IH, Arentsen-Peters ST, et al.: NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 27 (12): 2280-8, 2013.
- Masetti R, Pigazzi M, Togni M, et al.: CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 121 (17): 3469-72, 2013.
- Masetti R, Rondelli R, Fagioli F, et al.: Infants with acute myeloid leukemia treated according to the Associazione Italiana di Ematologia e Oncologia Pediatrica 2002/01 protocol have an outcome comparable to that of older children. Haematologica 99 (8): e127-9, 2014.
- de Rooij JD, Masetti R, van den Heuvel-Eibrink MM, et al.: Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective intergroup study. Blood 127 (26): 3424-30, 2016.
- Hara Y, Shiba N, Ohki K, et al.: Prognostic impact of specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down syndrome. Genes Chromosomes Cancer 56 (5): 394-404, 2017.
- Chisholm KM, Smith J, Heerema-McKenney AE, et al.: Pathologic, cytogenetic, and molecular features of acute myeloid leukemia with megakaryocytic differentiation: A report from the Children's Oncology Group. Pediatr Blood Cancer 70 (5): e30251, 2023.
- Eidenschink Brodersen L, Alonzo TA, Menssen AJ, et al.: A recurrent immunophenotype at diagnosis independently identifies high-risk pediatric acute myeloid leukemia: a report from Children's Oncology Group. Leukemia 30 (10): 2077-2080, 2016.
- Pardo LM, Voigt AP, Alonzo TA, et al.: Deciphering the Significance of CD56 Expression in Pediatric Acute Myeloid Leukemia: A Report from the Children's Oncology Group. Cytometry B Clin Cytom 98 (1): 52-56, 2020.
- Smith JL, Ries RE, Hylkema T, et al.: Comprehensive Transcriptome Profiling of Cryptic CBFA2T3-GLIS2 Fusion-Positive AML Defines Novel Therapeutic Options: A COG and TARGET Pediatric AML Study. Clin Cancer Res 26 (3): 726-737, 2020.
- Tang T, Le Q, Castro S, et al.: Targeting FOLR1 in high-risk CBF2AT3-GLIS2 pediatric AML with STRO-002 FOLR1-antibody-drug conjugate. Blood Adv 6 (22): 5933-5937, 2022.
- Le Q, Hadland B, Smith JL, et al.: CBFA2T3-GLIS2 model of pediatric acute megakaryoblastic leukemia identifies FOLR1 as a CAR T cell target. J Clin Invest 132 (22): , 2022.
- Takeda A, Yaseen NR: Nucleoporins and nucleocytoplasmic transport in hematologic malignancies. Semin Cancer Biol 27: 3-10, 2014.
- Bertrums EJM, Smith JL, Harmon L, et al.: Comprehensive molecular and clinical characterization of NUP98 fusions in pediatric acute myeloid leukemia. Haematologica 108 (8): 2044-2058, 2023.
- Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, et al.: NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 118 (13): 3645-56, 2011.
- Ostronoff F, Othus M, Gerbing RB, et al.: NUP98/NSD1 and FLT3/ITD coexpression is more prevalent in younger AML patients and leads to induction failure: a COG and SWOG report. Blood 124 (15): 2400-7, 2014.
- Panarello C, Rosanda C, Morerio C: Cryptic translocation t(5;11)(q35;p15.5) with involvement of the NSD1 and NUP98 genes without 5q deletion in childhood acute myeloid leukemia. Genes Chromosomes Cancer 35 (3): 277-81, 2002.
- Struski S, Lagarde S, Bories P, et al.: NUP98 is rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group with a poor prognosis. Leukemia 31 (3): 565-572, 2017.
- Cerveira N, Correia C, Dória S, et al.: Frequency of NUP98-NSD1 fusion transcript in childhood acute myeloid leukaemia. Leukemia 17 (11): 2244-7, 2003.
- McNeer NA, Philip J, Geiger H, et al.: Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia 33 (8): 1934-1943, 2019.
- van Zutven LJ, Onen E, Velthuizen SC, et al.: Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Genes Chromosomes Cancer 45 (5): 437-46, 2006.
- Noort S, Wander P, Alonzo TA, et al.: The clinical and biological characteristics of NUP98-KDM5A in pediatric acute myeloid leukemia. Haematologica 106 (2): 630-634, 2021.
- Espersen ADL, Noren-Nyström U, Abrahamsson J, et al.: Acute myeloid leukemia (AML) with t(7;12)(q36;p13) is associated with infancy and trisomy 19: Data from Nordic Society for Pediatric Hematology and Oncology (NOPHO-AML) and review of the literature. Genes Chromosomes Cancer 57 (7): 359-365, 2018.
- von Bergh AR, van Drunen E, van Wering ER, et al.: High incidence of t(7;12)(q36;p13) in infant AML but not in infant ALL, with a dismal outcome and ectopic expression of HLXB9. Genes Chromosomes Cancer 45 (8): 731-9, 2006.
- Tosi S, Harbott J, Teigler-Schlegel A, et al.: t(7;12)(q36;p13), a new recurrent translocation involving ETV6 in infant leukemia. Genes Chromosomes Cancer 29 (4): 325-32, 2000.
- Slater RM, von Drunen E, Kroes WG, et al.: t(7;12)(q36;p13) and t(7;12)(q32;p13)--translocations involving ETV6 in children 18 months of age or younger with myeloid disorders. Leukemia 15 (6): 915-20, 2001.
- Park J, Kim M, Lim J, et al.: Three-way complex translocations in infant acute myeloid leukemia with t(7;12)(q36;p13): the incidence and correlation of a HLXB9 overexpression. Cancer Genet Cytogenet 191 (2): 102-5, 2009.
- de Rooij J, Beuling E, Fornerod M, et al.: ETV6 Aberrations Are a Recurrent Event in Pediatric Acute Myeloid Leukemia with Poor Clinical Outcome. [Abstract] Blood 124 (21): 1012, 2014.
Also available online . Last accessed January 25, 2024. - Helton HL, Ries RE, Alonzo TA, et al.: Clinically Significant Mutations, Deletions and Translocations Involving ETV6 Identified by Whole Genome and Whole Exome Sequencing; Report From NCI/COG Target AML Initiative. [Abstract] Blood 120 (21): 125, 2012.
Also available online . Last accessed January 25, 2024. - Papadopoulou V, Schoumans J, Scarpelli I, et al.: Description of an Institutional Cohort of Myeloid Neoplasms Carrying ETV6-Locus Deletions or ETV6 Rearrangements. Acta Haematol 146 (5): 401-407, 2023.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Outcome of pediatric patients with acute myeloid leukemia (AML) and -5/5q- abnormalities from five pediatric AML treatment protocols: a report from the Children's Oncology Group. Pediatr Blood Cancer 60 (12): 2073-8, 2013.
- Stevens RF, Hann IM, Wheatley K, et al.: Marked improvements in outcome with chemotherapy alone in paediatric acute myeloid leukemia: results of the United Kingdom Medical Research Council's 10th AML trial. MRC Childhood Leukaemia Working Party. Br J Haematol 101 (1): 130-40, 1998.
- Hasle H, Alonzo TA, Auvrignon A, et al.: Monosomy 7 and deletion 7q in children and adolescents with acute myeloid leukemia: an international retrospective study. Blood 109 (11): 4641-7, 2007.
- Rasche M, von Neuhoff C, Dworzak M, et al.: Genotype-outcome correlations in pediatric AML: the impact of a monosomal karyotype in trial AML-BFM 2004. Leukemia 31 (12): 2807-2814, 2017.
- Blink M, Zimmermann M, von Neuhoff C, et al.: Normal karyotype is a poor prognostic factor in myeloid leukemia of Down syndrome: a retrospective, international study. Haematologica 99 (2): 299-307, 2014.
- Wlodarski MW, Sahoo SS, Niemeyer CM: Monosomy 7 in Pediatric Myelodysplastic Syndromes. Hematol Oncol Clin North Am 32 (4): 729-743, 2018.
- Abla O, Ries RE, Triche T, et al.: Structural variants involving MLLT10 fusion are associated with adverse outcomes in pediatric acute myeloid leukemia. Blood Adv 8 (8): 2005-2017, 2024.
- Mark C, Meshinchi S, Joyce B, et al.: Treatment outcomes of childhood PICALM::MLLT10 acute leukaemias. Br J Haematol 204 (2): 576-584, 2024.
- Schnittger S, Schoch C, Dugas M, et al.: Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 100 (1): 59-66, 2002.
- Thiede C, Steudel C, Mohr B, et al.: Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 99 (12): 4326-35, 2002.
- Iwai T, Yokota S, Nakao M, et al.: Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. The Children's Cancer and Leukemia Study Group, Japan. Leukemia 13 (1): 38-43, 1999.
- Meshinchi S, Stirewalt DL, Alonzo TA, et al.: Activating mutations of RTK/ras signal transduction pathway in pediatric acute myeloid leukemia. Blood 102 (4): 1474-9, 2003.
- Zwaan CM, Meshinchi S, Radich JP, et al.: FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood 102 (7): 2387-94, 2003.
- Voigt AP, Brodersen LE, Alonzo TA, et al.: Phenotype in combination with genotype improves outcome prediction in acute myeloid leukemia: a report from Children's Oncology Group protocol AAML0531. Haematologica 102 (12): 2058-2068, 2017.
- Berman JN, Gerbing RB, Alonzo TA, et al.: Prevalence and clinical implications of NRAS mutations in childhood AML: a report from the Children's Oncology Group. Leukemia 25 (6): 1039-42, 2011.
- Kühn MW, Radtke I, Bullinger L, et al.: High-resolution genomic profiling of adult and pediatric core-binding factor acute myeloid leukemia reveals new recurrent genomic alterations. Blood 119 (10): e67-75, 2012.
- Lion T, Haas OA: Acute megakaryocytic leukemia with the t(1;22)(p13;q13). Leuk Lymphoma 11 (1-2): 15-20, 1993.
- Duchayne E, Fenneteau O, Pages MP, et al.: Acute megakaryoblastic leukaemia: a national clinical and biological study of 53 adult and childhood cases by the Groupe Français d'Hématologie Cellulaire (GFHC). Leuk Lymphoma 44 (1): 49-58, 2003.
- Ma Z, Morris SW, Valentine V, et al.: Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat Genet 28 (3): 220-1, 2001.
- Mercher T, Coniat MB, Monni R, et al.: Involvement of a human gene related to the Drosophila spen gene in the recurrent t(1;22) translocation of acute megakaryocytic leukemia. Proc Natl Acad Sci U S A 98 (10): 5776-9, 2001.
- Bernstein J, Dastugue N, Haas OA, et al.: Nineteen cases of the t(1;22)(p13;q13) acute megakaryblastic leukaemia of infants/children and a review of 39 cases: report from a t(1;22) study group. Leukemia 14 (1): 216-8, 2000.
- Hammer ASB, Juul-Dam KL, Sandahl JD, et al.: Hypodiploidy has unfavorable impact on survival in pediatric acute myeloid leukemia: an I-BFM Study Group collaboration. Blood Adv 7 (6): 1045-1055, 2023.
- Umeda M, Ma J, Huang BJ, et al.: Integrated Genomic Analysis Identifies UBTF Tandem Duplications as a Recurrent Lesion in Pediatric Acute Myeloid Leukemia. Blood Cancer Discov 3 (3): 194-207, 2022.
- Ryland GL, Umeda M, Holmfeldt L, et al.: Description of a novel subtype of acute myeloid leukemia defined by recurrent CBFB insertions. Blood 141 (7): 800-805, 2023.
- Rungjirajittranon T, Siriwannangkul T, Kungwankiattichai S, et al.: Clinical Outcomes of Acute Myeloid Leukemia Patients Harboring the RUNX1 Mutation: Is It Still an Unfavorable Prognosis? A Cohort Study and Meta-Analysis. Cancers (Basel) 14 (21): , 2022.
- Yamato G, Shiba N, Yoshida K, et al.: RUNX1 mutations in pediatric acute myeloid leukemia are associated with distinct genetic features and an inferior prognosis. Blood 131 (20): 2266-2270, 2018.
- Sendker S, Awada A, Domagalla S, et al.: RUNX1 mutation has no prognostic significance in paediatric AML: a retrospective study of the AML-BFM study group. Leukemia 37 (7): 1435-1443, 2023.
- Paschka P, Marcucci G, Ruppert AS, et al.: Wilms' tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol 26 (28): 4595-602, 2008.
- Virappane P, Gale R, Hills R, et al.: Mutation of the Wilms' tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: the United Kingdom Medical Research Council Adult Leukaemia Working Party. J Clin Oncol 26 (33): 5429-35, 2008.
- Gaidzik VI, Schlenk RF, Moschny S, et al.: Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: a study of the German-Austrian AML Study Group. Blood 113 (19): 4505-11, 2009.
- Renneville A, Boissel N, Zurawski V, et al.: Wilms tumor 1 gene mutations are associated with a higher risk of recurrence in young adults with acute myeloid leukemia: a study from the Acute Leukemia French Association. Cancer 115 (16): 3719-27, 2009.
- Ho PA, Zeng R, Alonzo TA, et al.: Prevalence and prognostic implications of WT1 mutations in pediatric acute myeloid leukemia (AML): a report from the Children's Oncology Group. Blood 116 (5): 702-10, 2010.
- Hollink IH, van den Heuvel-Eibrink MM, Zimmermann M, et al.: Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood 113 (23): 5951-60, 2009.
- Ley TJ, Ding L, Walter MJ, et al.: DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363 (25): 2424-33, 2010.
- Yan XJ, Xu J, Gu ZH, et al.: Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet 43 (4): 309-15, 2011.
- Thol F, Damm F, Lüdeking A, et al.: Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol 29 (21): 2889-96, 2011.
- Ho PA, Kutny MA, Alonzo TA, et al.: Leukemic mutations in the methylation-associated genes DNMT3A and IDH2 are rare events in pediatric AML: a report from the Children's Oncology Group. Pediatr Blood Cancer 57 (2): 204-9, 2011.
- Green CL, Evans CM, Hills RK, et al.: The prognostic significance of IDH1 mutations in younger adult patients with acute myeloid leukemia is dependent on FLT3/ITD status. Blood 116 (15): 2779-82, 2010.
- Paschka P, Schlenk RF, Gaidzik VI, et al.: IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol 28 (22): 3636-43, 2010.
- Abbas S, Lugthart S, Kavelaars FG, et al.: Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 116 (12): 2122-6, 2010.
- Marcucci G, Maharry K, Wu YZ, et al.: IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 28 (14): 2348-55, 2010.
- Wagner K, Damm F, Göhring G, et al.: Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol 28 (14): 2356-64, 2010.
- Figueroa ME, Abdel-Wahab O, Lu C, et al.: Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18 (6): 553-67, 2010.
- Ward PS, Patel J, Wise DR, et al.: The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17 (3): 225-34, 2010.
- Dang L, White DW, Gross S, et al.: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462 (7274): 739-44, 2009.
- Damm F, Thol F, Hollink I, et al.: Prevalence and prognostic value of IDH1 and IDH2 mutations in childhood AML: a study of the AML-BFM and DCOG study groups. Leukemia 25 (11): 1704-10, 2011.
- Oki K, Takita J, Hiwatari M, et al.: IDH1 and IDH2 mutations are rare in pediatric myeloid malignancies. Leukemia 25 (2): 382-4, 2011.
- Pigazzi M, Ferrari G, Masetti R, et al.: Low prevalence of IDH1 gene mutation in childhood AML in Italy. Leukemia 25 (1): 173-4, 2011.
- Ho PA, Alonzo TA, Kopecky KJ, et al.: Molecular alterations of the IDH1 gene in AML: a Children's Oncology Group and Southwest Oncology Group study. Leukemia 24 (5): 909-13, 2010.
- Andersson AK, Miller DW, Lynch JA, et al.: IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia 25 (10): 1570-7, 2011.
- Maxson JE, Ries RE, Wang YC, et al.: CSF3R mutations have a high degree of overlap with CEBPA mutations in pediatric AML. Blood 127 (24): 3094-8, 2016.
- Germeshausen M, Kratz CP, Ballmaier M, et al.: RAS and CSF3R mutations in severe congenital neutropenia. Blood 114 (16): 3504-5, 2009.
- Skokowa J, Steinemann D, Katsman-Kuipers JE, et al.: Cooperativity of RUNX1 and CSF3R mutations in severe congenital neutropenia: a unique pathway in myeloid leukemogenesis. Blood 123 (14): 2229-37, 2014.
- Melnick A, Licht JD: Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 93 (10): 3167-215, 1999.
- Sanz MA, Grimwade D, Tallman MS, et al.: Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 113 (9): 1875-91, 2009.
- Falini B, Flenghi L, Fagioli M, et al.: Immunocytochemical diagnosis of acute promyelocytic leukemia (M3) with the monoclonal antibody PG-M3 (anti-PML). Blood 90 (10): 4046-53, 1997.
- Gomis F, Sanz J, Sempere A, et al.: Immunofluorescent analysis with the anti-PML monoclonal antibody PG-M3 for rapid and accurate genetic diagnosis of acute promyelocytic leukemia. Ann Hematol 83 (11): 687-90, 2004.
- Dimov ND, Medeiros LJ, Kantarjian HM, et al.: Rapid and reliable confirmation of acute promyelocytic leukemia by immunofluorescence staining with an antipromyelocytic leukemia antibody: the M. D. Anderson Cancer Center experience of 349 patients. Cancer 116 (2): 369-76, 2010.
- Zelent A, Guidez F, Melnick A, et al.: Translocations of the RARalpha gene in acute promyelocytic leukemia. Oncogene 20 (49): 7186-203, 2001.
- Yan W, Zhang G: Molecular Characteristics and Clinical Significance of 12 Fusion Genes in Acute Promyelocytic Leukemia: A Systematic Review. Acta Haematol 136 (1): 1-15, 2016.
- Rego EM, Ruggero D, Tribioli C, et al.: Leukemia with distinct phenotypes in transgenic mice expressing PML/RAR alpha, PLZF/RAR alpha or NPM/RAR alpha. Oncogene 25 (13): 1974-9, 2006.
- Licht JD, Chomienne C, Goy A, et al.: Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood 85 (4): 1083-94, 1995.
- Guidez F, Ivins S, Zhu J, et al.: Reduced retinoic acid-sensitivities of nuclear receptor corepressor binding to PML- and PLZF-RARalpha underlie molecular pathogenesis and treatment of acute promyelocytic leukemia. Blood 91 (8): 2634-42, 1998.
- Grimwade D, Biondi A, Mozziconacci MJ, et al.: Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Groupe Français de Cytogénétique Hématologique, Groupe de Français d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action "Molecular Cytogenetic Diagnosis in Haematological Malignancies". Blood 96 (4): 1297-308, 2000.
- Sukhai MA, Wu X, Xuan Y, et al.: Myeloid leukemia with promyelocytic features in transgenic mice expressing hCG-NuMA-RARalpha. Oncogene 23 (3): 665-78, 2004.
- Redner RL, Corey SJ, Rush EA: Differentiation of t(5;17) variant acute promyelocytic leukemic blasts by all-trans retinoic acid. Leukemia 11 (7): 1014-6, 1997.
- Wells RA, Catzavelos C, Kamel-Reid S: Fusion of retinoic acid receptor alpha to NuMA, the nuclear mitotic apparatus protein, by a variant translocation in acute promyelocytic leukaemia. Nat Genet 17 (1): 109-13, 1997.
- Wells RA, Hummel JL, De Koven A, et al.: A new variant translocation in acute promyelocytic leukaemia: molecular characterization and clinical correlation. Leukemia 10 (4): 735-40, 1996.
- Chen X, Wang F, Zhou X, et al.: Torque teno mini virus driven childhood acute promyelocytic leukemia: The third case report and sequence analysis. Front Oncol 12: 1074913, 2022.
- Sala-Torra O, Beppu LW, Abukar FA, et al.: TTMV-RARA fusion as a recurrent cause of AML with APL characteristics. Blood Adv 6 (12): 3590-3592, 2022.
- Callens C, Chevret S, Cayuela JM, et al.: Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia 19 (7): 1153-60, 2005.
- Gale RE, Hills R, Pizzey AR, et al.: Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood 106 (12): 3768-76, 2005.
- Arrigoni P, Beretta C, Silvestri D, et al.: FLT3 internal tandem duplication in childhood acute myeloid leukaemia: association with hyperleucocytosis in acute promyelocytic leukaemia. Br J Haematol 120 (1): 89-92, 2003.
- Noguera NI, Breccia M, Divona M, et al.: Alterations of the FLT3 gene in acute promyelocytic leukemia: association with diagnostic characteristics and analysis of clinical outcome in patients treated with the Italian AIDA protocol. Leukemia 16 (11): 2185-9, 2002.
- Tallman MS, Kim HT, Montesinos P, et al.: Does microgranular variant morphology of acute promyelocytic leukemia independently predict a less favorable outcome compared with classical M3 APL? A joint study of the North American Intergroup and the PETHEMA Group. Blood 116 (25): 5650-9, 2010.
- Iland HJ, Bradstock K, Supple SG, et al.: All-trans-retinoic acid, idarubicin, and IV arsenic trioxide as initial therapy in acute promyelocytic leukemia (APML4). Blood 120 (8): 1570-80; quiz 1752, 2012.
- Kutny MA, Moser BK, Laumann K, et al.: FLT3 mutation status is a predictor of early death in pediatric acute promyelocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 59 (4): 662-7, 2012.
- Kutny MA, Alonzo TA, Abla O, et al.: Assessment of Arsenic Trioxide and All-trans Retinoic Acid for the Treatment of Pediatric Acute Promyelocytic Leukemia: A Report From the Children's Oncology Group AAML1331 Trial. JAMA Oncol 8 (1): 79-87, 2022.
- Quintás-Cardama A, Cortes J: Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 113 (8): 1619-30, 2009.
- Loghavi S, Kanagal-Shamanna R, Khoury JD, et al.: Fifth Edition of the World Health Classification of Tumors of the Hematopoietic and Lymphoid Tissue: Myeloid Neoplasms. Mod Pathol 37 (2): 100397, 2024.
- Wang W, Cortes JE, Tang G, et al.: Risk stratification of chromosomal abnormalities in chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood 127 (22): 2742-50, 2016.
- Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 (11): 1334-40, 2015.
- Stieglitz E, Taylor-Weiner AN, Chang TY, et al.: The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47 (11): 1326-33, 2015.
- Murakami N, Okuno Y, Yoshida K, et al.: Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 131 (14): 1576-1586, 2018.
- Sakaguchi H, Okuno Y, Muramatsu H, et al.: Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet 45 (8): 937-41, 2013.
- Stieglitz E, Mazor T, Olshen AB, et al.: Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat Commun 8 (1): 2127, 2017.
- Hecht A, Meyer JA, Behnert A, et al.: Molecular and phenotypic diversity of CBL-mutated juvenile myelomonocytic leukemia. Haematologica 107 (1): 178-186, 2022.
- Helsmoortel HH, Bresolin S, Lammens T, et al.: LIN28B overexpression defines a novel fetal-like subgroup of juvenile myelomonocytic leukemia. Blood 127 (9): 1163-72, 2016.
- Schwartz JR, Ma J, Lamprecht T, et al.: The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun 8 (1): 1557, 2017.
- Pastor V, Hirabayashi S, Karow A, et al.: Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia 31 (3): 759-762, 2017.
- Collin M, Dickinson R, Bigley V: Haematopoietic and immune defects associated with GATA2 mutation. Br J Haematol 169 (2): 173-87, 2015.
- Wlodarski MW, Hirabayashi S, Pastor V, et al.: Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 127 (11): 1387-97; quiz 1518, 2016.
- Wlodarski MW, Collin M, Horwitz MS: GATA2 deficiency and related myeloid neoplasms. Semin Hematol 54 (2): 81-86, 2017.
- Davidsson J, Puschmann A, Tedgård U, et al.: SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 32 (5): 1106-1115, 2018.
- Schwartz JR, Wang S, Ma J, et al.: Germline SAMD9 mutation in siblings with monosomy 7 and myelodysplastic syndrome. Leukemia 31 (8): 1827-1830, 2017.
- Narumi S, Amano N, Ishii T, et al.: SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet 48 (7): 792-7, 2016.
- Chen DH, Below JE, Shimamura A, et al.: Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am J Hum Genet 98 (6): 1146-1158, 2016.
- Wong JC, Bryant V, Lamprecht T, et al.: Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight 3 (14): , 2018.
- Göhring G, Michalova K, Beverloo HB, et al.: Complex karyotype newly defined: the strongest prognostic factor in advanced childhood myelodysplastic syndrome. Blood 116 (19): 3766-9, 2010.
- Haase D, Germing U, Schanz J, et al.: New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood 110 (13): 4385-95, 2007.
- Arber DA, Vardiman JW, Brunning RD: Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 110-23.
- Hitzler JK, Cheung J, Li Y, et al.: GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood 101 (11): 4301-4, 2003.
- Mundschau G, Gurbuxani S, Gamis AS, et al.: Mutagenesis of GATA1 is an initiating event in Down syndrome leukemogenesis. Blood 101 (11): 4298-300, 2003.
- Massey GV, Zipursky A, Chang MN, et al.: A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481. Blood 107 (12): 4606-13, 2006.
- Groet J, McElwaine S, Spinelli M, et al.: Acquired mutations in GATA1 in neonates with Down's syndrome with transient myeloid disorder. Lancet 361 (9369): 1617-20, 2003.
- Rainis L, Bercovich D, Strehl S, et al.: Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood 102 (3): 981-6, 2003.
- Wechsler J, Greene M, McDevitt MA, et al.: Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet 32 (1): 148-52, 2002.
- Ge Y, Stout ML, Tatman DA, et al.: GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst 97 (3): 226-31, 2005.
- Sato T, Yoshida K, Toki T, et al.: Landscape of driver mutations and their clinical effects on Down syndrome-related myeloid neoplasms. Blood 143 (25): 2627-2643, 2024.
Tumores del sistema nervioso central
Los tumores del sistema nervioso central (SNC) son los gliomas (incluso astrocitomas), los tumores glioneuronales, los tumores neuronales, los tumores teratoides rabdoides atípicos del SNC, los meduloblastomas, los tumores embrionarios distintos al meduloblastoma, los tumores pineales y los ependimomas.
A continuación, se usa la terminología de la clasificación de la Organización Mundial de la Salud (OMS) de 2021 para los tumores del sistema nervioso central. En esta clasificación de 2021 se favorece la función del diagnóstico molecular en la clasificación de tumores del SNC e incluye muchos cambios importantes a la clasificación anterior de la OMS, que data de 2016.[
Gliomas (incluso astrocitomas) y tumores neuronales o glioneuronales
Esta categoría incluye, entre otros diagnósticos, los gliomas difusos de grado bajo de tipo pediátrico, los gliomas difusos de grado alto de tipo pediátrico, los gliomas astrocíticos circunscritos, los tumores glioneuronales y los tumores neuronales.
En el caso de los gliomas difusos de tipo pediátrico, los reordenamientos de la familia MYB de factores de transcripción (MYB y MYBL1) son las alteraciones genómicas que se notifican con mayor frecuencia en los tumores de grado bajo.[
La categoría del glioma difuso de línea media con alteración H3 K27M incluye los tumores que antes se clasificaban como gliomas pontinos intrínsecos difusos (GPID o DIPG); la mayoría de los datos provienen de experiencias con GPID. Esta categoría también incluye los gliomas que tienen una variante H3 K27M y surgen en las estructuras de la línea media como el tálamo.
Síndromes de susceptibilidad al cáncer seleccionados relacionados con los gliomas infantiles
Neurofibromatosis de tipo 1
Los niños con neurofibromatosis de tipo 1 (NF1) tienen una mayor propensión a presentar gliomas de grado bajo, en especial en la vía óptica. Hasta el 20 % de los pacientes con NF1 presentarán un glioma de la vía óptica. La mayoría de los niños con gliomas del nervio óptico relacionados con la NF1 son asintomáticos o tienen síntomas no progresivos y no requieren tratamiento antitumoral. Por lo general, no está indicada la detección sistemática con imágenes por resonancia magnética (IRM) en pacientes asintomáticos con NF1, aunque algunos investigadores obtienen una IRM al inicio para niños pequeños que no se pueden someter a exámenes oftalmológicos detallados.[
El diagnóstico a menudo se basa en los hallazgos clínicos compatibles y las características de las imágenes. La confirmación histológica casi nunca es necesaria en el momento del diagnóstico. Cuando se realizan biopsias, los tumores que se encuentran son de manera predominante astrocitomas pilocíticos.[
Las indicaciones para el tratamiento varían y, a menudo, se basan en el objetivo de preservar la visión.
En muy pocas ocasiones, los pacientes con NF1 presentan gliomas de grado alto. A veces, estos tumores son el resultado de una transformación de un tumor de grado inferior.[
Esclerosis tuberosa
Los pacientes con esclerosis tuberosa son más propensos a presentar astrocitoma subependimario de células gigantes (SEGA). Las variantes de TSC1 o TSC2 causan la activación constitutiva de la vía de señalización del complejo 1 del blanco de la rapamicina en los mamíferos (mTORC1), lo que aumenta la proliferación. Los SEGA responden a los abordajes molecularmente dirigidos con inhibidores de la vía mTORC1.[
Características moleculares y alteraciones genómicas recurrentes
Las alteraciones genómicas recurrentes que provocan la activación constitutiva de la vía de la proteína cinasa activada por mitógenos (MAPK), que con mayor frecuencia afecta al gen BRAF, representan el principal (y a menudo único) iniciador oncogénico en la gran mayoría de los gliomas infantiles de grado bajo, como los astrocitomas pilocíticos o pilomixoides, los gangliogliomas y otros.[
Los genomas tumorales más complejos son característicos de los gliomas difusos de grado alto de tipo pediátrico. Estos genomas complejos incluyen alteraciones genómicas recurrentes en genes codificadores de histonas H3 (por ejemplo, H3F3A, HIST1H3B), genes de las vías de reparación del daño al DNA (por ejemplo, TP53, PPM1D, ATM, MDM2), genes modificadores de la cromatina (por ejemplo, ATRX, BCOR, SETD2), genes de las vías del ciclo celular (por ejemplo, CDKN2A, CDKN2B, RB1) o presentan amplificaciones oncogénicas (PDGFR, VEGFR2, KIT, MYC, MYCN).[
Un subconjunto infrecuente de gliomas de grado alto de tipo pediátrico que surgen en pacientes con deficiencia hereditaria y bialélica en la reparación de los errores de emparejamiento (bMMRD) se caracteriza por una carga mutacional extremadamente alta. La identificación correcta de estos pacientes en el momento del diagnóstico es fundamental debido a la resistencia intrínseca a la temozolomida y la sensibilidad al tratamiento con inhibidores de puntos de control inmunitario.[
BRAF::KIAA1549
En el astrocitoma pilocítico, la activación de BRAF sucede con mayor frecuencia por una fusión génica BRAF::KIAA1549, que genera una proteína de fusión sin el dominio autorregulador de BRAF.[
La presencia de la fusión BRAF::KIAA1549 se relaciona con una mejora del desenlace clínico (supervivencia sin progresión [SSP] y supervivencia general [SG]) en pacientes con astrocitoma pilocítico.[
Variantes deBRAF
Las variantes puntuales activadoras de BRAF, con mayor frecuencia BRAF V600E, están presentes en un subconjunto de gliomas infantiles y tumores glioneuronales que abarcan varios tipos histológicos, como el xantoastrocitoma pleomórfico, el astrocitoma pilocítico, el ganglioglioma, el astrocitoma o el ganglioglioma desmoplásico infantil (de lactantes), entre otros.[
En estudios clínicos retrospectivos se observó lo siguiente:
- En una serie retrospectiva de más de 400 niños con gliomas de grado bajo, el 17 % de los tumores exhibían una variante BRAF V600E. La tasa de SSP a 10 años fue del 27 % en los pacientes que tenían tumores con variante BRAF V600E, en comparación con el 60 % en los pacientes cuyos tumores no albergaban esa variante. Otros factores relacionados con este pronóstico precario fueron la resección subtotal y la deleción de CDKN2A.[
17 ][Nivel de evidencia C2] Incluso en los pacientes sometidos a una resección macroscópica total, se observó recidiva en un tercio de ellos, lo que indica que los tumores que tienen la variante BRAF V600E tienen un fenotipo más invasivo que otras variantes de glioma de grado bajo. - En un análisis similar, los niños con astrocitomas diencefálicos de grado bajo con una variante BRAF V600E tuvieron una tasa de SSP a 5 años del 22 %, en comparación con una tasa de SSP del 52 % en los niños con BRAF natural.[
18 ][Nivel de evidencia C2] - La frecuencia de la variante BRAF V600E en casos pediátricos fue significativamente superior en los gliomas de grado bajo que se transformaron en gliomas de grado alto (8 de 18 casos), en comparación con la frecuencia de la variante en los casos sin transformación (10 de 167 casos).[
15 ]
Variantes deNF1
Las alteraciones somáticas en NF1 se observan con mayor frecuencia en niños con NF1 y se relacionan con alteraciones de la línea germinal en el supresor tumoral NF1. La pérdida de heterocigosis para NF1 representa la alteración somática más común en estos pacientes seguida de variantes inactivadoras del segundo alelo de NF1, y es compatible con la lesión secundaria ("segundo golpe") necesaria para la carcinogénesis. Si bien la mayoría de los pacientes con gliomas de grado bajo que presentan alteraciones en NF1 tienen un pronóstico excelente a largo plazo, es posible que se produzca una transformación secundaria a un glioma de grado alto en un subgrupo pequeño de estos casos. Desde el punto de vista genómico, la transformación se vincula con la adquisición de otros iniciadores oncogénicos, como alteraciones funcionales de pérdida de función en CDKN2A, CDKN2B o ATRX. Los pacientes con NF1 también presentan gliomas primarios de grado alto, pero esto es muy infrecuente. Las alteraciones genómicas fuera de NF1 que afectan la vía de señalización MAPK son muy infrecuentes en los gliomas que se presentan en niños con NF1.[
Fusiones de los genesALK,NTRK1,NTRK2,NTRK3oROS1
Los gliomas de grado alto con características moleculares típicas surgen en lactantes, por lo general se diagnostican durante el primer año de vida.[
Las fusiones génicas de ROS1 también se han notificado en los gliomas que se presentan en niños de más edad y en adultos. En un metanálisis retrospectivo, en el que se incluyeron 40 niños mayores de 1 año, se encontró que las fusiones génicas de ROS1 se presentaron en gliomas de diferentes tipos histológicos, incluso en gliomas difusos de grado alto y de grado bajo, así como en tumores glioneuronales.[
Otras alteraciones genómicas
Además de la activación de BRAF o la pérdida de NF1, se han observado otras alteraciones primarias iniciadoras oncogénicas en la vía de señalización MAPK en astrocitomas pilocíticos y otros gliomas de tipo pediátrico. Estos incluyen variantes oncogénicas o fusiones que afectan FGFR1, FGFR2, PTPN11, RAF1, NTRK2 y otros genes.[
Los gliomas de grado bajo con reordenamientos en la familia de factores de transcripción MYB [
Gliomas angiocéntricos
Los gliomas angiocéntricos habitualmente surgen en niños y adultos jóvenes como tumores encefálicos que causan convulsiones.[
En dos informes de 2016 se identificaron alteraciones en el gen MYB presentes en casi todos los casos diagnosticados como gliomas angiocéntricos; el gen QKI fue el principal compañero de fusión en los casos en los que fue posible obtener pruebas sobre la pareja de fusión.[
Astroblastomas con alteración deMN1
Los astroblastomas se definen según sus características histológicas como neoplasias gliales compuestas de células positivas para GFAP que contienen pseudorosetas astroblásticas, a menudo con esclerosis. Los astroblastomas se diagnostican de manera principal durante la niñez y hasta el comienzo de la edad adulta.[
En los siguientes estudios se caracterizaron las alteraciones genómicas asociadas con el astroblastoma:
- En un informe se detalló una clasificación molecular de los tumores neuroectodérmicos primitivos (TNEP) del SNC y se descubrió una entidad nueva llamada tumor neuroepitelial de grado alto del SNC con alteración de MN1 (CNS HGNET-MN1) que se caracterizó por la presencia de fusiones génicas que afectan el gen MN1.[
27 ] La mayoría de los tumores con diagnóstico histológico de astroblastoma (16 de 23) pertenecían a esta entidad definida por sus características moleculares. - En un informe de 27 casos de astroblastomas definidos por sus características histológicas se encontraron 10 casos con reordenamientos de MN1, 7 casos con reordenamientos de BRAF y 2 casos con reordenamientos de RELA.[
28 ] Con el análisis de matriz de metilación se observó que los casos con reordenamientos de MN1 se agruparon con el CNS HGNET-MN1, los casos con alteraciones de BRAF se agruparon con los xantoastrocitomas pleomórficos, y los casos con alteración en RELA se agruparon con los ependimomas. - En la evaluación genómica de 8 casos de astroblastomas se detectaron 4 casos con alteraciones de MN1. De los 4 casos restantes, 2 presentaron alteraciones genómicas compatibles con glioma de grado alto y 2 casos no se pudieron clasificar según sus características moleculares.[
29 ] - En un estudio se caracterizaron 8 casos de astroblastoma. Los 5 casos sometidos a análisis de hibridación fluorescente in situ mostraron reordenamientos de MN1.[
30 ]
En estos informes se indica que el diagnóstico histológico del astroblastoma abarca un grupo heterogéneo de entidades definidas por sus características genómicas. Los astroblastomas con fusiones de MN1 representan un subconjunto diferenciado de los casos diagnosticados por sus características histológicas.[
Variantes deIDH1eIDH2
Los tumores con alteraciones de IDH1 e IDH2 ocurren en la población pediátrica como los gliomas de grado bajo (grado 2 de la OMS), los gliomas de grado alto (grado 3 y 4 de la OMS), y los oligodendrogliomas con codeleción de 1p y 19q. Para obtener más información sobre los gliomas con alteración en IDH1 y IDH2, consultar la sección
Características moleculares de los gliomas de grado alto de tipo pediátrico
Los gliomas infantiles de grado alto son diferentes, desde el punto de vista biológico, de los que surgen en adultos.[
Subgrupos definidos mediante los patrones de metilación del DNA
Los gliomas de grado alto de tipo pediátrico se pueden separar en subgrupos característicos a partir de patrones epigenéticos (metilación del DNA). Estos subgrupos exhiben ganancias o pérdidas del número de copias cromosómicas y variantes génicas en el tumor que son características.[
Los siguientes subgrupos de glioma de grado alto de tipo pediátrico se identificaron a partir de sus patrones de metilación del DNA y muestran características clínicas y moleculares distintivas:[
Alteraciones genómicas asociadas con los gliomas difusos de línea media
Variantes de histonas en K27: variantes de H3.3 (H3F3A) y H3.1 (HIST1H3By, con menor frecuencia,HIST1H3C) en K27 y variantes de EZHIP
Los casos con alteraciones de histonas en K27 se presentan sobre todo en la mitad de la niñez (mediana de edad, alrededor de 10 años), surgen casi exclusivamente en la línea media (tálamo, tronco encefálico y médula espinal) y acarrean un pronóstico muy precario. En la clasificación de la OMS de 2021 se agrupan estos cánceres en una sola entidad: glioma difuso de línea media con alteración H3 K27. Sin embargo, hay diferencias clínicas y biológicas entre los casos con variantes de H3.3 y H3.1, como se describe a continuación.[
Los casos de glioma difuso de línea media con alteración H3 K27 se definen por la pérdida de la trimetilación de H3 K27 debido a una variante H3 K27M, o con menor frecuencia, por sobreexpresión de EZHIP. Esta entidad incluye la mayoría de los gliomas de grado alto del hipotálamo, la protuberancia (gliomas pontinos intrínsecos difusos [GPID]) y la médula espinal, de predominio en niños, pero que también se presenta en adultos.[
Gliomas con H3.3 K27M: estos casos surgen por toda la línea media y la protuberancia; corresponden a cerca del 60 % de los casos en estos sitios y, por lo común, aparecen entre los 5 y 10 años de edad.[
Gliomas con H3.1 K27M: los casos con la alteración H3.1 K27M son casi 5 veces menos comunes que los casos con la alteración H3.3 K27M. Surgen de manera primaria en la protuberancia y se presentan a una edad más temprana que los otros casos del subtipo H3.3 K27M (mediana de edad, 5 vs. 6–10 años). Estos pacientes tienen un pronóstico algo más favorable que los casos del subtipo H3.3 K27M (mediana de supervivencia, 15 vs. 11 meses). Las variantes de ACVR1, que también es la variante observada en la afección genética fibrodisplasia osificante progresiva, están presentes en una proporción alta de casos con la alteración H3.1 K27M.[
Gliomas con H3.2 K27M: en escasas ocasiones, también se han identificado variantes K27M en casos con alteración en H3.2 (HIST2H3C).[
Un subgrupo de tumores con variantes H3 K27 presentarán al mismo tiempo una variante BRAF V600E o una variante de FGFR1. En una cohorte retrospectiva de 29 tumores, que se combinó con 31 casos que se habían publicado en el pasado, se demostró una predisposición mayor por la ubicación en el tálamo. Estos casos exhibían un grupo de metilación del DNA singular que se diferencia de otros subgrupos de glioma difusos de línea media y subtipos de glioma con alteraciones de BRAF o FGFR1. La mediana de supervivencia de estos pacientes superó los 3 años.[
Sobrexpresión de EZHIP: la pequeña minoría de pacientes con gliomas difusos de línea media que carecen de variantes de la histona H3 a menudo exhiben sobrexpresión de EZHIP.[
Variante de H3.3 (H3F3A) en G34
Los subtipos con alteración H3.3 G34 se presentan por variantes de H3.3 cuando cambia la glicina en posición 34 por arginina o valina (G34R/V).[
Los pacientes con variantes de H3F3A tienen un riesgo alto de fracaso del tratamiento,[
Variantes deIDH1eIDH2
Los tumores con alteraciones de IDH1 y de IDH2 se presentan en la población pediátrica como gliomas de grado bajo (grado 2 de la OMS), gliomas de grado alto (grados 3 y 4 de la OMS) y oligodendrogliomas con codeleción de 1p y 19q.[
- Las variantes de IDH1 son mucho más comunes que las variantes de IDH2 y se encuentran en alrededor del 90 % de los tumores del SNC con alteración de IDH.
- Los gliomas de grado bajo con alteración de IDH son más comunes que los gliomas de grado alto con alteración de IDH, y representan alrededor de tres cuartas partes de los casos de gliomas pediátricos con alteración de IDH.
- Los oligodendrogliomas con variantes de IDH representan cerca del 20 % de los tumores pediátricos en el SNC con variantes de IDH.
- La mediana de edad en el momento del diagnóstico para los pacientes pediátricos con tumores que exhiben alteración de IDH es de alrededor de 16 años, y los tumores en el SNC con alteración de IDH son muy poco frecuentes en niños de 10 años o menos.
- Al igual que en los astrocitomas con variantes de IDH que se presentan en los adultos, los que se observan en los niños con frecuencia exhiben variantes de TP53 (cerca del 90 % de los casos) y variantes de ATRX (cerca del 50 % de los casos).
- Del mismo modo que los gliomas de grado bajo con alteración de IDH, los tumores de grado bajo en pacientes pediátricos pueden progresar y convertirse en gliomas de grado alto.
Los casos con alteraciones de IDH1 representan una proporción baja de los gliomas de grado alto (cerca del 5–10 %) que se observan en el ámbito pediátrico. La mayoría de los casos corresponden a adolescentes mayores (mediana de edad en una población pediátrica, 16 años) con tumores hemisféricos.[
Los pacientes pediátricos con variantes de IDH1 tienen un pronóstico más favorable que aquellos con otros tipos de gliomas de grado alto.[
Se han notificado gliomas de grado alto raros, con alteración de IDH en niños con síndromes de deficiencia en la reparación de errores de emparejamiento (síndrome de Lynch o síndrome de deficiencia constitucional en la reparación de errores de emparejamiento).[
Tumor similar al xantoastrocitoma pleomórfico
Alrededor del 10 % de los gliomas infantiles de grado alto exhiben patrones de metilación del DNA que son similares a los del xantoastrocitoma pleomórfico (XAP).[
Astrocitoma de grado alto con características piloides
Esta entidad se incluyó en la clasificación de la OMS de 2016 (llamada astrocitoma pilocítico con anaplasia) para describir los tumores con características histológicas del astrocitoma pilocítico, aumento de la actividad mitótica y características adicionales de grado alto. La nomenclatura actual se adoptó en la clasificación de la OMS de 2021. En una publicación más reciente se describió una cohorte de 83 casos con estas características histológicas (tumores denominados astrocitomas anaplásicos con características piloides) que compartían un perfil de metilación del DNA común, distinto de los perfiles de metilación de otros gliomas. Estos tumores se presentaron con más frecuencia en adultos (mediana de edad, 41 años) y con frecuencia albergaban deleciones de CDKN2A/B, alteraciones en la vía MAPK (más a menudo en el gen NF1) y variantes o deleciones de ATRX. Se relacionan con una evolución clínica intermedia entre el astrocitoma pilocítico y el glioblastoma con IDH natural.[
Otras variantes
Los pacientes pediátricos con glioma de grado alto tipo glioblastoma multiforme cuyos tumores carecen de variantes de histonas y variantes de IDH1 representan alrededor del 40 % de los casos de glioblastoma multiforme infantil.[
Gliomas de grado alto en lactantes
Los lactantes y niños pequeños con gliomas de grado alto tienen tumores con características moleculares típicas [
En dos estudios de las características moleculares de los gliomas de grado alto en lactantes y niños pequeños se definió la naturaleza distintiva de los tumores que surgen en niños menores de 1 año. Un hallazgo clave de ambos estudios es la importancia de las fusiones de genes relacionados con tirosina–cinasas (por ejemplo, ALK, NTRK1, NTRK2, NTRK3 y ROS1) en pacientes de este grupo de edad. En ambos estudios también se encontró que los lactantes con gliomas de grado alto cuyos tumores tienen estas fusiones génicas tienen tasas de supervivencia mucho más altas que las de los niños mayores con gliomas de grado alto.[
En el primer estudio se presentaron datos de 118 niños menores de 1 año con diagnóstico de glioma de grado bajo o alto que tenían tejido tumoral disponible para la caracterización genómica.[
- Los tumores del grupo 1 están determinados por receptores tirosina–cinasas (RTK) y en su mayoría son de grado alto (83 %). Estos tumores albergan lesiones en ALK, ROS1, NTRK y MET. La mediana de edad en el momento del diagnóstico es de 3 meses, y las tasas de SG se acercan al 60 %.
- Los tumores del grupo 2 están determinados por RAS/MAPK; todos son gliomas hemisféricos de grado bajo, lo que representa un cuarto de los gliomas hemisféricos en lactantes. La alteración más común es la variante BRAF V600E, seguida de alteraciones en FGFR1 y fusiones de BRAF. En este grupo, la mediana de edad en el momento de la presentación inicial es de 8 meses y su pronóstico es el más favorable de todos (tasa de SG a 10 años del 93 %).
- Los tumores del grupo 3 están determinados por RAS/MAPK, su tipo histológico es de grado bajo y el sitio de origen es la línea media (alrededor del 80 % son gliomas de vía óptica y de hipotálamo). La mayoría de los tumores del grupo 3 exhiben fusiones de BRAF o alteraciones BRAF V600E. La mediana de edad en el momento del diagnóstico es de 7,5 meses. La tasa de supervivencia sin progresión (SSP) a los 5 años fue de cerca del 20 %, y la tasa de SG a los 10 años fue de alrededor del 50 % (muy inferior a la tasa de los gliomas de vía óptica o hipotálamo en niños >1 año).
El segundo estudio se centró en los tumores de niños menores de 4 años con un diagnóstico patológico de gliomas, astrocitomas o tumores glioneuronales de grado 2, 3 y 4 de la OMS. Entre los 191 tumores estudiados que cumplían los criterios de inclusión, 61 tenían perfiles de metilación compatibles con los subtipos de glioma que se presentan en niños mayores (por ejemplo, IDH1, glioma difuso de línea media con alteración H3 K27, SEGA, xantoastrocitoma pleomórfico, etc.). Los 130 casos restantes se denominaron conjunto intrínseco y fueron objeto de caracterización molecular adicional:[
- El conjunto intrínseco contenía a la mayoría de los pacientes diagnosticados antes de tener 1 año de edad (49 de 63 pacientes, 78 %) que tenían una mediana de edad de 7,2 meses. Con frecuencia, los tumores se encontraban en una ubicación hemisférica superficial, a menudo con compromiso de las meninges, pero con un borde bien definido con el encéfalo normal adyacente.
- El clasificador de metilación colocó la mayoría de estos casos en el subgrupo de ganglioglioma desmoplásico infantil o astrocitoma (DIG/DIA) o en el subgrupo de glioma hemisférico infantil.
- En el conjunto intrínseco, se disponía de tejido para la secuenciación del panel génico y del ARN de 41 tumores, entre ellos, 25 tumores presentaron fusiones que afectaban ALK (n = 10), NTRK1 (n = 2), NTRK2 (n = 2) , NTRK3 (n = 8 ), ROS1 (n = 2) o MET (n = 1). Se observaron variantes de BRAF (n = 3) en los casos con puntuación alta en la matriz de metilación para los subgrupos DIG/DIA o similar a DIG/DIA.
- En los pacientes del grupo intrínseco, la tasa de supervivencia a 5 años fue más alta para los pacientes cuyos tumores tenían fusiones génicas, en comparación con los pacientes cuyos tumores no tenían fusiones (80 vs. 60 %, respectivamente). Sin embargo, ambos grupos de pacientes tuvieron tasas de supervivencia mucho más altas que otros niños con gliomas de grado alto.
Gliomas de grado alto secundarios
Los gliomas infantiles de grado alto secundarios (glioma de grado alto precedido por un glioma de grado bajo) son poco comunes (2,9 % en un estudio de 886 pacientes). Ningún glioma infantil de grado bajo con la fusión BRAF::KIAA1549 se transformó en un glioma de grado alto, mientras que los gliomas de grado bajo con variantes BRAF V600E se relacionaron con un aumento del riesgo de transformación. De los 18 pacientes con glioma secundario de grado alto, 7 (aproximadamente el 40 %) presentaron variantes BRAF V600E, y 8 de 14 casos (57 %) presentaron alteraciones en CDKN2A.[
Características moleculares de los tumores glioneuronales y neuronales
Los tumores glioneuronales y los tumores neuronales por lo general son tumores de grado bajo. Algunos tipos histológicos reconocidos por la clasificación de la OMS de 2021 son los siguientes:[
- Ganglioglioma.
- Ganglioglioma desmoplásico infantil o astrocitoma desmoplásico infantil (de lactantes).
- Tumor neuroepitelial disembrioplásico.
- Tumor glioneuronal papilar.
- Tumor glioneuronal formador de rosetas.
- Gangliocitoma displásico cerebeloso (enfermedad de Lhermitte-Duclos).
- Gangliocitoma.
- Tumor glioneuronal leptomeníngeo difuso.
- Neurocitoma central.
- Neurocitoma extraventricular.
Ganglioglioma
El ganglioglioma se presenta en niños y adultos. Produce convulsiones y aparece con más frecuencia en la corteza cerebral, pero a veces surge en otros sitios, como la médula espinal.[
El elemento fundamental de la patogénesis molecular del ganglioglioma son las alteraciones genómicas que conducen a la activación de la vía MAPK.[
Astrocitoma desmoplásico infantil y ganglioglioma desmoplásico infantil
Los astrocitomas desmoplásicos infantiles (DIA) y los gangliogliomas desmoplásicos infantiles (DIG) se presentan con mayor frecuencia en el primer año de vida, es decir en lactantes, y muestran una apariencia característica en las imágenes donde se ve un nódulo sólido que se realza con el contraste y se acompaña de un componente quístico grande.[
Las alteraciones genómicas observadas con mayor frecuencia en el DIA y el DIG son variantes de BRAF que afectan V600. Las fusiones génicas que afectan genes de cinasas se observan con menos frecuencia.
- Entre 16 casos de DIA y DIG confirmados mediante análisis histológico y de perfil de metilación del DNA, se identificaron variantes de BRAF en 7 casos (43,8 %): 4 variantes BRAF V600E y 3 variantes BRAF V600D.[
56 ] Otro caso tenía una fusión EML4::ALK. Se presentaron variantes de BRAF en 4 de 12 (25 %) casos de DIG (3 de 4 casos con la alteración BRAF V600D) y en 3 de 4 (75 %) casos de DIA (3 casos con la alteración BRAF V600E). - En un estudio de 7 casos de DIG se identificaron alteraciones en la vía MAPK en 4 casos (57 %).[
57 ] Entre ellas, 3 alteraciones que afectaban el gen BRAF (V600E, V600D y una deleción o inserción en V600) y otra se trató de una fusión en el marco de lectura TPM3::NTRK1. Cabe aclarar que la frecuencia de variantes alélicas fue baja (8–27 %), lo que indica que el DIG se caracteriza por un componente no neoplásico abundante que se traduce en frecuencias bajas de alelos con variantes clonales oncoiniciadoras. - En otro informe también se identificó la variante BRAF V600D en un caso de DIG.[
58 ] Debido a que la variante V600D es mucho menos frecuente que la variante V600E en otros tipos de cáncer, la detección de esta variante en varios casos de DIG indica una relación de la variante con esta enfermedad.
Tumor neuroepitelial disembrioplásico
El tumor neuroepitelial disembrioplásico (TNED) se presenta en niños y adultos con una mediana de edad en el momento del diagnóstico entre la adolescencia media o tardía. Desde el punto histopatológico, se caracteriza por filas de células de apariencia oligodendroglial y células ganglionares corticales flotando en mucina.[
Se han notificado alteraciones en FGFR1 en el 60 % a 80 % de los TNED, entre ellas variantes puntuales activadoras de FGFR1, duplicaciones internas en tándem del dominio cinasa y fusiones génicas activadoras.[
Tumor glioneuronal papilar
El tumor glioneuronal papilar es una neoplasia bifásica de grado bajo con diferenciación astrocítica y neuronal que casi siempre aparece en el compartimiento supratentorial.[
La alteración genómica principal relacionada con el tumor glioneuronal papilar es una fusión génica, SLC44A1::PRKCA, que se relaciona con la translocación t(9:17)(q31;q24).[
Tumor glioneuronal formador de rosetas
El tumor glioneuronal formador de rosetas (TGNR) se presenta en adolescentes y adultos, a menudo se ubica a nivel infratentorial, pero también surge en las regiones mesencefálica y diencefálica.[
El perfil de metilación del DNA indica que el TGNR exhibe un perfil epigenético propio que lo diferencia de otras entidades tumorales glioneuronales o neurogliales de grado bajo.[
Tumor glioneuronal leptomeníngeo difuso
El tumor glioneuronal leptomeníngeo difuso (DLGNT) es un tipo raro de tumor del SNC que se caracteriza desde el punto de vista radiográfico por el realce leptomeníngeo en la IRM. Este tumor suele afectar la fosa posterior, la región del tronco encefálico y la médula espinal.[
El DLGNT exhibió un perfil epigenético específico en las matrices de metilación del DNA, y la agrupación no supervisada de datos de matrices aplicadas en 30 casos permitió determinar dos subtipos según el tipo de metilación: MC-1 (n = 17) y MC-2 (n = 13).[
- Los 30 casos exhibieron pérdida del cromosoma 1p, pero solo 6 de 17 casos de DLGNT-MC-1 presentaron ganancia adicional del cromosoma 1q, en comparación con todos los casos de DLGNT-MC-2.[
67 ] En otro informe se encontró que la ganancia del cromosoma 1q fue un factor de pronóstico adverso en pacientes con DLGNT (incluso casos con enfermedad localizada),[69 ] lo que es coherente con el desenlace inferior para los pacientes con DLGNT-MC-2. - Las codeleciones de 1p/19q fueron más frecuentes en el grupo de DLGNT-MC-1 (7 de 13, 54 %) que en el grupo de DLGNT-MC-2 (2 de 13, 15 %). En contraste con el oligodendroglioma, no se identificaron variantes de IDH1 y IDH2.[
67 ] - La activación de la vía MAPK es común en los casos de DLGNT.[
67 ] Se encontró la fusión KIAA1549::BRAF en 11 de 15 casos de DLGNT-MC-1 (65 %) y en 9 de 13 casos de DLGNT-MC-2 (69 %). En dos casos se encontraron fusiones que afectaron NTRK1, NTRK2 o NTRK3, y en otro caso se encontró una fusión TRIM33::RAF1.
Neurocitoma extraventricular
El neurocitoma extraventricular es similar, desde el punto de vista histológico, al neurocitoma central y contiene células pequeñas uniformes con diferenciación neuronal. Sin embargo, el neurocitoma extraventricular se presenta en el parénquima encefálico, en lugar del sistema ventricular.[
En un estudio de 40 tumores con clasificación histológica de neurocitoma extraventricular sometidos a análisis de matriz de metilación, solo 26 se agruparon según el tipo histológico en un grupo diferenciado al de los tumores de referencia de otros tipos histológicos.[
Para obtener información sobre el tratamiento de gliomas, tumores glioneuronales y tumores neuronales, consultar
Tumores teratoides rabdoides atípicos del sistema nervioso central
GenesSMARCB1ySMARCA4
El tumor teratoide rabdoide atípico (TTRA) fue el primer tumor encefálico primario infantil en el que se identificó el gen SMARCB1 y se lo propuso como gen oncosupresor.[
Los casos raros de tumores rabdoides que expresan SMARCB1 y carecen de variantes de SMARCB1 también se relacionaron con variantes somáticas o germinales de SMARCA4, otro miembro del complejo de reestructuración de la cromatina SWI/SNF.[
Con menor frecuencia, se describieron tumores negativos para SMARCA4 (pero que retienen SMARCB1).[
En la clasificación de 2021 de la OMS se define el TTRA por la presencia de alteraciones en SMARCB1 o SMARCA4. Los tumores con características histológicas de TTRA que no tienen estas alteraciones genómicas se denominan tumores embrionarios del SNC con características rabdoides.[
A pesar de la ausencia de alteraciones genómicas recurrentes diferentes a las de SMARCB1 y SMARCA4,[
- TTRA-tirosinasa (TYR). Este subconjunto representa casi un tercio de los casos y se caracterizó por una expresión alta de marcadores melanosómicos como TYR (gen que codifica la tirosinasa). Los casos en este subconjunto fueron sobre todo infratentoriales; la mayoría de los casos se presentaron en niños de 0 a 1 año y exhibían pérdida del cromosoma 22q.[
81 ] En los pacientes que tenían TTRA TYR, la media de supervivencia general (SG) fue de 37 meses en un grupo heterogéneo desde el punto de vista clínico (intervalo de confianza [IC] 95 %, 18–56 meses).[83 ] En una serie prospectiva del European Rhabdoid Registry (EU-RHAB), se observó que los pacientes de 1 año o más con TTRA TYR presentaban una tasa de SG a 5 años del 71 %, mientras que aquellos menores de 1 año con TTRA TYR tuvieron una tasa de supervivencia muy precaria.[84 ] - TTRA-erizo sónico (SHH). Este subconjunto representó cerca del 40 % de los casos y se caracterizó por una expresión alta de los genes de la vía del erizo sónico (sonic hedgehog [SHH]) (por ejemplo, GLI2 y MYCN). Los casos de este subconjunto se presentaron con una frecuencia similar a nivel supratentorial e infratentorial. Si bien la mayoría de los casos se manifestaron antes de los 2 años de edad, alrededor de un tercio de los casos se presentó entre los 2 y los 5 años.[
81 ] En los pacientes con TTRA SHH, la media de SG fue de 16 meses (IC 95 %, 8–25 meses).[83 ]En un estudio posterior, el subgrupo TTRA SHH se dividió a su vez en tres subtipos: SHH-1A, SHH-1B y SHH-2.[
85 ] Los niños mayores de 3 años que albergaban la firma SHH-1B experimentaron los resultados más favorables. - TTRA MYC. Este subconjunto representa casi un cuarto de los casos y se caracterizó por una expresión alta de MYC. Los casos de TTRA MYC tienden a presentarse en el compartimento supratentorial. Si bien la mayoría de los casos de TTRA MYC se presentaron a los 5 años de edad, el TTRA MYC representó el subconjunto más común diagnosticado a la edad de 6 años o más. Las deleciones focales en SMARCB1 fueron el mecanismo más común de pérdida de SMARCB1 en este subconjunto.[
81 ] En los pacientes con TTRA MYC, la media de SG fue de 13 meses (IC 95 %, 5–22 meses).[83 ]
La pérdida de la expresión de las proteínas SMARCB1 o SMARCA4 tiene importancia terapéutica, porque esta pérdida crea una dependencia de la actividad de EZH2 por parte de las células cancerosas.[
Para obtener información sobre el tratamiento de los tumores teratoides rabdoides atípicos del SNC, consultar
Meduloblastomas
Subtipos moleculares del meduloblastoma
Se han identificado múltiples subtipos de meduloblastoma mediante análisis molecular integrador.[
Es probable que diferentes áreas del mismo tumor presenten variantes genéticas dispares, lo que aumenta la complejidad al elaborar un tratamiento eficaz dirigido al nivel molecular.[
Es posible establecer nuevas divisiones dentro de estos subgrupos, lo que proporcionará aún más información pronóstica útil.[
Meduloblastoma con activación de WNT
Los tumores WNT son meduloblastomas con alteraciones en la vía de señalización WNT que representan alrededor del 10 % de todos los meduloblastomas.[
Se observan variantes de CTNNB1 en el 85 % al 90 % de los casos de meduloblastoma WNT, y se detectan variantes de APC en muchos de los casos que no tienen variantes de CTNNB1. Los pacientes de meduloblastoma WNT con tumores que tienen variantes de APC a menudo presentan el síndrome de Turcot (es decir, variantes patogénicas de la línea germinal en APC).[
El subconjunto WNT se observa de manera primaria en niños grandes, adolescentes y adultos, y no muestra un predominio por el sexo masculino. Se cree que el subconjunto tiene origen en el tronco encefálico, en la región del labio rómbico embrionario.[
Meduloblastoma con activación de SHH y alteración deTP53, y meduloblastoma con activación de SHH yTP53natural
Los tumores SHH son meduloblastomas con alteraciones en la vía de señalización SHH y representan alrededor del 25 % de todos los casos de meduloblastomas.[
Las variantes patogénicas germinales heterocigotas deletéreas en el gen GPR161 se identificaron en cerca del 3 % de los casos de meduloblastoma SHH.[
Las variantes del tercer nucleótido (r.3A>G) de los RNA nucleares pequeños (snRNA) U1 de los empalmosomas son muy específicas del meduloblastoma SHH.[
Los meduloblastomas SHH exhiben una distribución etaria bimodal y se observan más a menudo en niños menores de 3 años, al final de la adolescencia y en la edad adulta. Se cree que los tumores surgen en la capa granular externa del cerebelo. La heterogeneidad en la edad de presentación esquematiza diferentes subgrupos identificados mediante caracterización molecular adicional, como se describe a continuación:
- El subgrupo de meduloblastoma más común en niños de 3 a 16 años, que se llama SHH-α (subgrupo provisional en la clasificación de meduloblastoma de 2021), tiene alteración de TP53 y exhibe enriquecimiento de amplificaciones de MYCN y GLI2.[
110 ,112 ] Las amplificaciones de PTCH1 y SUFU en ocasiones se presentan en este subtipo y son mutuamente excluyentes de las variantes de TP53 (a menudo de la línea germinal), mientras que la variante de SMO es infrecuente.[112 ,124 ,125 ] Las variantes de los snRNA U1 se presentan en cerca del 25 % de los casos de meduloblastoma SSH-α e indican un pronóstico muy precario.[123 ] - Se han descrito dos subtipos de meduloblastomas SHH que se presentan de manera primaria en niños menores de 3 años.[
110 ] Uno de estos subtipos, que se llama SHH-β, es metastásico con mayor frecuencia, y a menudo tiene amplificaciones focales.[126 ] El segundo de estos subtipos, que se llama SSH-γ, exhibe enriquecimiento del tipo histológico del meduloblastoma con nodularidad extensa (MBNE). Las variantes de la vía SHH en niños menores de 3 años con meduloblastoma incluyen las variantes de PTCH1 y SUFU.[112 ] Las variantes de SUFU casi no se observan en niños más grandes ni en adultos; en ellos, son comunes las alteraciones de la línea germinal.[124 ]En informes en los que se usaron matrices de metilación del DNA también se identificaron 2 subtipos de meduloblastoma SHH en niños jóvenes.[
126 ,127 ] Uno de estos subtipos abarcó todos los casos con variantes de SMO y se relacionó con un pronóstico favorable. El otro subtipo exhibió la mayoría de las variantes de SUFU y se relacionó con una tasa de supervivencia sin progresión (SSP) mucho más baja. Ambos subtipos exhibieron variantes de PTCH1. - Un cuarto subtipo de SHH, que se llama SHH-δ, incluye la mayoría de los casos de meduloblastomas SHH en adultos.[
110 ] Prácticamente todos los casos de meduloblastoma SHH-δ tienen la variante r.A>3 en los snRNA U1 [123 ] y cerca del 90 % de los casos exhiben una variante del promotor de TERT.[110 ] También se observan variantes de PTCH1 y SMO en adultos con meduloblastoma SHH.
El desenlace de los pacientes con meduloblastoma SHH no metastásico es relativamente favorable para niños menores de 3 años y adultos.[
Los pacientes con hallazgos moleculares desfavorables tienen un pronóstico desfavorable, menos del 50 % de los pacientes sobreviven después del tratamiento convencional.[
En la clasificación de 2021 de la OMS, el meduloblastoma SHH con variante de TP53 se considera una entidad diferenciada (meduloblastoma con activación de SHH y alteración de TP53).[
Meduloblastoma sin activación de WNT ni SHH
En la clasificación de la OMS se combinan los casos de meduloblastomas de los grupos 3 y 4 en una sola entidad, en parte por la ausencia de efecto clínico inmediato de esta diferenciación. Los meduloblastomas del grupo 3 representan cerca de un 25 % de los casos de meduloblastoma, mientras que los meduloblastoma del grupo 4 representan cerca de un 40 % de los casos de meduloblastoma.[
Se observan varias alteraciones genómicas en los meduloblastomas del grupo 3 y 4; no obstante, ninguna alteración única se presenta en más de un 10 % a un 20 % de los casos. Las alteraciones genómicas son las siguientes:
- La amplificación de MYC fue la alteración diferenciada más común notificada para los meduloblastomas del grupo 3, que se presenta en alrededor del 15 % de los casos.[
104 ,111 ] - La alteración genómica diferenciada más común descrita para el meduloblastoma del grupo 4 (alrededor de un 15 % de los casos) fue la activación de PRDM6 por secuestro del activador de transcripción como consecuencia de duplicación en tándem del gen adyacente SNCAIP.[
111 ] - Otras alteraciones genómicas que se observaron en casos de los grupos 3 y 4 fueron amplificación de MYCN y variantes estructurales que llevan a la sobreexpresión de GFI1 o GFI1B mediante secuestro del activador de transcripción.
- El isocromosoma 17q (i17q) es la anormalidad citogenética más común y se observa en un porcentaje alto de los casos del grupo 4, así como en los casos del grupo 3, pero pocas veces se observa en los meduloblastomas WNT y SHH.[
104 ,111 ] La presencia de i17q no parece afectar el pronóstico de los pacientes de los grupos 3 y 4.[137 ]
Los pacientes del grupo 3 con amplificación de MYC o sobreexpresión de MYC tienen un pronóstico precario.[
Los meduloblastomas del grupo 4 se presentan durante toda la infancia y la niñez, así como en la edad adulta. El pronóstico de los pacientes con meduloblastoma del grupo 4 es similar al de los pacientes con meduloblastoma sin activación de WNT y es posible que se vea afectado por factores adicionales como la presencia de enfermedad metastásica, pérdida del cromosoma 11q y pérdida del cromosoma 17p.[
Para los pacientes de riesgo estándar del grupo 3 y 4 (es decir, sin amplificación de MYC ni enfermedad metastásica), la ganancia o pérdida de cromosomas enteros acarrea un pronóstico favorable. Estos hallazgos se obtuvieron de datos de 91 pacientes con meduloblastoma sin activación de WNT ni SHH que participaron en el ensayo clínico SIOP-PNET-4 (NCT01351870) y se confirmaron en un grupo independiente de 70 niños con meduloblastoma sin activación de WNT ni SHH tratados entre 1990 y 2014.[
- La ganancia o pérdida de uno o más cromosomas enteros se relacionó con una tasa de supervivencia sin complicaciones (SSC) a 5 años del 93 % en comparación con el 64 % para la ausencia de ganancia o pérdida de cromosomas enteros.
- Las ganancias o pérdidas de cromosomas enteros más comunes son la ganancia del cromosoma 7 y la pérdida de los cromosomas 8 y 11.
- El discriminador pronóstico con desempeño óptimo es la presentación simultánea de dos o más de las siguientes alteraciones: ganancia del cromosoma 7, pérdida del cromosoma 8 y pérdida del cromosoma 11. Alrededor del 40 % de los pacientes de riesgo estándar de los grupos 3 y 4 tuvieron 2 o más de estas alteraciones cromosómicas; la tasa de SSC a 5 años fue del 100 % en comparación con el 68 % para los pacientes con menos de 2 de estas alteraciones.
- En una cohorte independiente, se confirmó la importancia pronóstica de 2 o más ganancias o pérdidas versus 0 o 1 ganancias o pérdidas de los cromosomas 7, 8 y 11 (tasa de SSC a 5 años, el 95 % de los pacientes con 2 o más vs. el 59 % de los pacientes con 1 o menos).
Es probable que la clasificación del meduloblastoma en cuatro subtipos principales se modifique en el futuro.[
No se sabe si la clasificación para los adultos con meduloblastoma tiene la misma capacidad pronóstica que para los niños.[
Para obtener información sobre el tratamiento del meduloblastoma infantil, consultar
Tumores embrionarios distintos del meduloblastoma
En esta sección se describen las características genómicas de los tumores embrionarios distintos del meduloblastoma y del tumor teratoide rabdoide atípico. En la clasificación de la OMS publicada en 2016 se eliminó el término tumores neuroectodérmicos primitivos (TNEP) del vocabulario diagnóstico.[
La clasificación de la OMS de 2021 categoriza los tumores embrionarios no meduloblastoma principalmente por sus características histológicas e inmunohistológicas y, en algunos casos, por hallazgos moleculares. La clasificación incluye los siguientes tumores:[
- Tumor teratoide rabdoide atípico (TTRA).
- Tumor neuroepitelial cribiforme.
- Tumor embrionario con rosetas de capas múltiples (ETMR).
- Neuroblastoma del SNC, con activación de FOXR2.
- Tumor del SNC con duplicaciones internas en tándem en BCOR.
- Tumor del SNC, sin clasificar (SC) o sin otra indicación (SAI).
El tumor SC se define como un tumor no clasificado en otro lugar. La nomenclatura SAI se usa para los tumores que no pueden clasificarse de otro modo. En el caso de muchas lesiones, existen características histológicas superpuestas, y la agrupación basada en la metilación es fundamental para lograr un diagnóstico específico.[
Subtipos moleculares de tumores embrionarios distintos del meduloblastoma
En los estudios en el que se aplicaron métodos de agrupamiento sin supervisión de los patrones de metilación del DNA a tumores embrionarios distintos del meduloblastoma, se observó que cerca de la mitad de los tumores diagnosticados como tumores embrionarios distintos del meduloblastoma exhibían perfiles moleculares característicos de otros tumores de encéfalo (cerebrales) infantiles conocidos (por ejemplo, glioma de grado alto).[
Entre los tumores diagnosticados como tumores embrionarios distintos del meduloblastoma, la caracterización molecular permitió identificar subtipos distintivos por sus características genómicas y biológicas. Estos subtipos son los siguientes:
- Tumor neuroepitelial cribiforme. Este subtipo, que representa menos del 50 % de los tumores embrionarios distintos del meduloblastoma, es un tumor no rabdoide que se presenta en las proximidades de cualquiera de los ventrículos (tercer, cuarto o laterales). Se caracteriza por presentar cordones y cintas cribiformes y exhibe la pérdida de expresión nuclear de SMARCB1. La mediana de edad en el momento del diagnóstico es de 20 meses. Este tumor se presenta con más frecuencia en los hombres, con una proporción hombre-mujer de 1,5 a 1.[
83 ] - Tumor embrionario con rosetas de capas múltiples (ETMR). Este subtipo, que representa hasta el 20 % de los tumores embrionarios distintos del meduloblastoma, abarca los tumores neuroepiteliales embrionarios de encéfalo que forman rosetas, que antes se clasificaban como tumores embrionarios con neurópilo abundante y rosetas verdaderas (ETANTR), ependimoblastomas o meduloepiteliomas.[
27 ,142 ] Los ETMR se presentan con mayor frecuencia en niños pequeños (mediana de edad en el momento del diagnóstico, 2-3 años), pero es posible que se presenten también en niños más grandes.[141 ,142 ,143 ,144 ,145 ,146 ]A nivel molecular, los ETMR se definen por el alto grado de amplificación del complejo de microRNA C19MC y por una fusión génica entre TTYH1 y C19MC.[
142 ,147 ,148 ] Esta fusión génica pone la expresión de C19MC bajo el control del promotor de TTYH1, lo que produce un grado alto de expresión anormal de los microRNA dentro del conglomerado. La Organización Mundial de la Salud (OMS) admite que se clasifiquen como ETMR, sin otra indicación (SAI), los tumores con características histológicas semejantes, pero sin alteraciones de C19MC. Es posible que esta subclase de tumores sin alteraciones de C19MC albergue variantes bialélicas de DICER1. - Neuroblastoma del sistema nervioso central (SNC) con activación de FOXR2 (NB-FOXR2 del SNC). Este tumor, que representa entre el 10 % y el 15 % de los tumores embrionarios distintos del meduloblastoma, tal vez se presente en niños menores de 3 años, pero es más frecuente en niños más grandes. Este subtipo se caracteriza por alteraciones genómicas que conducen a un aumento de la expresión del factor de transcripción FOXR2.[
27 ] El NB-FOXR2 del SNC se observa sobre todo en niños menores de 10 años; las características histológicas de estos tumores suelen ser las del neuroblastoma del SNC o el ganglioneuroblastoma del SNC.[27 ,149 ] No hay una alteración genómica única en los tumores NB-FOXR2 del SNC que conduzca a la sobreexpresión de FOXR2; se han identificado fusiones génicas en las que participan múltiples genes recíprocos de FOXR2.[27 ] La expresión proteica de SOX10 y ANKRD55 detectada mediante inmunohistoquímica se ha propuesto como un potencial biomarcador para diferenciar los tumores NB-FOXR2 del SNC de otros tipos de tumores relacionados.[149 ] - Tumor neuroepitelial de grado alto con alteración en BCOR (HGNET-BCOR del SNC). Este subtipo representa hasta el 3 % de los tumores embrionarios distintos del meduloblastoma y se caracteriza por duplicaciones internas en tándem en BCOR,[
27 ] una alteración genómica que también se encuentra en el sarcoma de células claras de riñón.[150 ,151 ] A pesar de que la mediana de edad en el momento del diagnóstico es de menos de 10 años, se presentan casos en la segunda década de la vida y más tarde.[27 ]
Aunque no figuran como entidades separadas en la clasificación de tumores del SNC de la OMS de 2021, también se han descrito otros tumores embrionarios distintos del meduloblastoma como entidades separadas, entre ellos los siguientes:
- Tumor de la familia de sarcomas de Ewing del SNC con alteración de CIC (EFT-CIC del SNC). Este subtipo de tumor representa entre el 2 % y el 4 % de los tumores embrionarios distintos del meduloblastoma; se caracteriza por alteraciones genómicas que afectan CIC (localizado en el cromosoma 19q13.2); y se ha identificado una fusión con NUTM1 en varios casos evaluados.[
27 ,141 ] Las fusiones del gen CIC también se identificaron en sarcomas similares al de Ewing fuera del SNC; además, la firma de expresión génica de los tumores EFT-CIC del SNC es similar al de estos sarcomas.[27 ] Los tumores EFT-CIC del SNC por lo general se presentan en niños menores de 10 años y se caracterizan por un fenotipo de células pequeñas, pero con características histológicas variables.[27 ] - Tumor neuroepitelial de grado alto del SNC con alteración en MN1 (HGNET-MN1 del SNC). Este subtipo representa entre el 1 % y el 3 % de los tumores embrionarios distintos del meduloblastoma y se caracteriza por fusiones génicas en las que participa MN1 (localizado en el cromosoma 22q12.3) con genes recíprocos de fusión como BEND2 y CXXC5.[
27 ,141 ] El subtipo HGNET-MN1 del SNC muestra un llamativo predominio en el sexo femenino y tiende a presentarse en la segunda década de vida.[27 ] Este subtipo representaba la mayoría de los casos diagnosticados como astroblastoma según el esquema de clasificación de la OMS de 2007.[27 ] Este subtipo no se ha agregado al vocabulario diagnóstico de la OMS. En otros 2 informes en los que se examinaron 35 casos de astroblastomas definidos mediante análisis histológico se observó que 14 exhibían perfiles de metilación compatibles con HGNET-MN1 del SNC o alteraciones de MN1 identificadas por hibridación fluorescente in situ.[28 ,29 ] - Meduloepitelioma. El meduloepitelioma con la clásica amplificación de C19MC se considera un ETMR con alteración de C19MC (consultar la información anterior sobre ETMR). No obstante, cuando un tumor tiene características histológicas de meduloblastoma, pero carece de la amplificación de C19MC, se identifica como un ETMR.[
152 ,153 ] Los tumores de meduloepiteliomas son poco frecuentes y tienden a presentarse con más frecuencia en lactantes y niños pequeños. Los meduloepiteliomas, que en el análisis histológico se asemejan al tubo neural embrionario, tienden a presentarse a nivel supratentorial, sobre todo dentro de los ventrículos, pero también aparecen a nivel infratentorial, en la cola de caballo e incluso en una ubicación extraneural, junto a las raíces de los nervios.[152 ,153 ] El meduloepitelioma intraocular es biológicamente diferente del meduloepitelioma intraaxial.[154 ,155 ] - Tumor embrionario del SNC con amplificación de PLAGL. En un análisis retrospectivo de más de 90 000 tumores encefálicos en niños y adultos se identificó un pequeño subconjunto de tumores embrionarios (n = 31) con perfiles de metilación característicos, con amplificación de grado alto y sobreexpresión de PLAGL1 o PLAGL2.[
156 ] En estos casos no se observaron las alteraciones genéticas recurrentes adicionales que se advirtieron en otros tipos de tumores pediátricos del SNC. Estos tumores se presentaron en todo el encéfalo y estaban compuestos, en la mayoría de los casos, por células primitivas de tipo embrionario sin marcadores de diferenciación glial o neuronal. En esta pequeña cohorte, se observaron diferencias en la edad en el momento del diagnóstico y en la supervivencia general (SG) a los 10 años entre los pacientes con amplificación de PLAGL1 (mediana de edad, 10,5 años; tasa de SG, 66 %), en comparación con amplificación de PLAGL2 (mediana de edad, 2 años; tasa de SG, 25 %).
Para obtener información sobre el tratamiento de los TNEP en los niños, consultar
Pineoblastoma
Características genómicas del pineoblastoma
En la actualidad, la Organización Mundial de la Salud (OMS) clasifica el pineoblastoma como un tumor de parénquima pineal, aunque antes era tradicional agruparlo con los tumores embrionarios. Debido a que el tratamiento del pineoblastoma es muy similar al de los tumores embrionarios, aquí se usa la tradición anterior de incluir el pineoblastoma con los tumores embrionarios del sistema nervioso central (SNC). El pineoblastoma se relaciona con variantes patogénicas germinales del gen RB1 y el gen DICER1, como se describe a continuación:
- El pineoblastoma se vincula con variantes patogénicas germinales de RB1; el término retinoblastoma trilateral se usa para el retinoblastoma ocular que se presenta de manera simultánea con un tumor de encéfalo con características histológicas similares que, por lo general, surge en la glándula pineal o en otras estructuras de la línea media. Tradicionalmente, se han notificado tumores intracraneales a entre el 5 % y el 15 % de los niños con retinoblastoma hereditario.[
157 ] Las tasas de pineoblastoma en los niños con retinoblastoma hereditario que se someten a los programas de tratamiento vigentes quizás sean más bajas que los cálculos históricos.[158 ,159 ,160 ] - También se han notificado variantes patogénicas germinales de DICER1 en pacientes con pineoblastoma.[
161 ] Entre 18 pacientes con pineoblastoma, se identificaron 3 pacientes con variantes patogénicas germinales de DICER1, y otros 3 pacientes de pineoblastoma se sabía que eran portadores de variantes patogénicas germinales de DICER1.[161 ] En pacientes de pineoblastoma, las variantes de DICER1 son variantes de pérdida de función; estas son diferentes a las variantes que se observan en los tumores relacionados con el síndrome de DICER1, como el blastoma pleuropulmonar.[161 ]
Se han aplicado métodos genómicos al estudio del pineoblastoma en un intento por conocer mejor las características biológicas de dicho tumor y orientar la futura clasificación molecular. En un metanálisis internacional retrospectivo se analizaron 221 niños y adultos diagnosticados con pineoblastoma y tumores de parénquima pineal de diferenciación intermedia.[
Para obtener información sobre el tratamiento del pineoblastoma infantil, consultar
Ependimomas
Subgrupos moleculares del ependimoma
En los estudios de caracterización molecular iniciales se identificaron 9 subgrupos moleculares de ependimoma, 6 de los cuales predominan en niños. Los subgrupos se identificaron a partir de los perfiles característicos de metilación del DNA y de expresión génica, y también por una variedad exclusiva de alteraciones genómicas (consultar la Figura 6).[
Se añadió un nuevo ependimoma definido a nivel molecular a la clasificación de tumores del sistema nervioso central de la Organización Mundial de la Salud (OMS) de 2021: ependimoma medular con amplificación de MYCN. En la clasificación de 2021 se describió con más detalle los tumores ependimarios definidos por la localización anatómica y las características histológicas, pero no por la alteración molecular. Estos tumores se denominan ependimoma de fosa posterior (PF-EPN), ependimoma supratentorial (ST-EPN) y ependimoma medular (SP-EPN). Estos tumores contienen una alteración molecular exclusiva (sin clasificar), o bien su análisis molecular falló o no se obtuvo (sin especificar).[
- Tumores infratentoriales.
- Ependimoma de fosa posterior (PF-EPN).
- Ependimoma de fosa posterior A (PF-EPN-A) con pérdida de la marca de trimetilación de H3 K27.
- Ependimoma de fosa posterior B (PF-EPN-B) con retención de la marca de trimetilación de H3 K27.
- Tumores supratentoriales.
- Ependimoma supratentorial (ST-EPN).
- Ependimoma supratentorial positivo para fusiones de ZFTA (ST-EPN-ZFTA). Antes se denominaba ependimoma supratentorial positivo para fusiones de RELA (ST-EPN-RELA), pero se ha cambiado el nombre porque ZFTA es la nueva denominación de C11orf95 y ZFTA puede fusionarse con otro gen diferente a RELA.[
167 ] - Ependimoma positivo para fusiones de YAP1 (ST-EPN-YAP1).
- Tumores de médula espinal.
- Ependimoma medular (SP-EPN).
- Ependimoma medular con amplificación de MYCN (SP-EPN-MYCN).
- Ependimoma mixopapilar (SP-EPN-MPE).
El subependimoma (supratentorial, infratentorial o medular) incluye las otras tres variantes moleculares que son bastante infrecuentes en niños.
Tumores infratentoriales
Ependimoma de fosa posterior A
El ependimoma de fosa posterior A (PF-EPN-A) es el subgrupo más común que se caracteriza por los siguientes aspectos:
- Cuadro clínico inicial en niños pequeños (mediana de edad, 3 años).[
163 ,168 ] - Tasas bajas de variantes que afectan la estructura proteica, alrededor de 5 por genoma.[
164 ] - Ganancia del cromosoma 1q, un factor de pronóstico precario para pacientes con ependimoma,[
169 ] en alrededor del 25 % de los casos.[163 ,165 ,170 ] - Pérdida del cromosoma 6q, que se ha notificado como factor de pronóstico precario para los pacientes con PF-EPN-A, en el 8 % al 10 % de los casos.[
171 ] - Perfil cromosómico equilibrado con pocas ganancias o pérdidas cromosómicas.[
163 ,164 ] - Pérdida de la marca de trimetilación de H3 K27 y DNA con hipometilación general.[
172 ] En un estudio multiinstitucional prospectivo se analizó un grupo de 147 pacientes con ependimoma. En el estudio se notificaron sensibilidad y especificidad altas para la detección por análisis inmunohistoquímico de la pérdida de la marca de trimetilación H3 K27 para identificar los casos de PF-EPN-A.[173 ] La pérdida de esta marca se presenta mediante múltiples mecanismos, como los siguientes:- Variantes recurrentes de EZHIP en el 10 % de los casos, con expresión alta de mRNA de EZHIP en casi todos los PF-EPN-A.[
44 ,174 ] La expresión de EZHIP (con alteración o sin esta) produce la inhibición de la metiltransferasa EZH2, que conduce a la pérdida de la marca de trimetilación de H3 K27.[43 ,44 ] - Variantes recurrentes de K27M en genes de la histona H3 en una proporción baja de los casos.[
175 ,176 ] A diferencia de los gliomas de línea media difusos, las variantes de H3.1 (H3C2 y H3C3) son más frecuentes que las variantes de H3.3 (H3-3A).[174 ] Las variantes de las histonas son mutuamente excluyentes de la expresión alta de EZHIP,[174 ] y también conducen a la pérdida de la marca de trimetilación de H3 K27 mediante la inhibición de EZH2.
- Variantes recurrentes de EZHIP en el 10 % de los casos, con expresión alta de mRNA de EZHIP en casi todos los PF-EPN-A.[
En un estudio en el que se incluyeron más de 600 casos de PF-EPN-A, se usaron perfiles de matrices de metilación para dividir a la población en dos subgrupos característicos: PFA-1 y PFA-2.[
Ependimoma de fosa posterior B
En niños, el subgrupo de ependimoma de fosa posterior B (PF-EPN-B) es menos común que el subgrupo PF-EPN-A, representa entre 15 y 20 % de todos los ependimomas de fosa posterior, y se caracteriza por los siguientes aspectos:
- Cuadro clínico inicial predominante en adolescentes y adultos jóvenes (mediana de edad, 30 años).[
163 ,168 ] - Tasas bajas de variantes que afectan la estructura proteica (alrededor de 5 por genoma), sin variantes recurrentes.[
165 ] - Numerosas anomalías citogenéticas, sobre todo ganancias o pérdidas de cromosomas enteros.[
163 ,165 ] - Retención de la trimetilación de H3 K27.[
172 ] - La ganancia de 1q y la pérdida de 6q se producen en el PF-EPN-B, pero no se han notificado como factor pronóstico en este subgrupo (a diferencia del PF-EPN-A).[
177 ]
Tumores supratentoriales
Ependimomas supratentoriales con fusiones deZFTA
El subgrupo de ependimomas supratentoriales con fusiones de ZFTA (ST-EPN-ZFTA) es el tipo más numeroso de ependimoma supratentorial infantil que se caracteriza por fusiones génicas que afectan a RELA,[
- Representa alrededor de 70 % de los ependimomas supratentoriales en niños,[
178 ,179 ] y se presenta a una mediana de edad de 8 años.[163 ] - Presencia de fusiones de ZFTA que se producen por cromotripsis del cromosoma 11q13.1.[
178 ] - Tasas bajas de variantes que afectan la estructura proteica y casi ausencia de variantes recurrentes fuera de las fusiones ZFTA::RELA.[
178 ] - Indicios de activación de la vía NF-κB a nivel proteico o del RNA.[
178 ] - Ganancia del cromosoma 1q en cerca de un cuarto de los casos, y efecto indeterminado en la supervivencia.[
163 ] - El grado de concordancia fue elevado entre la prueba inmunohistoquímica del p65-RelA nuclear, la hibridación fluorescente in situ de ZFTA y RELA, y la clasificación según la metilación del DNA para definir el ST-EPN-ZFTA.[
180 ] - La deleción homocigota de CDKN2A se ha relacionado con un pronóstico precario en pacientes con ependimoma positivo para fusiones de ZFTA.[
181 ][Nivel de evidencia B4] La deleción de CDKN2A también se ha notificado como un evento secundario en el ependimoma recidivante.[182 ]
Ependimomas supratentoriales con fusiones deYAP1
El subgrupo de ependimomas supratentoriales con fusiones de YAP1 (ST-EPN-YAP1) es el segundo tipo menos común de ependimomas supratentoriales y exhibe fusiones que afectan el gen YAP1 en el cromosoma 11; se caracteriza por los siguientes aspectos:
- Mediana de edad en el momento del diagnóstico de 1,4 años.[
163 ] - Presencia de una fusión génica que afecta el gen YAP1, y el compañero de fusión más común es MAMLD1.[
163 ,178 ] - Un genoma relativamente estable con pocos cambios cromosómicos además de la fusión del gen YAP1.[
163 ]
Tumores que se confunden con ependimomas supratentoriales
Los ependimomas supratentoriales sin fusiones de ZFTA o YAP1 (en el cromosoma 11) son una entidad que no se ha definido y no se conoce su importancia. Mediante análisis de metilación del DNA, estas muestras a menudo se agrupan con otras entidades como los gliomas de grado alto y los tumores embrionarios. Por ejemplo, en un análisis de metilación retrospectivo de tumores de encéfalo supratentoriales se identificó un grupo de tumores diferente al de los ependimomas supratentoriales que albergan fusiones de PLAGL1 recurrentes.[
Ependimoma medular con amplificación deMYCN
El ependimoma medular con amplificación de MYCN (SP-EPN-MYCN) es poco frecuente; solo se han notificado 27 casos.[
- La mediana de edad en el momento de la presentación fue de 31 años (intervalo, 12–56 años).
- Se presentó un alto nivel de amplificación de MYCN en el momento del diagnóstico y en la recaída.
- SP-EPN-MYCN tiene un perfil de metilación único en comparación con otros ependimomas de la médula espinal, con amplificación de MYCN parecida a la del glioblastoma de tipo pediátrico y el neuroblastoma.
Para obtener información sobre el tratamiento del ependimoma infantil, consultar
Referencias:
- Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
- Zhang J, Wu G, Miller CP, et al.: Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45 (6): 602-12, 2013.
- Ramkissoon LA, Horowitz PM, Craig JM, et al.: Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci U S A 110 (20): 8188-93, 2013.
- Qaddoumi I, Orisme W, Wen J, et al.: Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 131 (6): 833-45, 2016.
- Pollack IF, Hamilton RL, Sobol RW, et al.: IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children's Oncology Group. Childs Nerv Syst 27 (1): 87-94, 2011.
- Packer RJ, Iavarone A, Jones DTW, et al.: Implications of new understandings of gliomas in children and adults with NF1: report of a consensus conference. Neuro Oncol 22 (6): 773-784, 2020.
- Ryall S, Zapotocky M, Fukuoka K, et al.: Integrated Molecular and Clinical Analysis of 1,000 Pediatric Low-Grade Gliomas. Cancer Cell 37 (4): 569-583.e5, 2020.
- D'Angelo F, Ceccarelli M, Tala, et al.: The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat Med 25 (1): 176-187, 2019.
- Franz DN, Agricola K, Mays M, et al.: Everolimus for subependymal giant cell astrocytoma: 5-year final analysis. Ann Neurol 78 (6): 929-38, 2015.
- Mackay A, Burford A, Carvalho D, et al.: Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 32 (4): 520-537.e5, 2017.
- Bouffet E, Larouche V, Campbell BB, et al.: Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol 34 (19): 2206-11, 2016.
- Das A, Tabori U, Sambira Nahum LC, et al.: Efficacy of Nivolumab in Pediatric Cancers with High Mutation Burden and Mismatch Repair Deficiency. Clin Cancer Res 29 (23): 4770-4783, 2023.
- Jones DT, Kocialkowski S, Liu L, et al.: Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68 (21): 8673-7, 2008.
- Hawkins C, Walker E, Mohamed N, et al.: BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res 17 (14): 4790-8, 2011.
- Mistry M, Zhukova N, Merico D, et al.: BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol 33 (9): 1015-22, 2015.
- López GY, Van Ziffle J, Onodera C, et al.: The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol 137 (1): 139-150, 2019.
- Lassaletta A, Zapotocky M, Mistry M, et al.: Therapeutic and Prognostic Implications of BRAF V600E in Pediatric Low-Grade Gliomas. J Clin Oncol 35 (25): 2934-2941, 2017.
- Ho CY, Mobley BC, Gordish-Dressman H, et al.: A clinicopathologic study of diencephalic pediatric low-grade gliomas with BRAF V600 mutation. Acta Neuropathol 130 (4): 575-85, 2015.
- Guerreiro Stucklin AS, Ryall S, Fukuoka K, et al.: Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun 10 (1): 4343, 2019.
- Clarke M, Mackay A, Ismer B, et al.: Infant High-Grade Gliomas Comprise Multiple Subgroups Characterized by Novel Targetable Gene Fusions and Favorable Outcomes. Cancer Discov 10 (7): 942-963, 2020.
- Meredith DM, Cooley LD, Dubuc A, et al.: ROS1 Alterations as a Potential Driver of Gliomas in Infant, Pediatric, and Adult Patients. Mod Pathol 36 (11): 100294, 2023.
- Jones DT, Hutter B, Jäger N, et al.: Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45 (8): 927-32, 2013.
- Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.
- Bandopadhayay P, Ramkissoon LA, Jain P, et al.: MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet 48 (3): 273-82, 2016.
- D'Aronco L, Rouleau C, Gayden T, et al.: Brainstem angiocentric gliomas with MYB-QKI rearrangements. Acta Neuropathol 134 (4): 667-669, 2017.
- Chan E, Bollen AW, Sirohi D, et al.: Angiocentric glioma with MYB-QKI fusion located in the brainstem, rather than cerebral cortex. Acta Neuropathol 134 (4): 671-673, 2017.
- Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.
- Lehman NL, Usubalieva A, Lin T, et al.: Genomic analysis demonstrates that histologically-defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol Commun 7 (1): 42, 2019.
- Wood MD, Tihan T, Perry A, et al.: Multimodal molecular analysis of astroblastoma enables reclassification of most cases into more specific molecular entities. Brain Pathol 28 (2): 192-202, 2018.
- Hirose T, Nobusawa S, Sugiyama K, et al.: Astroblastoma: a distinct tumor entity characterized by alterations of the X chromosome and MN1 rearrangement. Brain Pathol 28 (5): 684-694, 2018.
- Lucas CG, Solomon DA, Perry A: A review of recently described genetic alterations in central nervous system tumors. Hum Pathol 96: 56-66, 2020.
- Paugh BS, Qu C, Jones C, et al.: Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol 28 (18): 3061-8, 2010.
- Bax DA, Mackay A, Little SE, et al.: A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin Cancer Res 16 (13): 3368-77, 2010.
- Ward SJ, Karakoula K, Phipps KP, et al.: Cytogenetic analysis of paediatric astrocytoma using comparative genomic hybridisation and fluorescence in-situ hybridisation. J Neurooncol 98 (3): 305-18, 2010.
- Sturm D, Witt H, Hovestadt V, et al.: Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22 (4): 425-37, 2012.
- Korshunov A, Ryzhova M, Hovestadt V, et al.: Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol 129 (5): 669-78, 2015.
- Castel D, Kergrohen T, Tauziède-Espariat A, et al.: Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol 139 (6): 1109-1113, 2020.
- Rodriguez Gutierrez D, Jones C, Varlet P, et al.: Radiological Evaluation of Newly Diagnosed Non-Brainstem Pediatric High-Grade Glioma in the HERBY Phase II Trial. Clin Cancer Res 26 (8): 1856-1865, 2020.
- Buczkowicz P, Hoeman C, Rakopoulos P, et al.: Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46 (5): 451-6, 2014.
- Taylor KR, Mackay A, Truffaux N, et al.: Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46 (5): 457-61, 2014.
- Auffret L, Ajlil Y, Tauziède-Espariat A, et al.: A new subtype of diffuse midline glioma, H3 K27 and BRAF/FGFR1 co-altered: a clinico-radiological and histomolecular characterisation. Acta Neuropathol 147 (1): 2, 2023.
- Williams EA, Brastianos PK, Wakimoto H, et al.: A comprehensive genomic study of 390 H3F3A-mutant pediatric and adult diffuse high-grade gliomas, CNS WHO grade 4. Acta Neuropathol 146 (3): 515-525, 2023.
- Jain SU, Do TJ, Lund PJ, et al.: PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat Commun 10 (1): 2146, 2019.
- Hübner JM, Müller T, Papageorgiou DN, et al.: EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol 21 (7): 878-889, 2019.
- Chen CCL, Deshmukh S, Jessa S, et al.: Histone H3.3G34-Mutant Interneuron Progenitors Co-opt PDGFRA for Gliomagenesis. Cell 183 (6): 1617-1633.e22, 2020.
- Mackay A, Burford A, Molinari V, et al.: Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell 33 (5): 829-842.e5, 2018.
- Yeo KK, Alexandrescu S, Cotter JA, et al.: Multi-institutional study of the frequency, genomic landscape, and outcome of IDH-mutant glioma in pediatrics. Neuro Oncol 25 (1): 199-210, 2023.
- Suwala AK, Stichel D, Schrimpf D, et al.: Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. Acta Neuropathol 141 (1): 85-100, 2021.
- Reinhardt A, Stichel D, Schrimpf D, et al.: Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol 136 (2): 273-291, 2018.
- Korshunov A, Schrimpf D, Ryzhova M, et al.: H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol 134 (3): 507-516, 2017.
- Becker AJ: Ganglioglioma. In: Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016, pp 138-41.
- Blumcke I, Spreafico R, Haaker G, et al.: Histopathological Findings in Brain Tissue Obtained during Epilepsy Surgery. N Engl J Med 377 (17): 1648-1656, 2017.
- Pekmezci M, Villanueva-Meyer JE, Goode B, et al.: The genetic landscape of ganglioglioma. Acta Neuropathol Commun 6 (1): 47, 2018.
- Bianchi F, Tamburrini G, Massimi L, et al.: Supratentorial tumors typical of the infantile age: desmoplastic infantile ganglioglioma (DIG) and astrocytoma (DIA). A review. Childs Nerv Syst 32 (10): 1833-8, 2016.
- Trehan G, Bruge H, Vinchon M, et al.: MR imaging in the diagnosis of desmoplastic infantile tumor: retrospective study of six cases. AJNR Am J Neuroradiol 25 (6): 1028-33, 2004 Jun-Jul.
- Wang AC, Jones DTW, Abecassis IJ, et al.: Desmoplastic Infantile Ganglioglioma/Astrocytoma (DIG/DIA) Are Distinct Entities with Frequent BRAFV600 Mutations. Mol Cancer Res 16 (10): 1491-1498, 2018.
- Blessing MM, Blackburn PR, Krishnan C, et al.: Desmoplastic Infantile Ganglioglioma: A MAPK Pathway-Driven and Microglia/Macrophage-Rich Neuroepithelial Tumor. J Neuropathol Exp Neurol 78 (11): 1011-1021, 2019.
- Greer A, Foreman NK, Donson A, et al.: Desmoplastic infantile astrocytoma/ganglioglioma with rare BRAF V600D mutation. Pediatr Blood Cancer 64 (6): , 2017.
- Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016.
- Stone TJ, Keeley A, Virasami A, et al.: Comprehensive molecular characterisation of epilepsy-associated glioneuronal tumours. Acta Neuropathol 135 (1): 115-129, 2018.
- Rivera B, Gayden T, Carrot-Zhang J, et al.: Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol 131 (6): 847-63, 2016.
- Matsumura N, Nobusawa S, Ito J, et al.: Multiplex ligation-dependent probe amplification analysis is useful for detecting a copy number gain of the FGFR1 tyrosine kinase domain in dysembryoplastic neuroepithelial tumors. J Neurooncol 143 (1): 27-33, 2019.
- Pages M, Lacroix L, Tauziede-Espariat A, et al.: Papillary glioneuronal tumors: histological and molecular characteristics and diagnostic value of SLC44A1-PRKCA fusion. Acta Neuropathol Commun 3: 85, 2015.
- Bridge JA, Liu XQ, Sumegi J, et al.: Identification of a novel, recurrent SLC44A1-PRKCA fusion in papillary glioneuronal tumor. Brain Pathol 23 (2): 121-8, 2013.
- Hou Y, Pinheiro J, Sahm F, et al.: Papillary glioneuronal tumor (PGNT) exhibits a characteristic methylation profile and fusions involving PRKCA. Acta Neuropathol 137 (5): 837-846, 2019.
- Sievers P, Appay R, Schrimpf D, et al.: Rosette-forming glioneuronal tumors share a distinct DNA methylation profile and mutations in FGFR1, with recurrent co-mutation of PIK3CA and NF1. Acta Neuropathol 138 (3): 497-504, 2019.
- Deng MY, Sill M, Chiang J, et al.: Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol 136 (2): 239-253, 2018.
- Chiang JCH, Harreld JH, Orr BA, et al.: Low-grade spinal glioneuronal tumors with BRAF gene fusion and 1p deletion but without leptomeningeal dissemination. Acta Neuropathol 134 (1): 159-162, 2017.
- Chiang J, Dalton J, Upadhyaya SA, et al.: Chromosome arm 1q gain is an adverse prognostic factor in localized and diffuse leptomeningeal glioneuronal tumors with BRAF gene fusion and 1p deletion. Acta Neuropathol 137 (1): 179-181, 2019.
- Sievers P, Stichel D, Schrimpf D, et al.: FGFR1:TACC1 fusion is a frequent event in molecularly defined extraventricular neurocytoma. Acta Neuropathol 136 (2): 293-302, 2018.
- Biegel JA, Tan L, Zhang F, et al.: Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8 (11): 3461-7, 2002.
- Biegel JA, Kalpana G, Knudsen ES, et al.: The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res 62 (1): 323-8, 2002.
- Schneppenheim R, Frühwald MC, Gesk S, et al.: Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet 86 (2): 279-84, 2010.
- Hasselblatt M, Gesk S, Oyen F, et al.: Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 35 (6): 933-5, 2011.
- Hasselblatt M, Nagel I, Oyen F, et al.: SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128 (3): 453-6, 2014.
- WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
- Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012.
- Kieran MW, Roberts CW, Chi SN, et al.: Absence of oncogenic canonical pathway mutations in aggressive pediatric rhabdoid tumors. Pediatr Blood Cancer 59 (7): 1155-7, 2012.
- Hasselblatt M, Isken S, Linge A, et al.: High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer 52 (2): 185-90, 2013.
- Torchia J, Picard D, Lafay-Cousin L, et al.: Molecular subgroups of atypical teratoid rhabdoid tumours in children: an integrated genomic and clinicopathological analysis. Lancet Oncol 16 (5): 569-82, 2015.
- Johann PD, Erkek S, Zapatka M, et al.: Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29 (3): 379-93, 2016.
- Upadhyaya SA, Robinson GW, Onar-Thomas A, et al.: Relevance of Molecular Groups in Children with Newly Diagnosed Atypical Teratoid Rhabdoid Tumor: Results from Prospective St. Jude Multi-institutional Trials. Clin Cancer Res 27 (10): 2879-2889, 2021.
- Johann PD, Hovestadt V, Thomas C, et al.: Cribriform neuroepithelial tumor: molecular characterization of a SMARCB1-deficient non-rhabdoid tumor with favorable long-term outcome. Brain Pathol 27 (4): 411-418, 2017.
- Frühwald MC, Hasselblatt M, Nemes K, et al.: Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro Oncol 22 (7): 1006-1017, 2020.
- Federico A, Thomas C, Miskiewicz K, et al.: ATRT-SHH comprises three molecular subgroups with characteristic clinical and histopathological features and prognostic significance. Acta Neuropathol 143 (6): 697-711, 2022.
- Wilson BG, Wang X, Shen X, et al.: Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18 (4): 316-28, 2010.
- Knutson SK, Warholic NM, Wigle TJ, et al.: Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110 (19): 7922-7, 2013.
- Kurmasheva RT, Sammons M, Favours E, et al.: Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 64 (3): , 2017.
- Italiano A, Soria JC, Toulmonde M, et al.: Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19 (5): 649-659, 2018.
- Onvani S, Etame AB, Smith CA, et al.: Genetics of medulloblastoma: clues for novel therapies. Expert Rev Neurother 10 (5): 811-23, 2010.
- Dubuc AM, Northcott PA, Mack S, et al.: The genetics of pediatric brain tumors. Curr Neurol Neurosci Rep 10 (3): 215-23, 2010.
- Thompson MC, Fuller C, Hogg TL, et al.: Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24 (12): 1924-31, 2006.
- Kool M, Koster J, Bunt J, et al.: Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 3 (8): e3088, 2008.
- Tabori U, Baskin B, Shago M, et al.: Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol 28 (8): 1345-50, 2010.
- Pfister S, Remke M, Benner A, et al.: Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol 27 (10): 1627-36, 2009.
- Ellison DW, Onilude OE, Lindsey JC, et al.: beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol 23 (31): 7951-7, 2005.
- Polkinghorn WR, Tarbell NJ: Medulloblastoma: tumorigenesis, current clinical paradigm, and efforts to improve risk stratification. Nat Clin Pract Oncol 4 (5): 295-304, 2007.
- Giangaspero F, Wellek S, Masuoka J, et al.: Stratification of medulloblastoma on the basis of histopathological grading. Acta Neuropathol 112 (1): 5-12, 2006.
- Northcott PA, Korshunov A, Witt H, et al.: Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29 (11): 1408-14, 2011.
- Pomeroy SL, Tamayo P, Gaasenbeek M, et al.: Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415 (6870): 436-42, 2002.
- Jones DT, Jäger N, Kool M, et al.: Dissecting the genomic complexity underlying medulloblastoma. Nature 488 (7409): 100-5, 2012.
- Peyrl A, Chocholous M, Kieran MW, et al.: Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr Blood Cancer 59 (3): 511-7, 2012.
- Taylor MD, Northcott PA, Korshunov A, et al.: Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123 (4): 465-72, 2012.
- Kool M, Korshunov A, Remke M, et al.: Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123 (4): 473-84, 2012.
- Pietsch T, Schmidt R, Remke M, et al.: Prognostic significance of clinical, histopathological, and molecular characteristics of medulloblastomas in the prospective HIT2000 multicenter clinical trial cohort. Acta Neuropathol 128 (1): 137-49, 2014.
- Cho YJ, Tsherniak A, Tamayo P, et al.: Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29 (11): 1424-30, 2011.
- Gajjar A, Bowers DC, Karajannis MA, et al.: Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J Clin Oncol 33 (27): 2986-98, 2015.
- Morrissy AS, Cavalli FMG, Remke M, et al.: Spatial heterogeneity in medulloblastoma. Nat Genet 49 (5): 780-788, 2017.
- Wang X, Dubuc AM, Ramaswamy V, et al.: Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol 129 (3): 449-57, 2015.
- Cavalli FMG, Remke M, Rampasek L, et al.: Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 31 (6): 737-754.e6, 2017.
- Northcott PA, Buchhalter I, Morrissy AS, et al.: The whole-genome landscape of medulloblastoma subtypes. Nature 547 (7663): 311-317, 2017.
- Schwalbe EC, Lindsey JC, Nakjang S, et al.: Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol 18 (7): 958-971, 2017.
- Hicks D, Rafiee G, Schwalbe EC, et al.: The molecular landscape and associated clinical experience in infant medulloblastoma: prognostic significance of second-generation subtypes. Neuropathol Appl Neurobiol 47 (2): 236-250, 2021.
- Northcott PA, Jones DT, Kool M, et al.: Medulloblastomics: the end of the beginning. Nat Rev Cancer 12 (12): 818-34, 2012.
- Korshunov A, Sahm F, Zheludkova O, et al.: DNA methylation profiling is a method of choice for molecular verification of pediatric WNT-activated medulloblastomas. Neuro Oncol 21 (2): 214-221, 2019.
- Gibson P, Tong Y, Robinson G, et al.: Subtypes of medulloblastoma have distinct developmental origins. Nature 468 (7327): 1095-9, 2010.
- Ellison DW, Dalton J, Kocak M, et al.: Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol 121 (3): 381-96, 2011.
- Gajjar A, Chintagumpala M, Ashley D, et al.: Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol 7 (10): 813-20, 2006.
- Michalski JM, Janss AJ, Vezina LG, et al.: Children's Oncology Group Phase III Trial of Reduced-Dose and Reduced-Volume Radiotherapy With Chemotherapy for Newly Diagnosed Average-Risk Medulloblastoma. J Clin Oncol 39 (24): 2685-2697, 2021.
- Goschzik T, Mynarek M, Doerner E, et al.: Genetic alterations of TP53 and OTX2 indicate increased risk of relapse in WNT medulloblastomas. Acta Neuropathol 144 (6): 1143-1156, 2022.
- Begemann M, Waszak SM, Robinson GW, et al.: Germline GPR161 Mutations Predispose to Pediatric Medulloblastoma. J Clin Oncol 38 (1): 43-50, 2020.
- Shuai S, Suzuki H, Diaz-Navarro A, et al.: The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 574 (7780): 712-716, 2019.
- Suzuki H, Kumar SA, Shuai S, et al.: Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 574 (7780): 707-711, 2019.
- Kool M, Jones DT, Jäger N, et al.: Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 25 (3): 393-405, 2014.
- Kolodziejczak AS, Guerrini-Rousseau L, Planchon JM, et al.: Clinical outcome of pediatric medulloblastoma patients with Li-Fraumeni syndrome. Neuro Oncol 25 (12): 2273-2286, 2023.
- Robinson GW, Rudneva VA, Buchhalter I, et al.: Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol 19 (6): 768-784, 2018.
- Lafay-Cousin L, Bouffet E, Strother D, et al.: Phase II Study of Nonmetastatic Desmoplastic Medulloblastoma in Children Younger Than 4 Years of Age: A Report of the Children's Oncology Group (ACNS1221). J Clin Oncol 38 (3): 223-231, 2020.
- Leary SE, Zhou T, Holmes E, et al.: Histology predicts a favorable outcome in young children with desmoplastic medulloblastoma: a report from the children's oncology group. Cancer 117 (14): 3262-7, 2011.
- Giangaspero F, Perilongo G, Fondelli MP, et al.: Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg 91 (6): 971-7, 1999.
- Rutkowski S, von Hoff K, Emser A, et al.: Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. J Clin Oncol 28 (33): 4961-8, 2010.
- Garrè ML, Cama A, Bagnasco F, et al.: Medulloblastoma variants: age-dependent occurrence and relation to Gorlin syndrome--a new clinical perspective. Clin Cancer Res 15 (7): 2463-71, 2009.
- von Bueren AO, von Hoff K, Pietsch T, et al.: Treatment of young children with localized medulloblastoma by chemotherapy alone: results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro Oncol 13 (6): 669-79, 2011.
- Shih DJ, Northcott PA, Remke M, et al.: Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol 32 (9): 886-96, 2014.
- Schwalbe EC, Williamson D, Lindsey JC, et al.: DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol 125 (3): 359-71, 2013.
- Zhukova N, Ramaswamy V, Remke M, et al.: Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol 31 (23): 2927-35, 2013.
- Gajjar A, Robinson GW, Smith KS, et al.: Outcomes by Clinical and Molecular Features in Children With Medulloblastoma Treated With Risk-Adapted Therapy: Results of an International Phase III Trial (SJMB03). J Clin Oncol 39 (7): 822-835, 2021.
- Goschzik T, Schwalbe EC, Hicks D, et al.: Prognostic effect of whole chromosomal aberration signatures in standard-risk, non-WNT/non-SHH medulloblastoma: a retrospective, molecular analysis of the HIT-SIOP PNET 4 trial. Lancet Oncol 19 (12): 1602-1616, 2018.
- Gottardo NG, Hansford JR, McGlade JP, et al.: Medulloblastoma Down Under 2013: a report from the third annual meeting of the International Medulloblastoma Working Group. Acta Neuropathol 127 (2): 189-201, 2014.
- Louis DN, Perry A, Burger P, et al.: International Society Of Neuropathology--Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol 24 (5): 429-35, 2014.
- Sharma T, Schwalbe EC, Williamson D, et al.: Second-generation molecular subgrouping of medulloblastoma: an international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol 138 (2): 309-326, 2019.
- von Hoff K, Haberler C, Schmitt-Hoffner F, et al.: Therapeutic implications of improved molecular diagnostics for rare CNS embryonal tumor entities: results of an international, retrospective study. Neuro Oncol 23 (9): 1597-1611, 2021.
- Korshunov A, Sturm D, Ryzhova M, et al.: Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol 128 (2): 279-89, 2014.
- Picard D, Miller S, Hawkins CE, et al.: Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: an integrative genomic analysis. Lancet Oncol 13 (8): 838-48, 2012.
- Spence T, Sin-Chan P, Picard D, et al.: CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol 128 (2): 291-303, 2014.
- Juhnke BO, Gessi M, Gerber NU, et al.: Treatment of embryonal tumors with multilayered rosettes with carboplatin/etoposide induction and high-dose chemotherapy within the prospective P-HIT trial. Neuro Oncol 24 (1): 127-137, 2022.
- Khan S, Solano-Paez P, Suwal T, et al.: Clinical phenotypes and prognostic features of embryonal tumours with multi-layered rosettes: a Rare Brain Tumor Registry study. Lancet Child Adolesc Health 5 (11): 800-813, 2021.
- Kleinman CL, Gerges N, Papillon-Cavanagh S, et al.: Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet 46 (1): 39-44, 2014.
- Li M, Lee KF, Lu Y, et al.: Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell 16 (6): 533-46, 2009.
- Korshunov A, Okonechnikov K, Schmitt-Hoffner F, et al.: Molecular analysis of pediatric CNS-PNET revealed nosologic heterogeneity and potent diagnostic markers for CNS neuroblastoma with FOXR2-activation. Acta Neuropathol Commun 9 (1): 20, 2021.
- Ueno-Yokohata H, Okita H, Nakasato K, et al.: Consistent in-frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat Genet 47 (8): 861-3, 2015.
- Roy A, Kumar V, Zorman B, et al.: Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat Commun 6: 8891, 2015.
- Louis DN, Ohgaki H, Wiestler OD, et al.: The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114 (2): 97-109, 2007.
- Sharma MC, Mahapatra AK, Gaikwad S, et al.: Pigmented medulloepithelioma: report of a case and review of the literature. Childs Nerv Syst 14 (1-2): 74-8, 1998 Jan-Feb.
- Jakobiec FA, Kool M, Stagner AM, et al.: Intraocular Medulloepitheliomas and Embryonal Tumors With Multilayered Rosettes of the Brain: Comparative Roles of LIN28A and C19MC. Am J Ophthalmol 159 (6): 1065-1074.e1, 2015.
- Korshunov A, Jakobiec FA, Eberhart CG, et al.: Comparative integrated molecular analysis of intraocular medulloepitheliomas and central nervous system embryonal tumors with multilayered rosettes confirms that they are distinct nosologic entities. Neuropathology 35 (6): 538-44, 2015.
- Keck MK, Sill M, Wittmann A, et al.: Amplification of the PLAG-family genes-PLAGL1 and PLAGL2-is a key feature of the novel tumor type CNS embryonal tumor with PLAGL amplification. Acta Neuropathol 145 (1): 49-69, 2023.
- de Jong MC, Kors WA, de Graaf P, et al.: Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol 15 (10): 1157-67, 2014.
- Ramasubramanian A, Kytasty C, Meadows AT, et al.: Incidence of pineal gland cyst and pineoblastoma in children with retinoblastoma during the chemoreduction era. Am J Ophthalmol 156 (4): 825-9, 2013.
- Abramson DH, Dunkel IJ, Marr BP, et al.: Incidence of pineal gland cyst and pineoblastoma in children with retinoblastoma during the chemoreduction era. Am J Ophthalmol 156 (6): 1319-20, 2013.
- Turaka K, Shields CL, Meadows AT, et al.: Second malignant neoplasms following chemoreduction with carboplatin, etoposide, and vincristine in 245 patients with intraocular retinoblastoma. Pediatr Blood Cancer 59 (1): 121-5, 2012.
- de Kock L, Sabbaghian N, Druker H, et al.: Germ-line and somatic DICER1 mutations in pineoblastoma. Acta Neuropathol 128 (4): 583-95, 2014.
- Liu APY, Li BK, Pfaff E, et al.: Clinical and molecular heterogeneity of pineal parenchymal tumors: a consensus study. Acta Neuropathol 141 (5): 771-785, 2021.
- Pajtler KW, Witt H, Sill M, et al.: Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 27 (5): 728-43, 2015.
- Witt H, Mack SC, Ryzhova M, et al.: Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 20 (2): 143-57, 2011.
- Mack SC, Witt H, Piro RM, et al.: Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506 (7489): 445-50, 2014.
- Pajtler KW, Mack SC, Ramaswamy V, et al.: The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol 133 (1): 5-12, 2017.
- Zschernack V, Jünger ST, Mynarek M, et al.: Supratentorial ependymoma in childhood: more than just RELA or YAP. Acta Neuropathol 141 (3): 455-466, 2021.
- Ramaswamy V, Hielscher T, Mack SC, et al.: Therapeutic Impact of Cytoreductive Surgery and Irradiation of Posterior Fossa Ependymoma in the Molecular Era: A Retrospective Multicohort Analysis. J Clin Oncol 34 (21): 2468-77, 2016.
- Korshunov A, Witt H, Hielscher T, et al.: Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol 28 (19): 3182-90, 2010.
- Merchant TE, Bendel AE, Sabin ND, et al.: Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma. J Clin Oncol 37 (12): 974-983, 2019.
- Baroni LV, Sundaresan L, Heled A, et al.: Ultra high-risk PFA ependymoma is characterized by loss of chromosome 6q. Neuro Oncol 23 (8): 1360-1370, 2021.
- Panwalkar P, Clark J, Ramaswamy V, et al.: Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol 134 (5): 705-714, 2017.
- Chapman RJ, Ghasemi DR, Andreiuolo F, et al.: Optimizing biomarkers for accurate ependymoma diagnosis, prognostication, and stratification within International Clinical Trials: A BIOMECA study. Neuro Oncol 25 (10): 1871-1882, 2023.
- Pajtler KW, Wen J, Sill M, et al.: Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol 136 (2): 211-226, 2018.
- Gessi M, Capper D, Sahm F, et al.: Evidence of H3 K27M mutations in posterior fossa ependymomas. Acta Neuropathol 132 (4): 635-7, 2016.
- Ryall S, Guzman M, Elbabaa SK, et al.: H3 K27M mutations are extremely rare in posterior fossa group A ependymoma. Childs Nerv Syst 33 (7): 1047-1051, 2017.
- Cavalli FMG, Hübner JM, Sharma T, et al.: Heterogeneity within the PF-EPN-B ependymoma subgroup. Acta Neuropathol 136 (2): 227-237, 2018.
- Parker M, Mohankumar KM, Punchihewa C, et al.: C11orf95-RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 506 (7489): 451-5, 2014.
- Pietsch T, Wohlers I, Goschzik T, et al.: Supratentorial ependymomas of childhood carry C11orf95-RELA fusions leading to pathological activation of the NF-κB signaling pathway. Acta Neuropathol 127 (4): 609-11, 2014.
- Pagès M, Pajtler KW, Puget S, et al.: Diagnostics of pediatric supratentorial RELA ependymomas: integration of information from histopathology, genetics, DNA methylation and imaging. Brain Pathol 29 (3): 325-335, 2019.
- Jünger ST, Andreiuolo F, Mynarek M, et al.: CDKN2A deletion in supratentorial ependymoma with RELA alteration indicates a dismal prognosis: a retrospective analysis of the HIT ependymoma trial cohort. Acta Neuropathol 140 (3): 405-407, 2020.
- Milde T, Pfister S, Korshunov A, et al.: Stepwise accumulation of distinct genomic aberrations in a patient with progressively metastasizing ependymoma. Genes Chromosomes Cancer 48 (3): 229-38, 2009.
- Sievers P, Henneken SC, Blume C, et al.: Recurrent fusions in PLAGL1 define a distinct subset of pediatric-type supratentorial neuroepithelial tumors. Acta Neuropathol 142 (5): 827-839, 2021.
- Fukuoka K, Kanemura Y, Shofuda T, et al.: Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol Commun 6 (1): 134, 2018.
- Ghasemi DR, Sill M, Okonechnikov K, et al.: MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol 138 (6): 1075-1089, 2019.
- Swanson AA, Raghunathan A, Jenkins RB, et al.: Spinal Cord Ependymomas With MYCN Amplification Show Aggressive Clinical Behavior. J Neuropathol Exp Neurol 78 (9): 791-797, 2019.
- Scheil S, Brüderlein S, Eicker M, et al.: Low frequency of chromosomal imbalances in anaplastic ependymomas as detected by comparative genomic hybridization. Brain Pathol 11 (2): 133-43, 2001.
- Raffeld M, Abdullaev Z, Pack SD, et al.: High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathol Commun 8 (1): 101, 2020.
Sarcomas
Osteosarcoma
Características moleculares del osteosarcoma
El panorama genómico del osteosarcoma es distinto al de otros tipos de cáncer infantil. Se caracteriza por un número inusual de variantes estructurales y un número relativamente pequeño de variantes de un solo nucleótido, en comparación con muchos cánceres en adultos.[
Las observaciones clave relacionadas con el panorama genómico del osteosarcoma son las siguientes:
- El número de variantes estructurales observadas en el osteosarcoma es alto: más de 200 variantes estructurales por genoma;[
1 ,2 ] por lo tanto, el osteosarcoma exhibe el genoma más caótico de los cánceres infantiles. Los diagramas Circos que se presentan en la Figura ilustran los números excepcionalmente altos de translocaciones intra e intercromosómicas que caracterizan los genomas del osteosarcoma.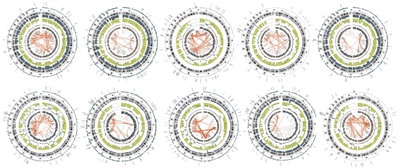
- La carga mutacional del tumor (TMB) en niños y adolescentes con osteosarcoma es de alrededor de 2 mutaciones por megabase y es más alta que la de otros tipos de cáncer infantil (por ejemplo, sarcoma de Ewing o tumores rabdoides).[
1 ,2 ] Sin embargo, esa tasa es muy inferior a la de los cánceres en adultos, como el melanoma o el cáncer de pulmón de células no pequeñas, que responden a los inhibidores de puntos de control. - En lugar de variantes activadoras en los oncogenes y variantes inactivadoras de los genes supresores de tumores, como se observa en muchos tipos de cáncer, el panorama genómico del osteosarcoma se debe a la ganancia o amplificación del número de copias en regiones cromosómicas que incluyen pérdida (deleciones) de oncogenes y número de copias en regiones cromosómicas que incluyen genes supresores de tumores. A continuación, se describen las ganancias y pérdidas recurrentes del número de copias que afectan los oncogenes y los genes supresores de tumores conocidos, respectivamente.
Las estimaciones de la frecuencia con la que se presentan alteraciones genómicas específicas en el osteosarcoma varían de un informe a otro. Este hallazgo quizás se deba a las diferentes definiciones que se usan para describir las alteraciones en el número de copias, los distintos métodos que se utilizan para su detección o las diferencias en las características biológicas del tumor entre las poblaciones de pacientes (por ejemplo, recién diagnosticado versus en recaída, localizado versus metastásico o pediátrico versus adulto).
- En la mayoría de los casos de osteosarcoma hay alteraciones genómicas en el gen TP53 que conducen a la pérdida de función de TP53.[
1 ] Una forma característica de inactivación de TP53 se produce por variantes estructurales en el primer intrón de TP53 que causan la alteración del gen TP53.[1 ] También se observan otros mecanismos de inactivación de TP53, como las mutaciones de aminoácido y las mutaciones interruptoras, así como las deleciones del gen TP53.[1 ,2 ] La combinación de varios de estos mecanismos que originan la pérdida de función de TP53 produce una inactivación bialélica en la mayoría de los casos de osteosarcoma. Debido a que muchas de las variaciones estructurales que conducen a la inactivación de TP53 se detectan mejor a través de la secuenciación del genoma completo, los resultados con base en paneles de pruebas genómicas clínicas tal vez muestren tasas más bajas de alteraciones de TP53 porque no detectan dichos cambios.[3 ] - La amplificación de MDM2, que es otra alteración genómica que conduce a la pérdida de función de TP53, se observa en una minoría de casos de osteosarcoma (alrededor de un 5 %).[
1 ,2 ,3 ,4 ] - El gen RB1 se inactiva con frecuencia en el osteosarcoma, a veces por variantes deletéreas, pero de manera más habitual por deleción cromosómica de la región del cromosoma 13q14 que incluye RB1.[
1 ,2 ,5 ] - Las deleciones cromosómicas que incluyen al cromosoma 9p21 llevan a la deleción del gen CDN2A en alrededor del 20 % de los casos de osteosarcoma.[
1 ,2 ,5 ] - Entre los oncogenes tumorales, el gen MYC en el cromosoma 8q24 muestra ganancia o amplificación en cerca del 10 % de los pacientes.[
3 ,5 ,6 ] En un estudio se relacionó la ganancia o amplificación de MYC con un pronóstico inferior. En un segundo estudio, la ganancia o amplificación de MYC fue más frecuente en niños que en adultos.[6 ] - El gen CCNE1 en el cromosoma 19q12 es otro oncogén tumoral que muestra ganancia o amplificación en alrededor del 10 % de los pacientes.[
3 ,5 ,6 ] Otras regiones cromosómicas con oncogén que muestran ganancia o amplificación cromosómica en una minoría de casos de osteosarcoma son la región que alberga el gen CDK4 en el cromosoma 12q14,[4 ,5 ,7 ] las regiones que albergan los genes VEGFA y CCND3 en el cromosoma 6p12,[3 ,4 ,5 ,7 ] la región que alberga el gen CCND1 en el cromosoma 11q13[4 ] y las regiones que albergan los genes PDGFRA, KIT y KDR en el cromosoma 4q12.[3 ,4 ,5 ] - El alargamiento alternativo de los telómeros (ALT) es el mecanismo de mantenimiento de telómeros empleado por la mayoría de los tumores de osteosarcoma.[
1 ,8 ,9 ] Las variantes inactivadoras del gen ATRX y las deleciones génicas se relacionan con el mecanismo de mantenimiento de telómeros ALT. Las alteraciones genómicas de ATRX están presentes en un subconjunto de tumores de osteosarcoma que usan este mecanismo de mantenimiento de telómeros.[1 ,3 ,9 ] - Muchas de las alteraciones genómicas notificadas para los tumores de osteosarcoma en el momento del diagnóstico no proporcionan dianas terapéuticas evidentes, ya que reflejan la pérdida de genes supresores de tumores (por ejemplo, los genes TP53, RB1 o PTEN), en lugar de la activación de oncogenes que pueden servir como dianas terapéuticas. Además, el éxito ha sido limitado en los diagnósticos de cáncer al usar las ganancias o amplificaciones de los oncogenes relevantes en el osteosarcoma para identificar a los pacientes que quizás se beneficien de la terapia dirigida.
Predisposición genética al osteosarcoma
Las variantes germinales de varios genes se relacionan con la susceptibilidad al osteosarcoma. En el Cuadro 5 se resumen los síndromes y los genes asociados con estas afecciones. En un reciente estudio genómico multiinstitucional de más de 1200 pacientes con osteosarcoma, se detectaron variantes germinales patógenas, o probablemente patógenas, en genes de susceptibilidad al cáncer de herencia autosómica dominante en un 18 % de los pacientes. Estos genes de susceptibilidad al cáncer fueron más frecuentes en niños de 10 años o menores.[
Variantes deTP53
Las variantes del gen TP53 son las alteraciones más comunes de la línea germinal relacionadas con el osteosarcoma. Se encuentran mutaciones en este gen en cerca del 70 % de los pacientes con síndrome de Li-Fraumeni (SLF), que se relaciona con un mayor riesgo de osteosarcoma, cáncer de mama, varios tipos de cáncer de encéfalo (incluso el cerebro), sarcomas de tejido blando y otros cánceres. Aunque el rabdomiosarcoma es el sarcoma más común en pacientes de 5 años o menos que tienen SLF relacionado con TP53, el osteosarcoma es el sarcoma más común en niños y adolescentes de 6 a 19 años.[
Variantes deRECQL4
Los investigadores analizaron datos de secuenciación del exoma completo de la línea germinal de 4435 pacientes con cáncer pediátrico del St. Jude Children's Research Hospital y 1127 pacientes de la base de datos National Cancer Institute's Therapeutically Applicable Research to Generate Effective Treatment (TARGET). Se identificaron 24 pacientes (0,43 %) que albergaban variantes de pérdida de función de RECQL4, entre ellos, 5 de 249 pacientes (2,0 %) con osteosarcoma.[
| Síndrome | Descripción | Localización | Gen | Función |
|---|---|---|---|---|
| LMA = leucemia mieloide aguda; IL-1 = interleucina-1; SMD = síndrome mielodisplásico; RANKL = ligando del receptor activador del factor nuclear κ β; FNT = factor de necrosis tumoral. | ||||
| a Adaptación de Kansara et al.[ |
||||
| Síndrome de Bloom[ |
Es un trastorno hereditario raro caracterizado por estatura baja y cambios por fotosensibilidad cutánea. A menudo se manifiesta con cara alargada y angosta, maxilar inferior pequeño, nariz grande y orejas prominentes. | 15q26.1 | BLM | Helicasa de DNA |
| Anemia de Diamond-Blackfan[ |
Aplasia pura de células rojas hereditaria. Pacientes en riesgo de SMD y LMA. Se relaciona con anomalías esqueléticas, como rasgos faciales anormales (dorso nasal plano e hipertelorismo ocular). | Proteínas ribosómicas | Producción ribosómica[ |
|
| Síndrome de Li-Fraumeni[ |
Variante hereditaria del genTP53. Los familiares afectados tienen un aumento del riesgo de tumores óseos, cáncer de mama, leucemia, tumores de encéfalo y sarcomas. | 17p13.1 | TP53 | Reacción al daño del DNA |
| Enfermedad de Paget[ |
Osteolisis excesiva con formación y remodelación de hueso anormal, lo que produce dolor por debilidad y deformidad óseas. | 18q21-qa22 | LOH18CR1 | Señalización IL-1/FNT; vía de señalización RANKL |
| 5q31 | ||||
| 5q35-qter | ||||
| Retinoblastoma[ |
Tumor maligno de retina Cerca del 66 % de los pacientes recibe el diagnóstico a los 2 años de edad y el 95 % lo recibe a los 3 años. Los pacientes con variantes hereditarias en las células germinales presentan un riesgo más alto de neoplasias subsiguientes. | 13q14.2 | RB1 | Punto de control del ciclo celular |
| Síndrome de Rothmund-Thomson (también llamado poiquilodermia congénita)[ |
Afección autosómica recesiva. Presenta manifestaciones cutáneas (atrofia, telangiectasias y pigmentación), vello escaso, cataratas, estatura baja y anormalidades esqueléticas. Aumento en la incidencia de osteosarcoma a menor edad. | 8q24.3 | RECQL4 | Helicasa de DNA |
| Síndrome de Werner[ |
Los pacientes a menudo tienen estatura baja, y un poco después de los 20 años aparecen signos de envejecimiento, como encanecimiento y esclerosis cutánea. Más adelante presentan otros problemas propios del envejecimiento como cataratas, úlceras cutáneas y aterosclerosis. | 8p12-p11.2 | WRN | Helicasa de DNA; actividad de exonucleasa |
Para obtener más información sobre estos síndromes genéticos, consulte los siguientes resúmenes:
-
Genética del cáncer de piel (síndrome de Bloom ,síndrome de Rothmund-Thomson ysíndrome de Werner ).
El siguiente resumen solo está disponible en inglés:
-
Genetics of Breast and Gynecologic Cancers (Li-Fraumeni syndrome [LFS] ).
Para obtener información sobre el tratamiento del osteosarcoma, consultar
Sarcoma de Ewing
Características moleculares del sarcoma de Ewing
La Organización Mundial de la Salud identificó la presencia de una fusión génica que involucra EWSR1 o FUS y un gen de la familia ETS como un elemento que define el sarcoma de Ewing.[
Una translocación EWSR1::FLI1 que se relaciona con el sarcoma de Ewing puede ocurrir en varios sitios de ruptura posibles en cada uno de los genes que se unen para formar el nuevo segmento de DNA. A pesar de que en algún momento se pensó que era importante,[
Además de las anomalías sistemáticas que comprometen el gen EWSR1, en la mayoría de los casos de sarcoma de Ewing se observan anomalías numéricas y estructurales secundarias. Las ganancias cromosómicas son más comunes que las pérdidas cromosómicas, y también se observan desequilibrios cromosómicos estructurales.[
- Ganancia de un cromosoma 8 completo (trisomía 8). La trisomía 8 es la alteración cromosómica más frecuente en el sarcoma de Ewing y se presenta en casi el 50 % de los tumores.[
28 ,29 ] La ganancia del cromosoma 8 no presenta importancia pronóstica.[28 ,37 ] - Ganancia del cromosoma 1q y pérdida del cromosoma 16q. Estas anomalías ocurren en aproximadamente el 20 % de los pacientes y, a menudo, se presentan juntas. La ganancia del cromosoma 1q o la deleción del cromosoma 16q se ha relacionado con un pronóstico más precario en los pacientes con sarcoma de Ewing de varias cohortes.[
28 ,38 ,39 ] Estas dos alteraciones cromosómicas a menudo se presentan juntas en varios tipos de cáncer, entre ellos, el sarcoma de Ewing.[40 ] Es probable que la presencia simultánea se deba a que su causa es una translocación t(1;16) desequilibrada que produce ganancia del cromosoma 1q junto con pérdida de material cromosómico de 16q.[37 ,41 ]
El panorama genómico del sarcoma de Ewing se caracteriza por un genoma relativamente inactivo, con una escasez de variantes en las vías que podrían ser susceptibles de tratamiento con terapias dirigidas nuevas.[
- Variantes de STAG2. Las variantes de STAG2, un miembro del complejo de la cohesina, se presentan en cerca del 15 % al 20 % de los casos.[
28 ,29 ,30 ] Estas variantes conducen a la pérdida de la expresión y el funcionamiento de STAG2 en las células tumorales.[30 ] Se observó pérdida de la expresión de STAG2 (detectada mediante pruebas inmunohistoquímicas [IHQ]) en tumores en los que no se puede detectar una variante de STAG2. En un informe, la pérdida de expresión de STAG2, comprobada mediante pruebas IHQ, se relacionó con un pronóstico inferior.[42 ] - Deleciones de CDKN2A. Se observaron deleciones de CDKN2A en un 12 % a un 22 % de los casos.[
28 ,29 ,30 ] - Variantes de TP53. Se identificaron variantes de TP53 en cerca de un 6 % a un 7 % de los casos de sarcoma de Ewing notificados por equipos de investigación en pediatría.[
28 ,29 ,30 ] Se han descrito tasas más elevadas de variantes de TP53 (hasta en un 19 %) en cohortes de una sola institución que contienen porcentajes más altos de pacientes adultos.[43 ,44 ] En un informe retrospectivo, la coexistencia de variantes de STAG2 y TP53 se relacionó con un desenlace clínico precario. - Alteraciones de ERF. Se notificaron alteraciones genómicas de ERF que condujeron a la pérdida de función (mutación en el marco de lectura, mutación de cambio de sentido y deleción importante) en el 7 % de los tumores de sarcoma de Ewing.[
43 ] En un segundo informe, se observaron alteraciones de ERF a una tasa del 3 % en otra cohorte de sarcoma de Ewing.[29 ] - Otros genes con alteraciones genómicas recurrentes en el sarcoma de Ewing. Se notificaron alteraciones genómicas recurrentes en menos del 5 % de los pacientes con sarcoma de Ewing: EZH2,[
28 ,44 ]BCOR,[28 ]SMARCA4,[44 ]CREBBP,[44 ]TERT y FGFR1.[43 ]
Todas las traslocaciones del sarcoma de Ewing se pueden encontrar mediante análisis citogenético estándar. En la actualidad, con frecuencia se realiza un análisis rápido de hibridación fluorescente in situ (FISH) en busca de una ruptura del gen EWSR1 para confirmar el diagnóstico molecular de sarcoma de Ewing.[
| Miembro de la familia FET | Fusión con un oncogén recíproco similar a ETS | Translocación | Comentario |
|---|---|---|---|
| a Estos oncogenes recíprocos no son miembros de la familia de oncogenes ETS; por lo tanto, estos tumores no se clasifican como sarcoma de Ewing. | |||
| EWSR1 | EWSR1::FLI1 | t(11;22)(q24;q12) | Más común; alrededor del 85 % al 90 % de los casos |
| EWSR1::ERG | t(21;22)(q22;q12) | Más común en segundo término; alrededor del 10 % de los casos | |
| EWSR1::ETV1 | t(7;22)(p22;q12) | Poco frecuente | |
| EWSR1::ETV4 | t(17;22)(q12;q12) | Poco frecuente | |
| EWSR1::FEV | t(2;22)(q35;q12) | Poco frecuente | |
| EWSR1::NFATC2a | t(20;22)(q13;q12) | Poco frecuente | |
| EWSR1::POU5F1a | t(6;22)(p21;q12) | ||
| EWSR1::SMARCA5a | t(4;22)(q31;q12) | Poco frecuente | |
| EWSR1::PATZ1a | t(6;22)(p21;q12) | ||
| EWSR1::SP3a | t(2;22)(q31;q12) | Poco frecuente | |
| FUS | FUS::ERG | t(16;21)(p11;q22) | Poco frecuente |
| FUS::FEV | t(2;16)(q35;p11) | Poco frecuente | |
Para obtener información sobre el tratamiento del sarcoma de Ewing, consultar
Rabdomiosarcoma
Características genómicas del rabdomiosarcoma
Las 4 categorías histológicas reconocidas en la 5a edición de la clasificación de la Organización Mundial de la Salud (OMS) de tumores de tejidos blandos y hueso exhiben alteraciones genómicas específicas que se resumen a continuación.[
- Rabdomiosarcoma embrionario: se caracteriza por la pérdida de heterocigosis en 11p15 y por la elevada frecuencia de variantes de genes de la vía RAS. Para los fines de esta sección, se considera que los pacientes con rabdomiosarcoma embrionario no presentan las fusiones génicas PAX3::FOXO1 y PAX7::FOXO1 (es decir, rabdomiosarcoma negativo para fusiones).
- Rabdomiosarcoma alveolar: se caracteriza por fusiones génicas de FOXO1 con PAX3 o PAX7 (es decir, rabdomiosarcoma positivo para fusiones de FOXO1). Los casos con rabdomiosarcoma de tipo histológico alveolar sin fusiones génicas de FOXO1 son similares a los casos con rabdomiosarcoma embrionario en cuanto al comportamiento clínico, la distribución de alteraciones génicas y el perfil transcriptómico. Por lo tanto, el análisis que sigue se enfoca solo en el rabdomiosarcoma alveolar con fusiones génicas de FOXO1.[
49 ,50 ,51 ,52 ,53 ] - Rabdomiosarcoma de células fusiformes o esclerosante: se caracteriza por variantes de MYOD1 en pacientes mayores, y por reordenamientos génicos en VGLL2 y NCOA2 en niños pequeños.
- Rabdomiosarcoma pleomórfico: se caracteriza por cariotipos complejos con cambios numéricos y cambios estructurales no equilibrados que son indistinguibles de los cariotipos del sarcoma pleomórfico indiferenciado.
La distribución de las variantes génicas y las amplificaciones génicas (de CDK4 y MYCN) difiere entre los pacientes con un tipo histológico embrionario sin fusiones génicas PAX::FOXO1 (rabdomiosarcoma negativo para fusiones) y los pacientes con fusiones génicas PAX::FOXO1 (rabdomiosarcoma positivo para fusiones). Consultar el Cuadro a continuación y el texto que sigue. Estas cifras de distribución se derivan de una cohorte combinada de pacientes con rabdomiosarcoma del Children's Oncology Group (COG) y del Reino Unido (n = 641).[
| Gen | % de casos negativos para fusiones que presentan la alteración génica | % de casos positivos para fusiones que presentan la alteración génica |
|---|---|---|
| a Adaptación de Shern et al.[ |
||
| NRAS | 17 % | 1 % |
| KRAS | 9 % | 1 % |
| HRAS | 8 % | 2 % |
| FGFR4 | 13 % | 0 % |
| NF1 | 15 % | 4 % |
| BCOR | 15 % | 6 % |
| TP53 | 13 % | 4 % |
| CTNNB1 | 6 % | 0 % |
| CDK4 | 0 % | 13 % |
| MYCN | 0 % | 10 % |
A continuación se describe información detallada sobre las alteraciones genómicas que predominan en cada categoría histológica de la OMS.
- Rabdomiosarcoma negativo para fusiones (tipo histológico embrionario): los tumores de rabdomiosarcoma embrionario a menudo exhiben pérdida de heterocigosis en 11p15 y ganancias en el cromosoma 8.[
55 ,56 ,57 ,58 ] Los tumores embrionarios, en comparación con los tumores de rabdomiosarcoma alveolar, tienen mayor tasa de variantes de fondo y también mayor tasa de variantes de un solo nucleótido. Además, el número de variantes somáticas aumenta con la edad en el momento del diagnóstico.[58 ,59 ] Los genes con variantes recurrentes más frecuentes son los de la vía RAS (por ejemplo, NRAS, KRAS, HRAS y NF1), que en conjunto se detectan en alrededor de la mitad de los casos.[54 ] Las variantes de NRAS son las variantes génicas más frecuentes de la vía RAS en niños mayores de 1 año, mientras que las variantes de HRAS predominan durante la lactancia.[54 ] La presencia de una variante de RAS no tiene importancia pronóstica.Entre los genes de la vía RAS, las variantes germinales de NF1 y HRAS predisponen al rabdomiosarcoma. En un estudio de 615 niños con rabdomiosarcoma, 347 tenían tumores del tipo histológico embrionario. De este grupo, 9 pacientes tenía variantes germinales de NF1 y 5 pacientes tenía variantes germinales de HRAS, lo que representa un 2,6 % y un 1,4 % de los casos de tipo histológico embrionario, respectivamente.[
60 ]Otros genes con variantes recurrentes en los tumores de rabdomiosarcoma negativos para fusiones son FGFR4, PIK3CA, CTNNB1, FBXW7 y BCOR, que en conjunto se detectan en menos del 15 % de los casos.[
54 ,58 ,59 ]Variantes de TP53: las variantes del gen TP53 se observan en el 10 % al 15 % de los pacientes con rabdomiosarcoma negativo para fusiones y son menos frecuentes (alrededor de un 4 %) en pacientes con rabdomiosarcoma alveolar.[
54 ] En otros tipos de cáncer infantil (por ejemplo, el tumor de Wilms) las variantes de TP53 se asocian con un tipo histológico anaplásico,[61 ] igual que para el rabdomiosarcoma embrionario. En un estudio de 146 pacientes de rabdomiosarcoma con variante de TP53 conocida, cerca de dos tercios de los tumores con variantes de TP53 mostraban anaplasia (69 %), pero solo un cuarto de los tumores con anaplasia tenía variantes de TP53.[62 ]En una cohorte de rabdomiosarcoma del COG y el Reino Unido, la presencia de variantes de TP53 se vinculó con disminución en la SSC tanto en el análisis estratificado por riesgo como en el análisis que no se estratificó según el riesgo.[
54 ] El pronóstico precario asociado a las variantes de TP53 se ha observado en pacientes con tumores embrionarios y alveolares. A partir de estos resultados, el COG planea clasificar la variante de TP53 como una característica definitoria de riesgo alto en futuros ensayos.[63 ]El rabdomiosarcoma es uno de los tipos de cáncer infantil que se asocian con el síndrome de Li-Fraumeni. En un estudio de 614 pacientes pediátricos con rabdomiosarcoma, 11 pacientes (1,7 %) presentaban variantes germinales de TP53. Las variantes fueron menos comunes en los pacientes con el tipo histológico alveolar (0,6 %) en comparación con los pacientes con tipos histológicos diferentes al alveolar (2,2 %).[
60 ] El rabdomiosarcoma con características morfológicas de anaplasia no alveolar quizás corresponda a un tipo de presentación en niños con el síndrome de Li-Fraumeni y variantes germinales de TP53.[64 ]- De 8 niños atendidos de manera consecutiva por rabdomiosarcoma con variantes germinales de TP53, todos exhibían características morfológicas de anaplasia. En otros 7 niños con rabdomiosarcoma anaplásico y estado desconocido de variante germinal de TP53, se encontró que 3 de ellos presentaban variantes germinales de TP53 con un efecto funcional de interés. La mediana de edad en el momento del diagnóstico de los 11 niños con estado conocido de variante germinal de TP53 fue de 40 meses (intervalo, 19–67 meses).[
64 ] - En otra serie, 26 de 31 pacientes con variantes germinales de TP53 tenían tumores con tipo histológico embrionario. De los 16 tumores que se enviaron a revisión patológica central, 12 tenían anaplasia focal o difusa. La mediana de edad de los pacientes de este grupo fue de 2,3 años.[
65 ]
Variantes de DICER1 en el rabdomiosarcoma embrionario: se observan variantes de DICER1 en un subgrupo pequeño de pacientes con rabdomiosarcoma embrionario; se presentan con mayor frecuencia en los tumores del aparato genitourinario femenino.[
54 ] De manera más específica, la mayoría de los casos de rabdomiosarcoma embrionario de cuello uterino,[66 ,67 ,68 ] que son más comunes en adolescentes y adultas jóvenes,[69 ,70 ] tienen variantes de DICER1. Por el contrario, las variantes de DICER1 son infrecuentes en pacientes con sitio primario en la vagina, una afección que se presenta sobre todo en niñas menores de 2 o 3 años.[67 ,69 ] Las variantes de DICER1 también son comunes en el rabdomiosarcoma embrionario que surge en el cuerpo uterino, pero esta presentación clínica se observa sobre todo en adultas.[67 ,71 ] El rabdomiosarcoma de cuello uterino suele mostrar características histológicas de sarcoma botrioide y muchos casos exhiben áreas de diferenciación cartilaginosa, un elemento que también se observa en otros tipos de tumores con variantes de DICER1.[69 ,70 ,72 ] Los casos de rabdomiosarcoma embrionario con variantes de DICER1 exhiben un patrón de metilación del DNA que es diferente al de otros tipos de rabdomiosarcoma embrionario, lo que apoya las características biológicas distintivas de este tipo de cáncer.[68 ] El diagnóstico de rabdomiosarcoma de cuello uterino es una indicación para realizar pruebas genéticas del síndrome de DICER1.[67 ,73 ] - De 8 niños atendidos de manera consecutiva por rabdomiosarcoma con variantes germinales de TP53, todos exhibían características morfológicas de anaplasia. En otros 7 niños con rabdomiosarcoma anaplásico y estado desconocido de variante germinal de TP53, se encontró que 3 de ellos presentaban variantes germinales de TP53 con un efecto funcional de interés. La mediana de edad en el momento del diagnóstico de los 11 niños con estado conocido de variante germinal de TP53 fue de 40 meses (intervalo, 19–67 meses).[
- Rabdomiosarcoma positivo para fusiones (tipo histológico alveolar): entre el 70 % y el 80 % de los tumores alveolares se caracterizan por presentar translocaciones entre el gen FOXO1 en el cromosoma 13 y el gen PAX3 en el cromosoma 2 (t(2;13)(q35;q14)) o el gen PAX7 en el cromosoma 1 (t(1;13)(p36;q14)).[
55 ,74 ,75 ] Otras fusiones infrecuentes son PAX3::NCOA1 y PAX3::INO80D.[58 ] Las translocaciones que afectan al gen PAX3 se encuentran en cerca del 60 % de los casos de rabdomiosarcoma alveolar, mientras que el gen PAX7 está afectado en cerca del 20 % de los casos.[74 ] Los pacientes que exhiben un tipo histológico alveolar de variante sólida presentan una incidencia más baja de fusiones génicas PAX::FOXO1 que quienes exhiben un tipo histológico alveolar clásico.[76 ] El tipo histológico alveolar asociado con el gen PAX7, en pacientes con enfermedad metastásica o sin esta, se presenta a una edad más temprana y a veces se asocia con tasas de SSC más prolongadas que las de los pacientes con reordenamientos del gen PAX3.[77 ,78 ,79 ,80 ,81 ,82 ] Los pacientes que exhiben el tipo histológico alveolar y el gen PAX3 son de más edad y presentan una incidencia más alta de tumor invasivo (T2). En cerca del 20 % de los casos que exhiben un tipo histológico alveolar no se detecta una translocación del gen PAX.[50 ,76 ] Estos pacientes exhiben un comportamiento clínico, un perfil de alteraciones génicas y un perfil transcripcional que se corresponde con el de pacientes que tienen rabdomiosarcoma embrionario, por lo que ahora se clasifican juntos dentro del grupo de rabdomiosarcoma negativo para fusiones.[49 ,50 ,51 ,52 ,53 ]Para el diagnóstico de rabdomiosarcoma alveolar es posible detectar el reordenamiento del gen FOXO1 mediante una prueba de hibridación fluorescente in situ o de reacción en cadena de la polimerasa con retrotranscripción, ambas con buena sensibilidad y especificidad.[
83 ]Además de los reordenamientos de FOXO1, los tumores alveolares se caracterizan por una carga mutacional más baja que los tumores negativos para fusiones; exhiben menos genes con mutaciones recurrentes.[
58 ,59 ] Las alteraciones que se observan con mayor frecuencia en los tumores positivos para fusiones son la amplificación focal de CDK4 (13 %) o MYCN (10 %), y un pequeño grupo de pacientes tienen variantes recurrentes de otros genes (por ejemplo, BCOR, 6 %; NF1, 4 %; TP53, 4 % y PIK3CA, 2 %).[54 ] Las variantes de TP53 en el rabdomiosarcoma alveolar confieren un riesgo alto de fracaso terapéutico.[54 ] - Tipo histológico de células fusiformes o esclerosante: en la clasificación de la OMS de los tumores de tejidos blandos y hueso, se propuso el rabdomiosarcoma de células fusiformes o esclerosante como una entidad separada.[
84 ] La categoría de rabdomiosarcoma de células fusiformes o esclerosante abarca varias entidades con características moleculares y clínicas diferenciadoras que se describen más adelante.Rabdomiosarcoma de células fusiformes congénito o infantil: en varios informes se han descrito casos de rabdomiosarcoma de células fusiformes congénito o infantil con fusiones génicas que afectan a VGLL2 y NCOA2 (por ejemplo, VGLL2::CITED2, TEAD1::NCOA2, VGLL2::NCOA2, SRF::NCOA2).[
85 ,86 ]- En un estudio se informó que 10 de 11 pacientes con rabdomiosarcoma de células fusiformes congénito o infantil exhibían genes de fusión recurrentes. La mayoría de estos pacientes presentaron tumores primarios de tronco y ninguno tenía tumores paratesticulares. Se observaron reordenamientos nuevos de VGLL2 en 7 pacientes (63 %), entre ellos, la fusión VGLL2::CITED2 en 4 pacientes, y la fusión VGLL2::NCOA2 en 2 pacientes.[
85 ] Se encontraron diferentes fusiones del gen NCOA2 en 3 pacientes (27 %), incluso TEAD1::NCOA2 en 2 pacientes y SRF::NCOA2 en 1 paciente. En este informe, todos los pacientes con rabdomiosarcoma de células fusiformes congénito o infantil positivo para fusiones sometidos a seguimiento a largo plazo estaban vivos y en buenas condiciones; ningún paciente presentó metástasis a distancia.[85 ] - Si bien la mayoría de los estudios de rabdomiosarcoma de células fusiformes congénito o infantil demuestran desenlaces favorables, se notificó que 4 pacientes presentaron enfermedad metastásica y 2 pacientes murieron. La progresión de la enfermedad se presentó al cabo de una mediana de 3,5 años desde el diagnóstico (intervalo, 1–8 años).[
87 ] Los 4 pacientes presentaron tumores irresecables y se trataron con quimioterapia. Sin embargo, en la mayoría de los casos notificados en la bibliografía médica se logró la extirpación quirúrgica. En el momento de progresión de la enfermedad, un paciente tenía un tumor con variante de TP53, y otro paciente tenía un tumor con deleción homocigota de CDKN2A y CDKN2B. - En un estudio de 40 pacientes con rabdomiosarcoma de células fusiformes congénito o infantil (definido como diagnóstico a una edad ≤12 meses) se encontró que casi todos los pacientes mostraba enfermedad localizada (n = 39) y que la mitad de quienes se hicieron pruebas moleculares (13 de 26) presentaba reordenamientos de NCOA2 o VGLL2.[
88 ] Debido a que las pruebas se limitaron a NCOA2 y VGLL2, es posible que un análisis genómico más amplio identificara una proporción más alta de pacientes con fusiones génicas relevantes. La tasa de SSC a 5 años de los 13 pacientes con fusiones de VGLL2 o NCOA2 fue del 90 % (IC 95 %, ±19 %), y la tasa de supervivencia general (SG) fue del 100 % (IC 95 %, ±9 %). - Se necesitan más estudios para precisar la prevalencia y la importancia pronóstica de los reordenamientos en VGLL2, NCOA2, y en otros genes relevantes en niños de corta edad con rabdomiosarcoma de células fusiformes congénito o infantil.
Rabdomiosarcoma de células fusiformes o esclerosante con variante de MYOD1: en una gran proporción de niños mayores y adultos con rabdomiosarcoma de células fusiformes o esclerosante, se ha encontrado una variante específica de MYOD1 (p.L122R).[
85 ,89 ,90 ,91 ] En una cohorte combinada de pacientes con rabdomiosarcoma del COG y el Reino Unido (n = 641), se encontraron variantes de MYOD1 en el 3 % (17 de 515) de todos los casos de rabdomiosarcoma negativo para fusiones, pero en ninguno de los casos positivos para fusiones. La edad de presentación clínica de los pacientes con variantes de MYOD1 fue de 10,8 años.[54 ] La mayoría de los casos de esta cohorte presentaban características de células fusiformes o esclerosantes, pero también se observaron células empaquetadas de manera densa que imitan la conformación densa del rabdomiosarcoma embrionario. La mayoría de los sitios primarios de los casos de esta cohorte (15 de 17, 88 %) estaban en la cabeza y el cuello o en la región parameníngea. Las variantes activadoras de PIK3CA se observan en cerca de la mitad de los casos que exhiben variantes de MYOD1.[54 ,92 ] La presencia de la variante de MYOD1 se asocia con aumento marcado del riesgo de fracaso terapéutico local y a distancia.[54 ,85 ,89 ,90 ]Rabdomiosarcoma de células fusiformes intraóseo: el rabdomiosarcoma intraóseo primario es un tipo de presentación muy infrecuente del rabdomiosarcoma. La mayoría de los casos exhiben reordenamientos génicos que afectan TFCP2 y forman fusiones con FUS o EWSR1.[
93 ,94 ,95 ,96 ,97 ] El rabdomiosarcoma con fusiones génicas FUS::TFCP2 o EWSR1::TFCP2 suele presentarse en adultos jóvenes, aunque se han notificado casos en niños mayores y adolescentes.[93 ,96 ,97 ] La localización del tumor más común es en los huesos craneofaciales, y es frecuente que en el análisis inmunohistoquímico se obtenga positividad para ALK y citoqueratinas. El perfil genómico complejo es otra característica de esta entidad, que en la mayoría de los casos exhibe deleción del gen supresor de tumores CDKN2A.[96 ] El rabdomiosarcoma de células fusiformes intraóseo con un gen de fusión FUS::TFCP2 o EWSR1::TFCP2 muestra un comportamiento clínico de gran malignidad. En un estudio la mediana de SG fue de solo 8 meses.[96 ] - En un estudio se informó que 10 de 11 pacientes con rabdomiosarcoma de células fusiformes congénito o infantil exhibían genes de fusión recurrentes. La mayoría de estos pacientes presentaron tumores primarios de tronco y ninguno tenía tumores paratesticulares. Se observaron reordenamientos nuevos de VGLL2 en 7 pacientes (63 %), entre ellos, la fusión VGLL2::CITED2 en 4 pacientes, y la fusión VGLL2::NCOA2 en 2 pacientes.[
En el ensayo del Instituto Nacional del Cáncer y el Children's Oncology Group conocido como NCI-COG Pediatric MATCH se sometieron a secuenciación tumoral los rabdomiosarcomas recidivantes y resistentes al tratamiento de pacientes pediátricos (n = 105) y adultos jóvenes (n = 15). Se observaron alteraciones genómicas aplicables en 53 de 120 tumores (44,2 %), y los pacientes con estas alteraciones fueron aptos para recibir tratamiento en los grupos del estudio MATCH.[
Para obtener información sobre el tratamiento del rabdomiosarcoma infantil, consultar
Referencias:
- Chen X, Bahrami A, Pappo A, et al.: Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 7 (1): 104-12, 2014.
- Perry JA, Kiezun A, Tonzi P, et al.: Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A 111 (51): E5564-73, 2014.
- Marinoff AE, Spurr LF, Fong C, et al.: Clinical Targeted Next-Generation Panel Sequencing Reveals MYC Amplification Is a Poor Prognostic Factor in Osteosarcoma. JCO Precis Oncol 7: e2200334, 2023.
- Suehara Y, Alex D, Bowman A, et al.: Clinical Genomic Sequencing of Pediatric and Adult Osteosarcoma Reveals Distinct Molecular Subsets with Potentially Targetable Alterations. Clin Cancer Res 25 (21): 6346-6356, 2019.
- Nacev BA, Sanchez-Vega F, Smith SA, et al.: Clinical sequencing of soft tissue and bone sarcomas delineates diverse genomic landscapes and potential therapeutic targets. Nat Commun 13 (1): 3405, 2022.
- De Noon S, Ijaz J, Coorens TH, et al.: MYC amplifications are common events in childhood osteosarcoma. J Pathol Clin Res 7 (5): 425-431, 2021.
- Parsons DW, Janeway KA, Patton DR, et al.: Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol 40 (20): 2224-2234, 2022.
- Sanders RP, Drissi R, Billups CA, et al.: Telomerase expression predicts unfavorable outcome in osteosarcoma. J Clin Oncol 22 (18): 3790-7, 2004.
- de Nonneville A, Salas S, Bertucci F, et al.: TOP3A amplification and ATRX inactivation are mutually exclusive events in pediatric osteosarcomas using ALT. EMBO Mol Med 14 (10): e15859, 2022.
- Mirabello L, Zhu B, Koster R, et al.: Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients With Osteosarcoma. JAMA Oncol 6 (5): 724-734, 2020.
- Ognjanovic S, Olivier M, Bergemann TL, et al.: Sarcomas in TP53 germline mutation carriers: a review of the IARC TP53 database. Cancer 118 (5): 1387-96, 2012.
- Mirabello L, Yeager M, Mai PL, et al.: Germline TP53 variants and susceptibility to osteosarcoma. J Natl Cancer Inst 107 (7): , 2015.
- Toguchida J, Yamaguchi T, Dayton SH, et al.: Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. N Engl J Med 326 (20): 1301-8, 1992.
- McIntyre JF, Smith-Sorensen B, Friend SH, et al.: Germline mutations of the p53 tumor suppressor gene in children with osteosarcoma. J Clin Oncol 12 (5): 925-30, 1994.
- Maciaszek JL, Oak N, Chen W, et al.: Enrichment of heterozygous germline RECQL4 loss-of-function variants in pediatric osteosarcoma. Cold Spring Harb Mol Case Stud 5 (5): , 2019.
- Kansara M, Thomas DM: Molecular pathogenesis of osteosarcoma. DNA Cell Biol 26 (1): 1-18, 2007.
- German J: Bloom's syndrome. XX. The first 100 cancers. Cancer Genet Cytogenet 93 (1): 100-6, 1997.
- Lipton JM, Federman N, Khabbaze Y, et al.: Osteogenic sarcoma associated with Diamond-Blackfan anemia: a report from the Diamond-Blackfan Anemia Registry. J Pediatr Hematol Oncol 23 (1): 39-44, 2001.
- Idol RA, Robledo S, Du HY, et al.: Cells depleted for RPS19, a protein associated with Diamond Blackfan Anemia, show defects in 18S ribosomal RNA synthesis and small ribosomal subunit production. Blood Cells Mol Dis 39 (1): 35-43, 2007 Jul-Aug.
- Li FP, Fraumeni JF, Mulvihill JJ, et al.: A cancer family syndrome in twenty-four kindreds. Cancer Res 48 (18): 5358-62, 1988.
- Grimer RJ, Cannon SR, Taminiau AM, et al.: Osteosarcoma over the age of forty. Eur J Cancer 39 (2): 157-63, 2003.
- Wong FL, Boice JD, Abramson DH, et al.: Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA 278 (15): 1262-7, 1997.
- Wang LL, Gannavarapu A, Kozinetz CA, et al.: Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst 95 (9): 669-74, 2003.
- Hicks MJ, Roth JR, Kozinetz CA, et al.: Clinicopathologic features of osteosarcoma in patients with Rothmund-Thomson syndrome. J Clin Oncol 25 (4): 370-5, 2007.
- Goto M, Miller RW, Ishikawa Y, et al.: Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 5 (4): 239-46, 1996.
- WHO Classification of Tumours Editorial Board: WHO Classification of Tumours. Volume 3: Soft Tissue and Bone Tumours. 5th ed., IARC Press, 2020.
- Schwartz JC, Cech TR, Parker RR: Biochemical Properties and Biological Functions of FET Proteins. Annu Rev Biochem 84: 355-79, 2015.
- Tirode F, Surdez D, Ma X, et al.: Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 4 (11): 1342-53, 2014.
- Crompton BD, Stewart C, Taylor-Weiner A, et al.: The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 4 (11): 1326-41, 2014.
- Brohl AS, Solomon DA, Chang W, et al.: The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 10 (7): e1004475, 2014.
- Hattinger CM, Rumpler S, Strehl S, et al.: Prognostic impact of deletions at 1p36 and numerical aberrations in Ewing tumors. Genes Chromosomes Cancer 24 (3): 243-54, 1999.
- Sankar S, Lessnick SL: Promiscuous partnerships in Ewing's sarcoma. Cancer Genet 204 (7): 351-65, 2011.
- de Alava E, Kawai A, Healey JH, et al.: EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing's sarcoma. J Clin Oncol 16 (4): 1248-55, 1998.
- van Doorninck JA, Ji L, Schaub B, et al.: Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 28 (12): 1989-94, 2010.
- Le Deley MC, Delattre O, Schaefer KL, et al.: Impact of EWS-ETS fusion type on disease progression in Ewing's sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol 28 (12): 1982-8, 2010.
- Roberts P, Burchill SA, Brownhill S, et al.: Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing's sarcoma family of tumors: a study by the United Kingdom Cancer Cytogenetics and the Children's Cancer and Leukaemia Group. Genes Chromosomes Cancer 47 (3): 207-20, 2008.
- Hattinger CM, Rumpler S, Ambros IM, et al.: Demonstration of the translocation der(16)t(1;16)(q12;q11.2) in interphase nuclei of Ewing tumors. Genes Chromosomes Cancer 17 (3): 141-50, 1996.
- Hattinger CM, Pötschger U, Tarkkanen M, et al.: Prognostic impact of chromosomal aberrations in Ewing tumours. Br J Cancer 86 (11): 1763-9, 2002.
- Mackintosh C, Ordóñez JL, García-Domínguez DJ, et al.: 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene 31 (10): 1287-98, 2012.
- Mrózek K, Bloomfield CD: Der(16)t(1;16) is a secondary chromosome aberration in at least eighteen different types of human cancer. Genes Chromosomes Cancer 23 (1): 78-80, 1998.
- Mugneret F, Lizard S, Aurias A, et al.: Chromosomes in Ewing's sarcoma. II. Nonrandom additional changes, trisomy 8 and der(16)t(1;16). Cancer Genet Cytogenet 32 (2): 239-45, 1988.
- Shulman DS, Chen S, Hall D, et al.: Adverse prognostic impact of the loss of STAG2 protein expression in patients with newly diagnosed localised Ewing sarcoma: A report from the Children's Oncology Group. Br J Cancer 127 (12): 2220-2226, 2022.
- Ogura K, Elkrief A, Bowman AS, et al.: Prospective Clinical Genomic Profiling of Ewing Sarcoma: ERF and FGFR1 Mutations as Recurrent Secondary Alterations of Potential Biologic and Therapeutic Relevance. JCO Precis Oncol 6: e2200048, 2022.
- Rock A, Uche A, Yoon J, et al.: Bioinformatic Analysis of Recurrent Genomic Alterations and Corresponding Pathway Alterations in Ewing Sarcoma. J Pers Med 13 (10): , 2023.
- Monforte-Muñoz H, Lopez-Terrada D, Affendie H, et al.: Documentation of EWS gene rearrangements by fluorescence in-situ hybridization (FISH) in frozen sections of Ewing's sarcoma-peripheral primitive neuroectodermal tumor. Am J Surg Pathol 23 (3): 309-15, 1999.
- Chen S, Deniz K, Sung YS, et al.: Ewing sarcoma with ERG gene rearrangements: A molecular study focusing on the prevalence of FUS-ERG and common pitfalls in detecting EWSR1-ERG fusions by FISH. Genes Chromosomes Cancer 55 (4): 340-9, 2016.
- Parham DM, Ellison DA: Rhabdomyosarcomas in adults and children: an update. Arch Pathol Lab Med 130 (10): 1454-65, 2006.
- Newton WA, Gehan EA, Webber BL, et al.: Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 76 (6): 1073-85, 1995.
- Davicioni E, Anderson JR, Buckley JD, et al.: Gene expression profiling for survival prediction in pediatric rhabdomyosarcomas: a report from the children's oncology group. J Clin Oncol 28 (7): 1240-6, 2010.
- Davicioni E, Anderson MJ, Finckenstein FG, et al.: Molecular classification of rhabdomyosarcoma--genotypic and phenotypic determinants of diagnosis: a report from the Children's Oncology Group. Am J Pathol 174 (2): 550-64, 2009.
- Williamson D, Missiaglia E, de Reyniès A, et al.: Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol 28 (13): 2151-8, 2010.
- Davicioni E, Finckenstein FG, Shahbazian V, et al.: Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res 66 (14): 6936-46, 2006.
- Skapek SX, Anderson J, Barr FG, et al.: PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: a children's oncology group report. Pediatr Blood Cancer 60 (9): 1411-7, 2013.
- Shern JF, Selfe J, Izquierdo E, et al.: Genomic Classification and Clinical Outcome in Rhabdomyosarcoma: A Report From an International Consortium. J Clin Oncol 39 (26): 2859-2871, 2021.
- Merlino G, Helman LJ: Rhabdomyosarcoma--working out the pathways. Oncogene 18 (38): 5340-8, 1999.
- Koufos A, Hansen MF, Copeland NG, et al.: Loss of heterozygosity in three embryonal tumours suggests a common pathogenetic mechanism. Nature 316 (6026): 330-4, 1985 Jul 25-31.
- Scrable H, Witte D, Shimada H, et al.: Molecular differential pathology of rhabdomyosarcoma. Genes Chromosomes Cancer 1 (1): 23-35, 1989.
- Shern JF, Chen L, Chmielecki J, et al.: Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov 4 (2): 216-31, 2014.
- Chen X, Stewart E, Shelat AA, et al.: Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell 24 (6): 710-24, 2013.
- Li H, Sisoudiya SD, Martin-Giacalone BA, et al.: Germline Cancer Predisposition Variants in Pediatric Rhabdomyosarcoma: A Report From the Children's Oncology Group. J Natl Cancer Inst 113 (7): 875-883, 2021.
- Ooms AH, Gadd S, Gerhard DS, et al.: Significance of TP53 Mutation in Wilms Tumors with Diffuse Anaplasia: A Report from the Children's Oncology Group. Clin Cancer Res 22 (22): 5582-5591, 2016.
- Shenoy A, Alvarez E, Chi YY, et al.: The prognostic significance of anaplasia in childhood rhabdomyosarcoma: A report from the Children's Oncology Group. Eur J Cancer 143: 127-133, 2021.
- Haduong JH, Heske CM, Allen-Rhoades W, et al.: An update on rhabdomyosarcoma risk stratification and the rationale for current and future Children's Oncology Group clinical trials. Pediatr Blood Cancer 69 (4): e29511, 2022.
- Hettmer S, Archer NM, Somers GR, et al.: Anaplastic rhabdomyosarcoma in TP53 germline mutation carriers. Cancer 120 (7): 1068-75, 2014.
- Pondrom M, Bougeard G, Karanian M, et al.: Rhabdomyosarcoma associated with germline TP53 alteration in children and adolescents: The French experience. Pediatr Blood Cancer 67 (9): e28486, 2020.
- de Kock L, Yoon JY, Apellaniz-Ruiz M, et al.: Significantly greater prevalence of DICER1 alterations in uterine embryonal rhabdomyosarcoma compared to adenosarcoma. Mod Pathol 33 (6): 1207-1219, 2020.
- Apellaniz-Ruiz M, McCluggage WG, Foulkes WD: DICER1-associated embryonal rhabdomyosarcoma and adenosarcoma of the gynecologic tract: Pathology, molecular genetics, and indications for molecular testing. Genes Chromosomes Cancer 60 (3): 217-233, 2021.
- Kommoss FKF, Stichel D, Mora J, et al.: Clinicopathologic and molecular analysis of embryonal rhabdomyosarcoma of the genitourinary tract: evidence for a distinct DICER1-associated subgroup. Mod Pathol 34 (8): 1558-1569, 2021.
- Dehner LP, Jarzembowski JA, Hill DA: Embryonal rhabdomyosarcoma of the uterine cervix: a report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol 25 (4): 602-14, 2012.
- Daya DA, Scully RE: Sarcoma botryoides of the uterine cervix in young women: a clinicopathological study of 13 cases. Gynecol Oncol 29 (3): 290-304, 1988.
- Bennett JA, Ordulu Z, Young RH, et al.: Embryonal rhabdomyosarcoma of the uterine corpus: a clinicopathological and molecular analysis of 21 cases highlighting a frequent association with DICER1 mutations. Mod Pathol 34 (9): 1750-1762, 2021.
- McCluggage WG, Foulkes WD: DICER1-associated sarcomas: towards a unified nomenclature. Mod Pathol 34 (6): 1226-1228, 2021.
- Schultz KAP, Williams GM, Kamihara J, et al.: DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin Cancer Res 24 (10): 2251-2261, 2018.
- Barr FG, Smith LM, Lynch JC, et al.: Examination of gene fusion status in archival samples of alveolar rhabdomyosarcoma entered on the Intergroup Rhabdomyosarcoma Study-III trial: a report from the Children's Oncology Group. J Mol Diagn 8 (2): 202-8, 2006.
- Dumont SN, Lazar AJ, Bridge JA, et al.: PAX3/7-FOXO1 fusion status in older rhabdomyosarcoma patient population by fluorescent in situ hybridization. J Cancer Res Clin Oncol 138 (2): 213-20, 2012.
- Parham DM, Qualman SJ, Teot L, et al.: Correlation between histology and PAX/FKHR fusion status in alveolar rhabdomyosarcoma: a report from the Children's Oncology Group. Am J Surg Pathol 31 (6): 895-901, 2007.
- Sorensen PH, Lynch JC, Qualman SJ, et al.: PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children's oncology group. J Clin Oncol 20 (11): 2672-9, 2002.
- Krsková L, Mrhalová M, Sumerauer D, et al.: Rhabdomyosarcoma: molecular diagnostics of patients classified by morphology and immunohistochemistry with emphasis on bone marrow and purged peripheral blood progenitor cells involvement. Virchows Arch 448 (4): 449-58, 2006.
- Kelly KM, Womer RB, Sorensen PH, et al.: Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J Clin Oncol 15 (5): 1831-6, 1997.
- Barr FG, Qualman SJ, Macris MH, et al.: Genetic heterogeneity in the alveolar rhabdomyosarcoma subset without typical gene fusions. Cancer Res 62 (16): 4704-10, 2002.
- Missiaglia E, Williamson D, Chisholm J, et al.: PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol 30 (14): 1670-7, 2012.
- Duan F, Smith LM, Gustafson DM, et al.: Genomic and clinical analysis of fusion gene amplification in rhabdomyosarcoma: a report from the Children's Oncology Group. Genes Chromosomes Cancer 51 (7): 662-74, 2012.
- Thway K, Wang J, Wren D, et al.: The comparative utility of fluorescence in situ hybridization and reverse transcription-polymerase chain reaction in the diagnosis of alveolar rhabdomyosarcoma. Virchows Arch 467 (2): 217-24, 2015.
- Nascimento AF, Barr FG: Spindle cell/sclerosing rhabdomyosarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn P, et al., eds.: WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. IARC Press, 2013, pp 134-5.
- Alaggio R, Zhang L, Sung YS, et al.: A Molecular Study of Pediatric Spindle and Sclerosing Rhabdomyosarcoma: Identification of Novel and Recurrent VGLL2-related Fusions in Infantile Cases. Am J Surg Pathol 40 (2): 224-35, 2016.
- Mosquera JM, Sboner A, Zhang L, et al.: Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer 52 (6): 538-50, 2013.
- Cyrta J, Gauthier A, Karanian M, et al.: Infantile Rhabdomyosarcomas With VGLL2 Rearrangement Are Not Always an Indolent Disease: A Study of 4 Aggressive Cases With Clinical, Pathologic, Molecular, and Radiologic Findings. Am J Surg Pathol 45 (6): 854-867, 2021.
- Whittle S, Venkatramani R, Schönstein A, et al.: Congenital spindle cell rhabdomyosarcoma: An international cooperative analysis. Eur J Cancer 168: 56-64, 2022.
- Kohsaka S, Shukla N, Ameur N, et al.: A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet 46 (6): 595-600, 2014.
- Agaram NP, Chen CL, Zhang L, et al.: Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for a common pathogenesis. Genes Chromosomes Cancer 53 (9): 779-87, 2014.
- Szuhai K, de Jong D, Leung WY, et al.: Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J Pathol 232 (3): 300-7, 2014.
- Agaram NP, LaQuaglia MP, Alaggio R, et al.: MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: an aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod Pathol 32 (1): 27-36, 2019.
- Watson S, Perrin V, Guillemot D, et al.: Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol 245 (1): 29-40, 2018.
- Dashti NK, Wehrs RN, Thomas BC, et al.: Spindle cell rhabdomyosarcoma of bone with FUS-TFCP2 fusion: confirmation of a very recently described rhabdomyosarcoma subtype. Histopathology 73 (3): 514-520, 2018.
- Agaram NP, Zhang L, Sung YS, et al.: Expanding the Spectrum of Intraosseous Rhabdomyosarcoma: Correlation Between 2 Distinct Gene Fusions and Phenotype. Am J Surg Pathol 43 (5): 695-702, 2019.
- Le Loarer F, Cleven AHG, Bouvier C, et al.: A subset of epithelioid and spindle cell rhabdomyosarcomas is associated with TFCP2 fusions and common ALK upregulation. Mod Pathol 33 (3): 404-419, 2020.
- Xu B, Suurmeijer AJH, Agaram NP, et al.: Head and neck rhabdomyosarcoma with TFCP2 fusions and ALK overexpression: a clinicopathological and molecular analysis of 11 cases. Histopathology 79 (3): 347-357, 2021.
Histiocitosis de células de Langerhans
Características genómicas de la histiocitosis de células de Langerhans
Variantes deBRAF,NRASyARAF
El fundamento genómico de la histiocitosis de células de Langerhans (HCL) avanzó gracias a un informe de 2010 sobre la detección de una variante activadora del oncogén BRAF (V600E) en 35 de 61 casos (57 %).[
En una serie de 100 pacientes, se estudió la variante BRAF V600E en sangre y médula ósea y se obtuvo un resultado positivo para la variante BRAF V600E en el 65 % de los pacientes cuando se usó un método de reacción en cadena de la polimerasa cuantitativa sensible.[
En pacientes de riesgo alto, el hallazgo en la médula ósea de células madre positivas para CD34 que además exhibían la variante confirmó el origen de la HCL en las células dendríticas mieloides. En los pacientes con enfermedad de riesgo bajo, la variante se identificó en células dendríticas mieloides más maduras, lo que indica que el estadio del desarrollo celular en el momento en que aparece la variante somática es determinante para definir la extensión de la enfermedad en la HCL.
En un principio se notificó que la HCL pulmonar en adultos no era clonal en cerca del 75 % de los casos,[
En un estudio de 117 pacientes con HCL, 83 pacientes adultos con HCL pulmonar se sometieron a análisis molecular. Cerca del 90 % de estos pacientes tenía variantes en la vía MAPK.[
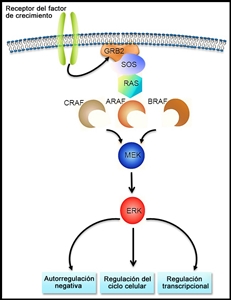
La vía de señalización RAS-MAPK (consultar la Figura 8) transmite señales desde un receptor de la superficie celular (por ejemplo, un factor de crecimiento) por la vía RAS (mediante una de las proteínas RAF [A, B o C]) de manera que se induce la fosforilación de MEK y después, de la cinasa regulada por señales extracelulares (ERK), lo que conduce a una señalización nuclear que afecta el ciclo celular y la regulación de la transcripción. La variante BRAF V600E produce fosforilación ininterrumpida y, por lo tanto, activación de MEK y ERK en ausencia de una señal externa. La activación de ERK ocurre por fosforilación, y esta ERK fosforilada se detecta en prácticamente todas las lesiones de HCL.[
En un modelo murino de HCL, se observó que la presencia de la variante BRAF V600E inhibe la migración de las células dendríticas mediada por un receptor de quimiocina (CCR7), lo que las obliga a acumularse en la lesión de la HCL.[
Otro modelo murino con la variante BRAF V600E bajo el control de los promotores de los genes Scl o Map17 añadió conocimientos adicionales sobre las características biológicas de la HCL neurodegenerativa.[
En resumen, la HCL ahora se considera una neoplasia mieloide impulsada principalmente por variantes activadoras de la vía MAPK. Entre el 50 % y el 60 % de las variantes activadoras son variantes BRAF V600E, que abundan en pacientes con HCL multisistémica con compromiso de órganos de riesgo y en pacientes que tienen enfermedad neurodegenerativa.[
Otras alteraciones en la vía RAS-MAPK
Debido a que es posible detectar la activación de la vía RAS-MAPK (elevación de ERK fosforilada) en todos los casos de HCL, incluso en aquellos sin variantes de BRAF, se consideró la posibilidad de que hubiera alteraciones genómicas en otros componentes de la vía. Se han identificado las siguientes alteraciones genómicas:
- Variantes de MAP2K1. La secuenciación del exoma completo en biopsias de tejido de HCL que exhiben alteraciones en el gen BRAF versus un gen BRAF natural reveló que 7 de 21 muestras con BRAF natural tenían variantes de MAP2K1; mientras que ninguna de las muestras con alteraciones en el gen BRAF tenía variantes de MAP2K1.[
13 ] Las variantes de MAP2K1 (que codifica MEK1) fueron activadoras, como se indica por la inducción de la fosforilación de ERK.[13 ]En otro estudio se detectaron variantes de MAP2K1 solo en 11 de 22 casos con gen BRAF natural.[
18 ] En un estudio se observó que la variante de MAP2K1 y otras variantes relacionadas con la HCL en niños y adultos son mutuamente excluyentes de la presencia de las variantes de BRAF.[19 ] Los autores encontraron diversas variantes en otras vías (por ejemplo, JNK, RAS-ERK y JAK-STAT) en niños y adultos con la variante BRAF V600E o variantes de MAP2K1. En otro estudio se evaluaron alteraciones de cinasas y variantes mieloides en 73 adultos con HCL.[20 ] Estos investigadores notificaron una mediana de 2 variantes por adulto, a diferencia de los niños que por lo general solo tienen 1 variante. Se encontró la variante BRAF V600E en el 31 % de los pacientes con HCL, una inserción y deleción (indel) en BRAF en el 29 % de ellos, y una variante de MAP2K1 en el 19 % de ellos. En el 89 % de los adultos con HCL se encontraron alteraciones en otras proteínas cinasas y vías relacionadas. Las variantes de MAP2K1 se presentaron en ausencia de variantes de BRAF. - Deleciones en el marco de lectura de BRAF y fusiones génicas FAM73A::BRAF. Se han encontrado deleciones en el marco de lectura del gen BRAF y fusiones génicas en el marco de lectura de FAM73A::BRAF en el grupo de casos que no tienen variantes BRAF V600E ni de MAP2K1.[
12 ]
En resumen, los estudios corroboran la activación generalizada de ERK en la HCL, La activación de ERK en la mayoría de los casos se explica a partir de las alteraciones de los genes BRAF y MAP2K1.[
Repercusiones clínicas
Las repercusiones clínicas de los hallazgos genómicos descritos son las siguientes:
- La HCL se agrupa con otros tumores del ámbito pediátrico que tienen variantes activadoras de BRAF, entre ellos, algunas afecciones benignas (por ejemplo, nevos benignos) [
21 ] y neoplasias malignas de grado bajo (por ejemplo, astrocitoma pilocítico).[22 ,23 ] Por lo general, todas estas afecciones tienen una evolución poco activa y en algunos casos se resuelven de manera espontánea. Esta evolución clínica característica quizás sea una manifestación de senescencia causada por oncogenes.[21 ,24 ] - En algunos estudios pediátricos, las variantes BRAF V600E se relacionaron con enfermedad multisistémica más grave, fracaso del tratamiento, aumento de las reactivaciones y mayor riesgo de neurodegeneración (ver más adelante).[
25 ] Estas correlaciones clínicas se investigaron hace poco con el fin de detectar otras variantes diferentes a la variante BRAF V600E. De manera similar a la cohorte con variantes BRAF V600E, todos los pacientes con HCL multisistémica y compromiso de órganos de riesgo exhibían variantes detectables en las células mononucleares de sangre periférica. De 7 pacientes con HCL multisistémica sin compromiso de órganos de riesgo, 4 tenían variantes detectables. Ningún paciente con enfermedad monosistémica presentó variantes detectables en las células mononucleares de sangre periférica. Los autores concluyeron que otras variantes en la vía MAPK se relacionan con el estado de riesgo, de manera similar a como lo hacen las variantes BRAF V600E.[17 ]Las variantes BRAF V600E son la diana terapéutica de los inhibidores de BRAF (por ejemplo, vemurafenib y dabrafenib) y de la combinación de inhibidores de BRAF con inhibidores de MEK (por ejemplo, dabrafenib y trametinib, o vemurafenib y cobimetinib). Estos fármacos y sus combinaciones están aprobadas para su uso en adultos con melanoma. En adultos, el tratamiento del melanoma con combinaciones de un inhibidor de BRAF y un inhibidor de MEK mostró una mejora significativa de los desenlaces de supervivencia sin progresión en comparación con el tratamiento con un inhibidor de BRAF solo.[
26 ,27 ]En varios informes de casos y en 2 series de casos también se observó eficacia de los inhibidores de BRAF para el tratamiento de la HCL en niños.[
28 ,29 ,30 ,31 ,32 ,33 ] No obstante, es difícil evaluar la función a largo plazo de este tratamiento porque la mayoría de los pacientes recaen al suspender los inhibidores. Para obtener más información, consultar las seccionesTratamiento de la histiocitosis de células de Langerhans multisistémica recidivante, resistente al tratamiento o progresiva de riesgo alto yTerapias dirigidas para el tratamiento de la enfermedad monosistémica y multisistémica . - Se encontraron células circulantes con variantes BRAF V600E en el 59 % de los pacientes con HCL y enfermedad neurodegenerativa en comparación con el 15 % de los pacientes con HCL sin enfermedad neurodegenerativa La detección de células circulantes que exhibe la variante tiene una sensibilidad de 0,59 y una especificidad de 0,86 para el desarrollo de enfermedad neurodegenerativa. Incluso después del tratamiento, algunos pacientes con HCL y enfermedad neurodegenerativa tienen células circulantes con variantes BRAF V600E.[
34 ] - Es posible que nuevas investigaciones permitan usar la detección de la variante BRAF V600E (o quizás las variantes de MAP2K1) en células circulantes o en el DNA extracelular circulante como una herramienta diagnóstica útil para diferenciar la enfermedad de riesgo alto de la enfermedad de riesgo bajo.[
2 ] Además, para los pacientes que tienen una variante somática, la persistencia de las células circulantes con la variante puede ser útil como marcador de enfermedad residual.[2 ]
Para obtener información sobre el tratamiento de la HCL infantil, consultar
Referencias:
- Badalian-Very G, Vergilio JA, Degar BA, et al.: Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 116 (11): 1919-23, 2010.
- Berres ML, Lim KP, Peters T, et al.: BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 211 (4): 669-83, 2014.
- Satoh T, Smith A, Sarde A, et al.: B-RAF mutant alleles associated with Langerhans cell histiocytosis, a granulomatous pediatric disease. PLoS One 7 (4): e33891, 2012.
- Sahm F, Capper D, Preusser M, et al.: BRAFV600E mutant protein is expressed in cells of variable maturation in Langerhans cell histiocytosis. Blood 120 (12): e28-34, 2012.
- Héritier S, Hélias-Rodzewicz Z, Chakraborty R, et al.: New somatic BRAF splicing mutation in Langerhans cell histiocytosis. Mol Cancer 16 (1): 115, 2017.
- Nelson DS, Quispel W, Badalian-Very G, et al.: Somatic activating ARAF mutations in Langerhans cell histiocytosis. Blood 123 (20): 3152-5, 2014.
- Héritier S, Hélias-Rodzewicz Z, Lapillonne H, et al.: Circulating cell-free BRAF(V600E) as a biomarker in children with Langerhans cell histiocytosis. Br J Haematol 178 (3): 457-467, 2017.
- Dacic S, Trusky C, Bakker A, et al.: Genotypic analysis of pulmonary Langerhans cell histiocytosis. Hum Pathol 34 (12): 1345-9, 2003.
- Roden AC, Hu X, Kip S, et al.: BRAF V600E expression in Langerhans cell histiocytosis: clinical and immunohistochemical study on 25 pulmonary and 54 extrapulmonary cases. Am J Surg Pathol 38 (4): 548-51, 2014.
- Mourah S, How-Kit A, Meignin V, et al.: Recurrent NRAS mutations in pulmonary Langerhans cell histiocytosis. Eur Respir J 47 (6): 1785-96, 2016.
- Jouenne F, Chevret S, Bugnet E, et al.: Genetic landscape of adult Langerhans cell histiocytosis with lung involvement. Eur Respir J 55 (2): , 2020.
- Chakraborty R, Burke TM, Hampton OA, et al.: Alternative genetic mechanisms of BRAF activation in Langerhans cell histiocytosis. Blood 128 (21): 2533-2537, 2016.
- Chakraborty R, Hampton OA, Shen X, et al.: Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood 124 (19): 3007-15, 2014.
- Hogstad B, Berres ML, Chakraborty R, et al.: RAF/MEK/extracellular signal-related kinase pathway suppresses dendritic cell migration and traps dendritic cells in Langerhans cell histiocytosis lesions. J Exp Med 215 (1): 319-336, 2018.
- Bigenwald C, Le Berichel J, Wilk CM, et al.: BRAFV600E-induced senescence drives Langerhans cell histiocytosis pathophysiology. Nat Med 27 (5): 851-861, 2021.
- Wilk CM, Cathomas F, Török O, et al.: Circulating senescent myeloid cells infiltrate the brain and cause neurodegeneration in histiocytic disorders. Immunity 56 (12): 2790-2802.e6, 2023.
- Milne P, Abhyankar H, Scull B, et al.: Cellular distribution of mutations and association with disease risk in Langerhans cell histiocytosis without BRAFV600E. Blood Adv 6 (16): 4901-4904, 2022.
- Brown NA, Furtado LV, Betz BL, et al.: High prevalence of somatic MAP2K1 mutations in BRAF V600E-negative Langerhans cell histiocytosis. Blood 124 (10): 1655-8, 2014.
- Durham BH, Lopez Rodrigo E, Picarsic J, et al.: Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med 25 (12): 1839-1842, 2019.
- Chen J, Zhao AL, Duan MH, et al.: Diverse kinase alterations and myeloid-associated mutations in adult histiocytosis. Leukemia 36 (2): 573-576, 2022.
- Michaloglou C, Vredeveld LC, Soengas MS, et al.: BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436 (7051): 720-4, 2005.
- Jones DT, Kocialkowski S, Liu L, et al.: Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68 (21): 8673-7, 2008.
- Pfister S, Janzarik WG, Remke M, et al.: BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 118 (5): 1739-49, 2008.
- Jacob K, Quang-Khuong DA, Jones DT, et al.: Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res 17 (14): 4650-60, 2011.
- Héritier S, Emile JF, Barkaoui MA, et al.: BRAF Mutation Correlates With High-Risk Langerhans Cell Histiocytosis and Increased Resistance to First-Line Therapy. J Clin Oncol 34 (25): 3023-30, 2016.
- Larkin J, Ascierto PA, Dréno B, et al.: Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 371 (20): 1867-76, 2014.
- Long GV, Stroyakovskiy D, Gogas H, et al.: Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 386 (9992): 444-51, 2015.
- Eckstein OS, Visser J, Rodriguez-Galindo C, et al.: Clinical responses and persistent BRAF V600E+ blood cells in children with LCH treated with MAPK pathway inhibition. Blood 133 (15): 1691-1694, 2019.
- Donadieu J, Larabi IA, Tardieu M, et al.: Vemurafenib for Refractory Multisystem Langerhans Cell Histiocytosis in Children: An International Observational Study. J Clin Oncol 37 (31): 2857-2865, 2019.
- Kolenová A, Schwentner R, Jug G, et al.: Targeted inhibition of the MAPK pathway: emerging salvage option for progressive life-threatening multisystem LCH. Blood Adv 1 (6): 352-356, 2017.
- Lee LH, Gasilina A, Roychoudhury J, et al.: Real-time genomic profiling of histiocytoses identifies early-kinase domain BRAF alterations while improving treatment outcomes. JCI Insight 2 (3): e89473, 2017.
- Héritier S, Jehanne M, Leverger G, et al.: Vemurafenib Use in an Infant for High-Risk Langerhans Cell Histiocytosis. JAMA Oncol 1 (6): 836-8, 2015.
- Váradi Z, Bánusz R, Csomor J, et al.: Effective BRAF inhibitor vemurafenib therapy in a 2-year-old patient with sequentially diagnosed Langerhans cell histiocytosis and Erdheim-Chester disease. Onco Targets Ther 10: 521-526, 2017.
- McClain KL, Picarsic J, Chakraborty R, et al.: CNS Langerhans cell histiocytosis: Common hematopoietic origin for LCH-associated neurodegeneration and mass lesions. Cancer 124 (12): 2607-2620, 2018.
Neuroblastoma
Características moleculares del neuroblastoma
Los niños con neuroblastoma se agrupan en subconjuntos con diferentes riesgos previstos de recaída de acuerdo con factores clínicos y marcadores biológicos presentes en el momento del diagnóstico.
- Pacientes con neuroblastoma de riesgo bajo o intermedio. Los pacientes clasificados con riesgo bajo o riesgo intermedio tienen un pronóstico favorable con tasas de supervivencia superiores al 95 %. El neuroblastoma de riesgo bajo o intermedio por lo general se presenta en niños menores de 18 meses. A menudo, estos tumores tienen ganancia de cromosomas enteros y son hiperdiploides cuando se los examina con citometría de flujo.[
1 ,2 ] - Pacientes con neuroblastoma de riesgo alto. El pronóstico de los pacientes con neuroblastoma de riesgo alto es más reservado con una tasa de supervivencia a largo plazo menor al 50 %. El neuroblastoma de riesgo alto suele presentarse en niños mayores de 18 meses, a menudo, con metástasis en hueso y médula ósea. En estos tumores, es habitual que se detecten anormalidades cromosómicas segmentarias (ganancias o pérdidas) o amplificación del gen MYCN. Según se observa en la medición con citometría de flujo, son casi diploides o casi tetraploides.[
1 ,2 ,3 ,4 ,5 ,6 ,7 ] Los tumores de riesgo alto pocas veces exhiben variantes exónicas, pero la mayoría de los tumores de riesgo alto carecen de dichas variantes génicas. Para obtener más información, consultar la sección Variantes exónicas en el neuroblastoma.
Las características genómicas clave del neuroblastoma de riesgo alto se describen a continuación:
- Anomalías cromosómicas segmentarias.
- Amplificaciones del gen MYCN.
- Activación de FOXR2.
- Tasas bajas de variantes exónicas; la alteración recurrente más común son las variantes activadoras de ALK.
- Alteraciones genómicas que promueven el mantenimiento de los telómeros.
Anomalías cromosómicas segmentarias
Las anomalías cromosómicas segmentarias, que a menudo se encuentran en 1p, 2p, 1q, 3p, 11q, 14q y 17p, se detectan mejor mediante hibridación genómica comparativa. Estas anomalías se encuentran en la mayoría de los tumores de neuroblastoma en estadio 4 o de riesgo alto.[
- Edad avanzada en el momento del diagnóstico.
- Estadio avanzado de la enfermedad.
- Riesgo más alto de recaída.
- Desenlace más precario.
En un análisis del neuroblastoma localizado, resecable y sin amplificación de MYCN, se evaluaron casos de dos estudios europeos consecutivos y una cohorte de Norte América (que incluyó casos en estadios 1, 2A y 2B de acuerdo al INSS) para detectar alteraciones cromosómicas segmentarias (a saber, ganancia de 1q, 2p y 17q, además de pérdida de 1p, 3p, 4p y 11q). En el estudio se descubrió que las características genómicas del tumor tenían una repercusión pronóstica diferente según la edad del paciente (<18 meses o >18 meses). Los pacientes se trataron solo con intervención quirúrgica, con independencia de la presencia de residuo tumoral.[
- La presencia de anomalías cromosómicas segmentarias, en particular la pérdida de 11q, redujo significativamente la supervivencia en los pacientes mayores de 18 meses con neuroblastoma en estadio 2, pero no en la cohorte de pacientes menores de 18 meses.
- La pérdida del cromosoma 1p es un factor de riesgo para la recaída, pero no para la disminución de la supervivencia general (SG) en los pacientes menores de 18 meses. La tasa de supervivencia sin complicaciones (SSC) a 5 años fue del 62 % en los pacientes con pérdida de 1p y del 87 % en aquellos sin pérdida de 1p (P = 0,019). La tasa de SG a 5 años fue del 92 % en los pacientes con pérdida de 1p y del 97 % en los pacientes sin pérdida de 1p.
- Las anomalías cromosómicas segmentarias (en particular, la pérdida de 11q) representan factores de riesgo para la reducción de la SSC y la SG en pacientes mayores de 18 meses. En los pacientes menores de 18 meses, solo las anomalías cromosómicas segmentarias condujeron a recaída y muerte; la pérdida de 11q fue el marcador más fuerte (pérdida de 11q: tasa de SSC a 5 años, 48 %; sin pérdida de 11q: tasa de SSC a 5 años, 85 %; P = 0,033; pérdida de 11q: tasa de SG a 5 años, 46 %; sin pérdida de 11q: tasa de SG a 5 años, 92 %; P = 0,038).
En un estudio de niños mayores de 12 meses con neuroblastomas primarios irresecables sin metástasis, se encontraron anomalías cromosómicas segmentarias en la mayoría de los pacientes. Los niños mayores fueron más propensos a presentarlas y tener más de estas anomalías por célula tumoral. En los niños de 12 a 18 meses, la presencia de anomalías cromosómicas segmentarias tuvo un efecto significativo en la SSC, pero no en la SG. Sin embargo, en los niños mayores de 18 meses, hubo una diferencia significativa en la SG entre aquellos con anomalías cromosómicas segmentarias (67 %) y aquellos sin anomalías cromosómicas segmentarias (100 %), con independencia de las características histológicas del tumor.[
Las anomalías cromosómicas segmentarias también permiten pronosticar la recidiva en lactantes con neuroblastoma metastásico o neuroblastoma localizado irresecable sin amplificación del gen MYCN.[
En un análisis de pacientes de riesgo intermedio en un estudio del Children's Oncology Group (COG), la pérdida de 11q, pero sin pérdida de 1p, se asoció a una SSC inferior; sin embargo, no afectó la SG (con pérdida de 11q y sin pérdida de 11q: tasas de SSC a 3 años, 68 % y 85 %, respectivamente P = 0,022; tasas de SG a 3 años, 88 % y 94 %, respectivamente; P = 0,09).[
En un análisis multivariante de 407 pacientes de 4 ensayos consecutivos del COG sobre alto riesgo, se observó que la pérdida de heterocigosidad de 11q es un factor de predicción significativo para la enfermedad progresiva, y que la ausencia de heterocigosidad de 11q se asociaba con tasas más altas de respuesta completa al final de la inducción y respuesta parcial al final de la inducción.[
En un estudio de colaboración internacional de 556 pacientes con neuroblastoma de riesgo alto se identificaron dos tipos de anomalías segmentarias en el número de copias que se relacionaron con desenlaces muy desfavorables. Se encontraron pérdidas distales de 6q en el 6 % de los pacientes y se relacionaron con una tasa de supervivencia a 10 años de solo el 3,4 %. Además de la amplificación de MYCN, se detectaron amplificaciones de regiones fuera del locus de MYCN en el 18 % de los pacientes y se relacionaron con una tasa de supervivencia a 10 años del 5,8 %.[
Amplificación del genMYCN
La amplificación de MYCN se detecta en el 16 % al 25 % de los tumores de neuroblastoma.[
De acuerdo a la mayoría de los análisis multivariantes de regresión de factores pronósticos, en todos los estadios de la enfermedad, la amplificación del gen MYCN permite predecir claramente un pronóstico más precario, tanto para el tiempo hasta la progresión del tumor como para la SG.[
En la cohorte de tumores localizados con amplificación de MYCN, los pacientes con tumores hiperdiploides tienen mejores desenlaces que aquellos con tumores diploides.[
Las características clínicas y biopatológicas más desfavorables se relacionan, en cierta medida, con la amplificación de MYCN. En un análisis multivariante de regresión logística en 7102 pacientes del estudio del Internacional Neuroblastoma Risk Group (INRG), las anomalías cromosómicas segmentarias agrupadas y las ganancias de 17q fueron las únicas características de pronóstico precario, aún cuando no estaban asociadas a la amplificación de MYCN. No obstante, las anomalías cromosómicas segmentarias en 11q, otra característica de pronóstico precario, son casi mutuamente excluyentes con la amplificación de MYCN.[
En una cohorte de 6223 pacientes de la base de datos del INRG con estado de MYCN conocido, el cociente de riesgos instantáneos (CRI) para la SG relacionada con la amplificación de MYCN fue de 6,3 (intervalo de confianza [IC] 95 %, 5,7–7,0; P < 0,001). El mayor efecto pronóstico adverso de la amplificación de MYCN en la SG se observó en los pacientes más jóvenes (<18 meses: CRI, 19,6; ≥18 meses: CRI, 3,0). Los pacientes cuyo desenlace se vio más afectado por el estado de MYCN fueron los que presentaban características favorables, como una edad menor de 18 meses, índice de mitosis cariorrexis alto y ferritina baja.[
La amplificación intratumoral heterogénea de MYCN (hetMNA) se refiere a la coexistencia de células tumorales con amplificación de MYCN (agrupadas o dispersas) y células tumorales sin amplificación de MYCN. La HetMNA se ha notificado de manera infrecuente. Es posible que se presente dentro del tumor, así como entre el tumor y la metástasis; al mismo tiempo o de forma transitoria durante la evolución de la enfermedad. El grupo de biología de la International Society of Paediatric Oncology Europe Neuroblastoma (SIOPEN) investigó la importancia pronóstica de este subtipo de neuroblastoma. Se analizó el tejido tumoral de 99 pacientes en quienes se identificó hetMNA y que recibieron el diagnóstico entre 1991 y 2015, para aclarar la importancia pronóstica de los clones con amplificación de MYCN en casos de neuroblastoma sin amplificación de MYCN. Los pacientes menores de 18 meses presentaron un desenlace más favorable en todos los estadios en comparación con los pacientes de más edad. Se estableció una correlación significativa de los antecedentes genómicos con la frecuencia de la recaída y la SG. No se presentaron recaídas en los casos que solo presentaban anomalías cromosómicas numéricas. Este estudio indica que los tumores con hetMNA se evalúan teniendo en cuenta las características genómicas del tumor y el cuadro clínico, incluso la edad del paciente y el estadio de la enfermedad. Se necesitan más estudios en pacientes menores de 18 meses que exhiban enfermedad localizada con hetMNA.[
Activación deFOXR2
La expresión del gen FOXR2 se observa en alrededor del 8 % de los casos de neuroblastoma. La expresión del gen FOXR2 por lo general es nula después del nacimiento, a excepción de los tejidos reproductivos en varones.[
El neuroblastoma con activación de FOXR2 se observa en tasas similares en los casos con riesgo alto y sin riesgo alto.[
Variantes exónicas en el neuroblastoma (incluso variantes y amplificaciones deALK)
En comparación con los cánceres en adultos, el neuroblastoma infantil exhibe un número bajo de variantes por genoma que afectan la secuencia de proteínas (10–20 por genoma).[
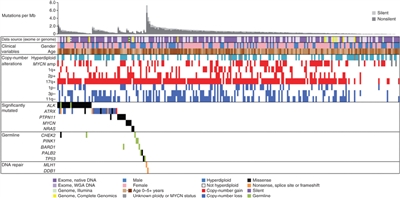
El gen ALK proporciona las instrucciones para la elaboración de un receptor tirosina–cinasa de superficie celular que se expresa en concentraciones importantes solo en los encéfalos embrionarios y neonatales en desarrollo. La variante exónica de ALK es la alteración que se encuentra con más frecuencia en el neuroblastoma. Las variantes germinales de ALK se identificaron como la causa principal del neuroblastoma hereditario. Se encontró que las variantes exónicas activadoras de ALK adquiridas de forma somática también son oncoiniciadoras del neuroblastoma.[
En 2 estudios de cohortes numerosas se examinaron los marcadores clínicos y la importancia pronóstica de las alteraciones en ALK. En un estudio del COG se analizó el estado de ALK en 1596 muestras de diagnóstico de neuroblastoma en todos los grupos de riesgo.[
- Las variantes de ALK que afectan el dominio de tirosina–cinasa se presentaron sobre todo en 3 puntos de gran actividad (posiciones F1174, R1275 y F1245); el 10 % al 15 % de las variantes ocurrieron en otras posiciones del dominio de cinasa.
- En la cohorte del COG la frecuencia de las variantes de ALK fue del 10 % en el grupo de neuroblastoma de riesgo alto, del 8 % en el grupo del neuroblastoma de riesgo intermedio y del 6 % en el grupo de neuroblastoma de riesgo bajo.
- En la población de riesgo alto de la SIOPEN, las variantes de ALK se dividieron en clonales (frecuencia de la variante del alelo [VAF] >20 %) y subclonales (VAF 0,1–20 %). Se observaron variantes clonales de ALK en un 10 % de los casos, y variantes subclonales en un 3,9 % de los pacientes. El 13,9 % de los casos presentaban variantes de ALK.
- Se encontraron tasas más altas de variantes de ALK en los pacientes con tumores con amplificación de MYCN en comparación con aquellos sin amplificación de MYCN: el 10,9 % versus el 7,2 %, respectivamente, en la cohorte del COG y el 14 % versus el 6,5 %, respectivamente, en la cohorte de la SIOPEN (para las variantes clonales de ALK).
- En los pacientes con neuroblastoma de riesgo alto, la amplificación de ALK se observó en cerca del 4 % de los casos en las cohortes del COG y de la SIOPEN. La amplificación de ALK se presentó casi de manera exclusiva en casos que también tenían amplificación de MYCN.
- Las alteraciones en ALK se relacionaron con un pronóstico inferior en los pacientes con neuroblastoma de riesgo alto en el estudio del COG y en el de la SIOPEN:
- En la cohorte de la SIOPEN, se observó una diferencia estadísticamente significativa en la SG entre los casos con amplificación de ALK (ALKa) o variante clonal de ALK (ALKm) versus ALKm subclonal o sin alteraciones en ALK (tasa de SG a 5 años: ALKa, 26 % [IC 95 %, 10–47 %]; ALKm clonal, 33 % [IC 95 %, 21–44 %]; ALKm subclonal, 48 % [IC 95 %, 26–67 %]; y sin alteración, 51 % [IC 95 %, 46–55 %], respectivamente P = 0,001). En un modelo multivariante, la amplificación en ALK (CRI, 2,38; P = 0,004) y la variante clonal de ALK (CRI, 1,77; P = 0,001) fueron factores independientes de predicción de desenlace precario.
- En la población de neuroblastoma de riesgo alto del COG se observaron pronósticos inferiores, similares a los observados en la cohorte de la SIOPEN, en los casos con variantes de ALK y amplificaciones en ALK.
En un estudio donde se compararon los datos genómicos de neuroblastomas de diagnóstico primario que se originaron en la glándula suprarrenal (n = 646) con los de neuroblastomas originados en los ganglios simpáticos torácicos (n = 118), el 16 % de los tumores torácicos albergaban variantes de ALK.[
Los inhibidores de molécula pequeña de la cinasa de ALK, como el loratinib (añadido a la terapia convencional), se están probando en pacientes de neuroblastoma con variante recurrente de ALK (NCT03107988) y en pacientes con neuroblastoma de riesgo alto recién diagnosticado con activación de ALK (COG ANBL1531).[
Evolución genómica de las variantes exónicas
Hay pocos datos sobre la evolución genómica de las variantes exónicas desde el diagnóstico hasta la recaída del neuroblastoma. Se aplicó la secuenciación del genoma completo a 23 muestras emparejadas de tumores de neuroblastoma obtenidas en el momento del diagnóstico y de la recaída con el fin de definir las alteraciones genéticas somáticas relacionadas con la recaída;[
- En el primer estudio, se encontró una mayor incidencia de variantes de los genes relacionados con la señalización de RAS-MAPK en tumores de recaída en comparación con los tumores del mismo paciente en el momento del diagnóstico: 15 de 23 muestras de recaída contenían variantes somáticas de los genes que participan en esta vía; además, cada variante fue compatible con la activación de la vía.[
33 ]Asimismo, en 3 muestras de recaída se encontraron alteraciones estructurales que comprometían genes de la vía MAPK compatibles con activación de la vía, es decir que se detectaron anomalías en esta vía en 18 de 23 (78 %) muestras de recaída. Se encontraron anomalías en ALK (n = 10) y en NF1 (n = 2), además de una anomalía en cada uno de los siguientes genes: NRAS, KRAS, HRAS, BRAF, PTPN11 y FGFR1. Incluso con una secuenciación masiva, 7 de las 18 alteraciones no fueron detectables en el tumor primario, esto subraya la evolución de las variantes que presumiblemente conducen a la recaída y la importancia de las evaluaciones genómicas de los tejidos obtenidos en el momento de la recaída.
- En el segundo estudio, no se observaron variantes de ALK en el momento del diagnóstico ni en la recaída, pero se detectaron variantes de un solo nucleótido recurrentes específicas de la recaída en 11 genes, incluso el presunto gen supresor de tumores del neuroblastoma CHD5 localizado en el cromosoma 1p36.[
34 ] - En un tercer estudio retrospectivo de secuenciación de variantes, se usaron datos de la Foundation Medicine para comparar las muestras de tumores de pacientes con neuroblastoma recién diagnosticado con las muestras de tumores de pacientes de neuroblastoma resistente al tratamiento y en recaída. En el estudio se encontró un porcentaje más alto de variantes susceptibles de tratamiento dirigido con fármacos actuales en el grupo de pacientes en recaída y resistente al tratamiento.[
35 ] - En un cuarto estudio se evaluó la frecuencia de las alteraciones en ALK en el momento del diagnóstico y de la recaída. Se presentaron tasas significativamente más altas de variantes de ALK en la recaída que en el diagnóstico (17,7 % en la recaída vs. 10,5 % en el diagnóstico). La tasa de amplificaciones en ALK no fue diferente entre el momento del diagnóstico y de la recaída.[
36 ]
Dada la naturaleza metastásica generalizada del neuroblastoma de riesgo alto y en recaída, el uso de métodos para medir el DNA tumoral circulante (ctDNA) quizás revele otras alteraciones genómicas que no se encuentran con las biopsias tumorales convencionales. Además, estos abordajes han demostrado la capacidad para detectar variantes de resistencia en pacientes con neuroblastoma tratados con inhibidores de ALK.[
En un estudio de secuenciación masiva, 276 muestras de neuroblastoma (en todos los estadios y de pacientes de todas las edades en el momento del diagnóstico) se evaluaron mediante secuenciación de solo 2 puntos de gran actividad de variantes de ALK, lo que reveló un 4,8 % de variantes clonales y otro 5 % de variantes subclonales. Este hallazgo indica que las variantes subclonales del gen ALK son comunes.[
Alteraciones genómicas que promueven el mantenimiento de los telómeros
El alargamiento de los telómeros, los extremos de los cromosomas, promueve la supervivencia celular. Por otra parte, los telómeros se acortan con cada replicación celular, lo que resulta finalmente en la incapacidad de la célula para replicarse. Los pacientes cuyos tumores carecen de mecanismos de mantenimiento de telómeros tienen un pronóstico excelente, mientras que los que albergan mecanismos de mantenimiento de telómeros tienen un pronóstico bastante más precario.[
- Los reordenamientos cromosómicos que comprometen una región cromosómica en 5p15.33 próxima al gen TERT, que codifica la unidad catalítica de la telomerasa, se presentan en el 20 % al 25 % de los casos de neuroblastoma de riesgo alto y son mutuamente excluyentes con las amplificaciones de MYCN y la activación del alargamiento alternativo de los telómeros.[
24 ,25 ,41 ] Los reordenamientos inducen el aumento regulado de la transcripción de TERT al yuxtaponer la secuencia codificante de TERT con fuertes elementos potenciadores. Los niños cuyos tumores presentan reordenamientos en TERT tienen un pronóstico precario, comparable con el pronóstico de los niños con tumores con amplificación de MYCN.[41 ] Es posible usar la secuenciación de última generación o la hibridación fluorescente in situ (FISH) para identificar estas alteraciones. En un estudio se identificaron reordenamientos en TERT mediante FISH en el 6 % de todos los pacientes con tumores neuroblásticos, con independencia del grupo de riesgo, y en el 12,4 % de los pacientes con tumores neuroblásticos de riesgo alto.[42 ] - Otro mecanismo que promueve la sobreexpresión de TERT es la amplificación de MYCN,[
43 ] que se relaciona con cerca del 40 % al 50 % de los casos de neuroblastoma de riesgo alto. - El alargamiento alternativo de los telómeros es un mecanismo adicional de mantenimiento de telómeros que utilizan los tumores de neuroblastoma. La activación del alargamiento alternativo de los telómeros se presenta en alrededor del 20 % al 25 % de los casos de riesgo alto recién diagnosticados, en comparación con cerca del 5 % al 12 % de los casos de riesgo bajo y riesgo intermedio.[
41 ,44 ,45 ] En comparación con los casos recién diagnosticados, la proporción de casos de neuroblastoma con tumores positivos para el alargamiento alternativo de los telómeros fue mayor en una cohorte de pacientes en recaída (10 % vs. 48 %, respectivamente). Es posible que este hallazgo refleje una evolución relativamente indolente de los tumores con activación del alargamiento alternativo de los telómeros tras la recaída, en comparación con la evolución clínica de otros tumores después de la recaída. Con el tiempo, la proporción de pacientes con neuroblastomas para el alargamiento alternativo de los telómeros en recaída (entre los pacientes con neuroblastoma) parece mayor que la de pacientes con otro tipo de tumor que recayeron (entre los pacientes diagnosticados con ese tumor).[44 ] Los casos de neuroblastoma con activación del alargamiento alternativo de los telómeros tienen expresión baja de TERT y se pueden identificar mediante prueba inmunohistoquímica por la presencia de cuerpos nucleares de leucemia promielocítica asociados a alargamiento alternativo de los telómeros. Estos casos también se identifican con una prueba de círculo C o con FISH al usar una sonda telomérica para observar los puntos brillantes de los telómeros.[44 ,45 ] Alrededor del 55 % al 60 % de los casos positivos para el alargamiento alternativo de los telómeros se caracterizan por presentar variantes deletéreas de ATRX.[26 ,44 ,45 ] Los casos que no tienen variantes de ATRX a menudo presentan expresión baja de la proteína ATRX.[44 ]Los tumores positivos para el alargamiento alternativo de los telómeros en las poblaciones infantiles casi nunca se presentan antes de los 18 meses de edad y surgen casi de forma exclusiva en niños más grandes (mediana de edad en el momento del diagnóstico, cerca de los 8 años).[
41 ,44 ] La proporción de casos de neuroblastoma con variantes de ATRX aumenta con la edad en las poblaciones de adolescentes y adultos jóvenes.[26 ]El pronóstico para la SSC de los pacientes de riesgo alto con activación del alargamiento alternativo de los telómeros es tan precario como el de los pacientes con amplificación de MYCN,[
41 ,44 ] sin embargo, la SG es superior en los pacientes con activación del alargamiento alternativo de los telómeros. La SG más favorable es consecuencia de una evolución un poco más prolongada de la enfermedad después de la recaída, pero con una supervivencia a largo plazo (10 a 15 años) tan baja como la de otros pacientes con neuroblastoma de riesgo alto.[41 ,44 ] En un informe, la SSC y la SG en los pacientes de riesgo intermedio y riesgo bajo con activación del alargamiento alternativo de los telómeros fueron similares a las observadas en pacientes positivos para el alargamiento alternativo de los telómeros con enfermedad de riesgo alto.[44 ]
Otros factores biológicos relacionados con el pronóstico
Expresión de MYC y MYCN
Con la inmunotinción de las proteínas MYC y MYCN en un subconjunto restringido de 357 tumores de neuroblastoma indiferenciado o poco diferenciado, se demostró que la expresión elevada de las proteínas MYC o MYCN es un factor pronóstico importante.[
- Los pacientes con tumores con características histológicas favorables sin expresión alta de MYCN o MYC tuvieron una supervivencia favorable (SSC a 3 años, 89,7 ± 5,5 %; SG a 3 años, 97 ± 3,2 %).
- Los pacientes con tumores con características histológicas indiferenciadas o poco diferenciadas sin expresión de MYCN o MYC tuvieron una tasa de SSC a 3 años del 63,1 % (± 13,6 %) y una tasa de SG a 3 años del 83,5 % (± 9,4 %).
- Las tasas de SSC a 3 años en pacientes con amplificación de MYCN, expresión alta de MYCN y expresión alta de MYC fueron del 48,1 % (± 11,5 %), del 46,2 % (± 12 %) y del 43,4 % (± 23,1 %), respectivamente. Las tasas de SG fueron del 65,8 % (± 11,1 %), del 63,2 % (± 12,1 %), y del 63,5 % (± 19,2 %), respectivamente.
- Además, cuando se hizo un análisis multivariante de la expresión alta de las proteínas MYC y MYCN y otros factores pronósticos, como la amplificación génica de MYC/MYCN, se concluyó que la expresión alta de las proteínas MYC y MYCN es independiente de otros marcadores pronósticos.
Cinasas receptoras de neurotrofina
La expresión de cinasas receptoras de neurotrofina y sus ligandos varía entre los tumores de riesgo alto y de riesgo bajo. El receptor TrkA se encuentra en tumores de riesgo bajo y se postula que la ausencia de su ligando NGF produce la remisión espontánea del tumor. En contraste, el receptor TrkB se encuentra en los tumores de riesgo alto que también expresan su ligando, BDNF, que promueve la proliferación y la supervivencia celular del neuroblastoma.[
Para obtener información sobre el tratamiento del neuroblastoma, consultar
Referencias:
- Cohn SL, Pearson AD, London WB, et al.: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27 (2): 289-97, 2009.
- Schleiermacher G, Mosseri V, London WB, et al.: Segmental chromosomal alterations have prognostic impact in neuroblastoma: a report from the INRG project. Br J Cancer 107 (8): 1418-22, 2012.
- Janoueix-Lerosey I, Schleiermacher G, Michels E, et al.: Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol 27 (7): 1026-33, 2009.
- Schleiermacher G, Michon J, Ribeiro A, et al.: Segmental chromosomal alterations lead to a higher risk of relapse in infants with MYCN-non-amplified localised unresectable/disseminated neuroblastoma (a SIOPEN collaborative study). Br J Cancer 105 (12): 1940-8, 2011.
- Carén H, Kryh H, Nethander M, et al.: High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc Natl Acad Sci U S A 107 (9): 4323-8, 2010.
- Schleiermacher G, Janoueix-Lerosey I, Ribeiro A, et al.: Accumulation of segmental alterations determines progression in neuroblastoma. J Clin Oncol 28 (19): 3122-30, 2010.
- Defferrari R, Mazzocco K, Ambros IM, et al.: Influence of segmental chromosome abnormalities on survival in children over the age of 12 months with unresectable localised peripheral neuroblastic tumours without MYCN amplification. Br J Cancer 112 (2): 290-5, 2015.
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.
- Ambros IM, Tonini GP, Pötschger U, et al.: Age Dependency of the Prognostic Impact of Tumor Genomics in Localized Resectable MYCN-Nonamplified Neuroblastomas. Report From the SIOPEN Biology Group on the LNESG Trials and a COG Validation Group. J Clin Oncol 38 (31): 3685-3697, 2020.
- Pinto N, Naranjo A, Ding X, et al.: Impact of Genomic and Clinical Factors on Outcome of Children ≥18 Months of Age with Stage 3 Neuroblastoma with Unfavorable Histology and without MYCN Amplification: A Children's Oncology Group (COG) Report. Clin Cancer Res 29 (8): 1546-1556, 2023.
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.
- Pinto N, Naranjo A, Hibbitts E, et al.: Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children's Oncology Group (COG). Eur J Cancer 112: 66-79, 2019.
- Depuydt P, Boeva V, Hocking TD, et al.: Genomic Amplifications and Distal 6q Loss: Novel Markers for Poor Survival in High-risk Neuroblastoma Patients. J Natl Cancer Inst 110 (10): 1084-1093, 2018.
- Ambros PF, Ambros IM, Brodeur GM, et al.: International consensus for neuroblastoma molecular diagnostics: report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br J Cancer 100 (9): 1471-82, 2009.
- Kreissman SG, Seeger RC, Matthay KK, et al.: Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): a randomised phase 3 trial. Lancet Oncol 14 (10): 999-1008, 2013.
- Bagatell R, Beck-Popovic M, London WB, et al.: Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol 27 (3): 365-70, 2009.
- Plantaz D, Vandesompele J, Van Roy N, et al.: Comparative genomic hybridization (CGH) analysis of stage 4 neuroblastoma reveals high frequency of 11q deletion in tumors lacking MYCN amplification. Int J Cancer 91 (5): 680-6, 2001.
- Maris JM, Hogarty MD, Bagatell R, et al.: Neuroblastoma. Lancet 369 (9579): 2106-20, 2007.
- Campbell K, Shyr D, Bagatell R, et al.: Comprehensive evaluation of context dependence of the prognostic impact of MYCN amplification in neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 66 (8): e27819, 2019.
- Berbegall AP, Bogen D, Pötschger U, et al.: Heterogeneous MYCN amplification in neuroblastoma: a SIOP Europe Neuroblastoma Study. Br J Cancer 118 (11): 1502-1512, 2018.
- Schmitt-Hoffner F, van Rijn S, Toprak UH, et al.: FOXR2 Stabilizes MYCN Protein and Identifies Non-MYCN-Amplified Neuroblastoma Patients With Unfavorable Outcome. J Clin Oncol 39 (29): 3217-3228, 2021.
- Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.
- Pugh TJ, Morozova O, Attiyeh EF, et al.: The genetic landscape of high-risk neuroblastoma. Nat Genet 45 (3): 279-84, 2013.
- Peifer M, Hertwig F, Roels F, et al.: Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526 (7575): 700-4, 2015.
- Valentijn LJ, Koster J, Zwijnenburg DA, et al.: TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet 47 (12): 1411-4, 2015.
- Cheung NK, Zhang J, Lu C, et al.: Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 307 (10): 1062-71, 2012.
- Molenaar JJ, Koster J, Zwijnenburg DA, et al.: Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483 (7391): 589-93, 2012.
- Sausen M, Leary RJ, Jones S, et al.: Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet 45 (1): 12-7, 2013.
- Bresler SC, Weiser DA, Huwe PJ, et al.: ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 26 (5): 682-94, 2014.
- Janoueix-Lerosey I, Lequin D, Brugières L, et al.: Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455 (7215): 967-70, 2008.
- Bellini A, Pötschger U, Bernard V, et al.: Frequency and Prognostic Impact of ALK Amplifications and Mutations in the European Neuroblastoma Study Group (SIOPEN) High-Risk Neuroblastoma Trial (HR-NBL1). J Clin Oncol 39 (30): 3377-3390, 2021.
- Oldridge DA, Truong B, Russ D, et al.: Differences in Genomic Profiles and Outcomes Between Thoracic and Adrenal Neuroblastoma. J Natl Cancer Inst 111 (11): 1192-1201, 2019.
- Eleveld TF, Oldridge DA, Bernard V, et al.: Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 47 (8): 864-71, 2015.
- Schramm A, Köster J, Assenov Y, et al.: Mutational dynamics between primary and relapse neuroblastomas. Nat Genet 47 (8): 872-7, 2015.
- Padovan-Merhar OM, Raman P, Ostrovnaya I, et al.: Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PLoS Genet 12 (12): e1006501, 2016.
- Rosswog C, Fassunke J, Ernst A, et al.: Genomic ALK alterations in primary and relapsed neuroblastoma. Br J Cancer 128 (8): 1559-1571, 2023.
- Bosse KR, Giudice AM, Lane MV, et al.: Serial Profiling of Circulating Tumor DNA Identifies Dynamic Evolution of Clinically Actionable Genomic Alterations in High-Risk Neuroblastoma. Cancer Discov 12 (12): 2800-2819, 2022.
- Berko ER, Witek GM, Matkar S, et al.: Circulating tumor DNA reveals mechanisms of lorlatinib resistance in patients with relapsed/refractory ALK-driven neuroblastoma. Nat Commun 14 (1): 2601, 2023.
- Bellini A, Bernard V, Leroy Q, et al.: Deep Sequencing Reveals Occurrence of Subclonal ALK Mutations in Neuroblastoma at Diagnosis. Clin Cancer Res 21 (21): 4913-21, 2015.
- Ackermann S, Cartolano M, Hero B, et al.: A mechanistic classification of clinical phenotypes in neuroblastoma. Science 362 (6419): 1165-1170, 2018.
- Roderwieser A, Sand F, Walter E, et al.: Telomerase is a prognostic marker of poor outcome and a therapeutic target in neuroblastoma. JCO Precis Oncol 3: 1-20, 2019.
- Yu Y, Zhang M, Yao X, et al.: Translational practice of fluorescence in situ hybridisation to identify neuroblastic tumours with TERT rearrangements. J Pathol Clin Res 9 (6): 475-487, 2023.
- Mac SM, D'Cunha CA, Farnham PJ: Direct recruitment of N-myc to target gene promoters. Mol Carcinog 29 (2): 76-86, 2000.
- Hartlieb SA, Sieverling L, Nadler-Holly M, et al.: Alternative lengthening of telomeres in childhood neuroblastoma from genome to proteome. Nat Commun 12 (1): 1269, 2021.
- Koneru B, Lopez G, Farooqi A, et al.: Telomere Maintenance Mechanisms Define Clinical Outcome in High-Risk Neuroblastoma. Cancer Res 80 (12): 2663-2675, 2020.
- Wang LL, Teshiba R, Ikegaki N, et al.: Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: a Children's Oncology Group study. Br J Cancer 113 (1): 57-63, 2015.
- Maris JM, Matthay KK: Molecular biology of neuroblastoma. J Clin Oncol 17 (7): 2264-79, 1999.
Tumores renales
Tumor de Wilms
Características moleculares del tumor de Wilms
El tumor de Wilms a veces surge durante la embriogénesis en el contexto de un riñón que por lo demás es normal desde el punto de vista genómico; en otras ocasiones, surge de lesiones precursoras genéticas somáticas no germinales presentes dentro de un tejido renal con características histológicas y funcionales normales. La hipermetilación de H19, un componente conocido de un subconjunto de tumores de Wilms, es una anomalía genética muy común que se encuentra en estas áreas de apariencia normal de las lesiones precursoras.[
En un estudio se obtuvo la secuenciación del genoma completo, la expresión de mRNA y miRNA, el número de copias de DNA y el análisis de metilación de117 tumores de Wilms; luego se hizo la secuenciación dirigida de 651 tumores de Wilms.[
- Los tumores de Wilms surgen a menudo como consecuencia de más de una alteración genética.
- Los tumores de Wilms con anomalías genéticas diferentes exhiben diferencias en la expresión génica y los patrones de metilación.
- En los tumores de Wilms hay un gran número de genes iniciadores propuestos como candidatos, y casi todos estos están alterados en menos del 5 % de los tumores de Wilms.
- Los tumores de Wilms exhiben variantes recurrentes de genes con funciones comunes; la mayoría de ellos participan en el desarrollo renal temprano o en la regulación epigenética (por ejemplo, modificaciones de la cromatina, elongación transcripcional y miRNA).
Alrededor de un tercio de los casos de tumor de Wilms tienen variantes de los genes WT1, CTNNB1 o AMER1 (WTX).[
Se observan tasas altas de tumor de Wilms en pacientes con una variedad de trastornos genéticos, como el síndrome WAGR (tumor de Wilms, aniridia, anormalidades genitourinarias y una variedad de alteraciones en el desarrollo neurológico), el síndrome de Beckwith-Wiedemann, la hemihipertrofia, el síndrome de Denys-Drash y el síndrome de Perlman.[
A continuación se resumen las características genómicas y genéticas del tumor de Wilms.
GenWT1
El gen WT1 se encuentra en el brazo corto del cromosoma 11 (11p13). WT1 es un factor de transcripción necesario para el desarrollo genitourinario normal y es fundamental para la diferenciación del blastema renal.[
El tumor de Wilms con variante de WT1 se caracteriza por los siguientes aspectos:
- A menudo presenta activación de la vía WNT por variantes activadoras del gen CTNNB1.[
13 ,14 ,15 ] - Frecuentemente se observa la pérdida de heterocigosidad (LOH) en 11p15, debido a que la disomía uniparental paterna en el cromosoma 11 representa un mecanismo común de pérdida del alelo de WT1 normal remanente.[
13 ,16 ] - Los restos nefrogénicos son centros benignos de células renales embrionarias que persisten de manera anormal durante el periodo posnatal. Se encuentran restos nefrogénicos intralobulares en casi el 20 % de los casos de tumor de Wilms. Estos restos se observan con bastante frecuencia en los casos de síndromes genéticos que tienen variantes de WT1, como los síndromes WAGR y de Denys-Drash.[
17 ] También se observan restos nefrogénicos intralobulares en casos con variantes esporádicas de los genes WT1 y MLLT1.[18 ,19 ] - Las variantes germinales de WT1 son infrecuentes (2–4 %) en el tumor de Wilms no sindrómico.[
20 ,21 ] - En un estudio de 56 pacientes que no habían recibido quimioterapia, las variantes de WT1 y la LOH de 11p15 se relacionaron con recidiva en pacientes con tumores de Wilms de riesgo muy bajo.[
22 ] Estos resultados requieren validación y quizás proporcionen biomarcadores para la clasificación de pacientes en el futuro.
Las variantes germinales de WT1 son más comunes en los niños que tienen tumor de Wilms y presentan una de las siguientes afecciones:
- Síndrome WAGR, síndrome de Denys-Drash [
23 ] o síndrome de Frasier.[24 ] - Anomalías genitourinarias, incluso hipospadias y criptorquidia.
- Tumor de Wilms bilateral.
- Tumor de Wilms unilateral con restos nefrogénicos en el riñón contralateral.
- Diferenciación estromal y rabdomiomatosa.
Las variantes germinales de un solo nucleótido de WT1 producen síndromes genéticos caracterizados por nefropatía, trastorno del desarrollo sexual 46XY y riesgos variables de tumor de Wilms.[
- Síndrome WAGR. Los niños con síndrome WAGR tienen un riesgo alto (cerca del 50 %) de presentar tumor de Wilms.[
27 ] El síndrome WAGR se produce por deleciones en el cromosoma 11p13 que afectan un conjunto de genes contiguos, entre ellos, los genes WT1 y PAX6.Las variantes inactivadoras o las deleciones del gen PAX6 producen aniridia, mientras que la deleción de WT1 aumenta el riesgo de tumor de Wilms. La pérdida del gen LMO2 se relacionó con una aparición más frecuente de tumor de Wilms en pacientes con aniridia congénita y deleciones en la región WAGR.[
28 ][Nivel de evidencia C1] La aniridia esporádica sin deleción de WT1 no se relaciona con aumento de riesgo de tumor de Wilms. En consecuencia, los niños con aniridia familiar (que por lo general afecta a muchas generaciones), pero sin anomalías renales, tienen un gen WT1 normal lo que no acarrea aumento del riesgo de tumor de Wilms.[29 ,30 ]El tumor de Wilms en niños con síndrome WAGR se caracteriza por un exceso de enfermedad bilateral, restos nefrogénicos intralobulares, edad temprana en el momento del diagnóstico y un tipo histológico de predominio estromal en tumores con características HF.[
31 ] Es posible que la discapacidad intelectual en el síndrome WAGR sea secundaria a la deleción de otros genes, como SLC1A2 o BDNF.[32 ]
- Síndrome de Denys-Drash. Esta afección se caracteriza por un síndrome nefrótico causado por esclerosis mesangial difusa, seudohermafroditismo XY y aumento del riesgo de tumor de Wilms (>90 %).
En este síndrome las variantes de WT1 por lo general son variantes de un aminoácido en los exones 8 y 9, que codifican la región de unión al DNA de WT1.[
23 ] - Síndrome de Frasier. Este síndrome se caracteriza por nefropatía progresiva causada por glomeruloesclerosis segmentaria focal, gonadoblastoma y seudohermafroditismo XY.
En este síndrome las variantes de WT1 suelen afectar el intrón 9 en el sitio KT, ello produce una variante de empalme alternativa, y por lo tanto, evitan la producción de la isoforma de WT1 +KTS que, por lo general, es más abundante.[
33 ]
En los estudios de evaluación de las correlaciones genotípicas y fenotípicas de las variantes de WT1, se observó que el riesgo de tumor de Wilms es superior cuando hay variantes interruptoras (14 de 17 casos, 82 %), y el riesgo es inferior cuando hay variantes de aminoácido (27 de 67 casos, 42 %). El riesgo mas bajo se presenta cuando hay variantes en el sitio de empalme KTS (1 de 27 casos, 4 %).[
GenCTNNB1
El gen CTNNB1 es uno de los genes alterados con mayor frecuencia en el tumor de Wilms; sus variantes se notifican en el 15 % de los pacientes con este tumor.[
GenAMER1(WTX) en el cromosoma X
El gen AMER1 está en el cromosoma X en Xq11.1 y se encuentra alterado en el 15 % al 20 % de los casos de tumor de Wilms.[
Las alteraciones en AMER1 se distribuyen por igual entre hombres y mujeres, y la inactivación de AMER1 no tiene un efecto aparente en el cuadro clínico ni en el pronóstico.[
Regiones de control de impronta en el cromosoma 11p15 (WT2) y síndrome de Beckwith-Wiedemann
Otro locus del tumor de Wilms, el de WT2, está en la región de impronta del cromosoma 11p15.5. Cuando este exhibe una variante germinal, produce el síndrome de Beckwith-Wiedemann. Alrededor del 3 % de los niños con tumor de Wilms tiene cambios epigenéticos o genéticos germinales en el locus del regulador del crecimiento 11p15.5, sin ninguna otra manifestación clínica de sobrecrecimiento. Del mismo modo que los niños con síndrome de Beckwith-Wiedemann, en estos niños hay mayor incidencia de tumor de Wilms bilateral o tumor de Wilms familiar.[
Alrededor de un quinto de los pacientes con síndrome de Beckwith-Wiedemann que presentan un tumor de Wilms exhiben enfermedad bilateral, y además se ha observado enfermedad bilateral metacrónica.[
Cerca del 80 % de los pacientes con síndrome de Beckwith-Wiedemann tienen un defecto molecular en el dominio 11p15.[
Hay varios genes candidatos en el locus de WT2 que abarca dos dominios de impronta independientes: IGF2 con H19; y CDKN1C con KCNQ1OT1.[
Se demostró una relación entre el epigenotipo y el fenotipo en el síndrome de Beckwith-Wiedemann, y una diferencia en la tasa de cáncer en este síndrome de acuerdo con el tipo de alteración en la región 11p15.[
Hay cuatro subtipos moleculares principales de síndrome de Beckwith-Wiedemann que se caracterizan por correlaciones genotípicas y fenotípicas específicas:
- Ganancia de metilación en ICR1 (ICR1-GoM). La ICR1-GoM telomérica causa entre el 5 % y el 10 % de los casos. Esta alteración produce expresión bialélica del gen IGF2 (que por lo general solo se expresa a partir del alelo paterno) acompañada de disminución de la expresión del gen oncosupresor H19. La incidencia de tumor de Wilms es del 22,8 %.[
47 ] - Pérdida de metilación en ICR2 (ICR2-LoM). La ICR2-LoM causa el 50 % de los casos de síndrome de Beckwith-Wiedemann; esta alteración produce disminución en la expresión del gen CDKN1C que por lo general se expresa solo a partir del cromosoma materno. La incidencia del tumor es muy baja (2,5 %).[
47 ] - Disomía uniparental (DUP). Se observa una expresión alterada de ambos conjuntos de genes de impronta en la DUP mosaica del cromosoma 11p15.5, que explica del 20 % al 25 % de los casos. La incidencia de tumor de Wilms es del 6,2 %, seguida de hepatoblastoma (4,7 %) y carcinoma de glándula suprarrenal (1,5 %).[
47 ] Menos del 1 % de los casos de síndrome de Beckwith-Wiedemann se producen a partir de reordenamientos cromosómicos que afectan la región 11p15. - Variantes de CDKN1C. Las variantes de pérdida de función de CDKN1C heredadas por línea materna explican casi el 5 % de los casos. Este tipo se vincula con una incidencia de neuroblastoma de un 4,3 %.[
47 ]
Se notificaron otros tumores como neuroblastoma o hepatoblastoma en pacientes con isodisomía paterna de 11p15.[
La pérdida de la impronta o la metilación génica es poco frecuente en otros locus, lo que respalda la especificidad de la pérdida de la impronta en 11p15.5.[
Otros genes y alteraciones cromosómicas
Otros genes y alteraciones cromosómicas que afectan la patogenia y las características biológicas del tumor de Wilms son los siguientes:
- 1q. La ganancia del cromosoma 1q se relaciona con un desenlace más precario y es el factor individual más poderoso para predecir el desenlace.[
53 ,54 ] La ganancia del cromosoma 1q es una de las anomalías citogenéticas más comunes en el tumor de Wilms y se observa en alrededor del 30 % de los tumores.En un análisis de tumores de Wilms con HF de 1114 pacientes participantes del NWTS-5 (COG-Q9401/NCT00002611) se encontró que el 28 % de los tumores exhibían ganancia de 1q.[
53 ]- La tasa de supervivencia sin complicaciones (SSC) a 8 años fue del 77 % en los pacientes con ganancia de 1q y del 90 % en aquellos sin ganancia de 1q (P < 0,001). En cada estadio de la enfermedad, la ganancia de 1q se relacionó con una SSC inferior.
- La tasa de supervivencia general (SG) a 8 años fue del 88 % en quienes tenían ganancia de 1q y del 96 % en aquellos sin ganancia de 1q (P < 0,001). La SG fue significativamente inferior en los casos con enfermedad en estadio I (P < 0,0015) y estadio IV (P = 0,011).
- También se notificaron resultados semejantes en el estudio WT 2001 de la International Society of Paediatric Oncology (SIOP) en 586 niños con tumor de Wilms.[
54 ]
Un estudio incluyó una cohorte de tumor de Wilms de tipo HF con muchos pacientes en recaída. En este se encontró que la prevalencia de ganancia de 1q fue más alta en muestras de tumor de Wilms en recaída (75 %) que en las muestras primarias emparejadas (47 %).[
55 ] La prevalencia en aumento de la ganancia de 1q en el momento de la recaída apoya su relación con un pronóstico precario y progresión de la enfermedad. - 16q y 1p. Es posible que en los cromosomas 16q y 1p se encuentren otros genes supresores de tumores o genes de progresión tumoral, como lo indica la LOH de estas regiones en el 17 % y el 11 % de los casos de tumor de Wilms, respectivamente.[
56 ]- En estudios grandes del NWTS, los pacientes con pérdida en estos locus tumorales específicos presentaron tasas de supervivencia sin recaída y tasas de SG significativamente más precarias. En un estudio en curso del Children's Oncology Group (COG), se utiliza la combinación de la pérdida de 1p y 16q para seleccionar a los pacientes de tumor de Wilms y HF con el fin de administrarles un tratamiento más intensivo. Sin embargo, en un estudio del Reino Unido con más de 400 pacientes, no se encontró una relación significativa entre la deleción de 1p y un pronóstico adverso; pero se encontró un vínculo entre la LOH de 16q y un pronóstico adverso.[
57 ] - En un estudio italiano de 125 pacientes, se administró un tratamiento muy similar al del estudio del COG y se encontró un pronóstico significativamente más precario en los pacientes con deleciones de 1p, pero sin deleciones de 16q.[
58 ]
Estos resultados contradictorios quizás se expliquen por la mayor repercusión pronóstica de la ganancia de 1q descrita antes. La LOH de 16q y 1p son menos relevantes como marcadores pronósticos independientes cuando hay una ganancia de 1q. Sin embargo, la ganancia de 1q y la LOH de 16q y 1p conservan su repercusión negativa sobre el pronóstico.[
53 ] La LOH de 16q y 1p se originan por alteraciones cromosómicas complejas que conducen a LOH de 1q o ganancia de 1q. La alteración genética oncógena relevante es el cambio en 1q.[59 ] - En estudios grandes del NWTS, los pacientes con pérdida en estos locus tumorales específicos presentaron tasas de supervivencia sin recaída y tasas de SG significativamente más precarias. En un estudio en curso del Children's Oncology Group (COG), se utiliza la combinación de la pérdida de 1p y 16q para seleccionar a los pacientes de tumor de Wilms y HF con el fin de administrarles un tratamiento más intensivo. Sin embargo, en un estudio del Reino Unido con más de 400 pacientes, no se encontró una relación significativa entre la deleción de 1p y un pronóstico adverso; pero se encontró un vínculo entre la LOH de 16q y un pronóstico adverso.[
- miRNAPG. Se observaron variantes de determinados miRNAPG en alrededor del 20 % de los casos de tumor de Wilms, lo que al parecer perpetúa el estado del progenitor.[
2 ,5 ,6 ,7 ,8 ] Los productos de estos genes dirigen la maduración de los miRNA desde los transcritos pre-miRNA hasta los miRNA funcionales citoplasmáticos (consultar la Figura 10).[60 ] El miRNAPG que está alterado con mayor frecuencia es DROSHA, que presenta una variante recurrente (E1147K) que afecta un residuo de enlace metálico en el dominio IIIb de la RNasa, y que explica cerca del 80 % de los tumores con alteraciones en DROSHA. Otros miRNAPG que se encuentran alterados en el tumor de Wilms son DGCR8, DICER1, TARBP2, DIS3L2 y XPO5. Por lo general, estas variantes son mutuamente excluyentes, deletéreas y alteran la expresión de los miRNA oncosupresores. Un sesgo sexual llamativo se señaló para las variantes de DGCR8 (ubicado en el cromosoma 22q11), pues 38 de 43 casos (88 %) surgieron en niñas.[5 ,6 ]Se observaron variantes germinales de los miRNAPG de los genes DICER1 y DIS3L2; las variantes del primer gen causan el síndrome DICER1 y las variantes del segundo gen producen el síndrome de Perlman.
- El síndrome DICER1 a menudo surge debido a variantes interruptoras hereditarias del gen DICER1. Los tumores se forman después de que se adquiere una variante de cambio de sentido en un dominio del alelo remanente de DICER1 (dominio IIIb de la RNasa) responsable del procesamiento de los miRNA derivados de los brazos 5p de los pre-miRNA.[
61 ] Los tumores relacionados con el síndrome DICER1 son el blastoma pleuropulmonar, el nefroma quístico, los tumores estromales de cordón sexual de ovario, el bocio multinodular y el rabdomiosarcoma embrionario.[61 ] El tumor de Wilms es una manifestación infrecuente del síndrome DICER1. En un estudio de 3 familias con síndrome DICER1 que incluían niños con tumor de Wilms, se encontró que 2 casos de tumor de Wilms exhibían la variante secundaria típica de DICER1 en el dominio IIIb de la RNasa.[62 ] En otro estudio, se encontraron variantes de DICER1 en 2 de 48 familias con tumor de Wilms familiar.[63 ] En extensos estudios de secuenciación de cohortes de casos de tumor de Wilms, también se observaron casos ocasionales con variantes de DICER1.[6 ,7 ] - El síndrome de Perlman es un trastorno autosómico recesivo de sobrecrecimiento raro producido por variantes de DIS3L2, que codifica la ribonucleasa responsable de degradar el pre-let-7 miRNA.[
64 ,65 ] Las inactivaciones germinales heterocigotas de DIS3L2 también se relacionan con la presentación de tumor de Wilms.[66 ] El pronóstico de los pacientes con síndrome de Perlman es precario, con una tasa de mortalidad neonatal alta. En una investigación de casos publicados de síndrome de Perlman (N = 28) en lactantes que sobrevivieron el periodo neonatal, se encontró que casi dos tercios presentaron tumor de Wilms y todos padecieron retraso del desarrollo. Las manifestaciones frecuentes son macrosomía fetal, ascitis y polihidramnios.[67 ]
- El síndrome DICER1 a menudo surge debido a variantes interruptoras hereditarias del gen DICER1. Los tumores se forman después de que se adquiere una variante de cambio de sentido en un dominio del alelo remanente de DICER1 (dominio IIIb de la RNasa) responsable del procesamiento de los miRNA derivados de los brazos 5p de los pre-miRNA.[
- SIX1 y SIX2. Los genes SIX1 y SIX2 codifican factores de transcripción altamente homólogos que cumplen una función básica en el desarrollo renal temprano y que se expresan en el mesénquima metanéfrico en donde controlan la población mesenquimatosa progenitora. En los pacientes con tumor de Wilms, la frecuencia de las variantes de SIX1 es del 3 % al 4 %, y la frecuencia de las variantes de SIX2 es del 1 % al 3 %.[
5 ,6 ]- Prácticamente todas las variantes de SIX1 y SIX2 se ubican en el exón 1 y producen una variante caracterizada por un cambio de glutamina a arginina en la posición 177 (Q177R).
- Las variantes de los genes WT1, AMER1 y CTNNB1 son infrecuentes en los casos con variantes de los genes SIX1, SIX2 o de miRNAPG. Por el contrario, las variantes de SIX1 o SIX2 y las variantes de miRNAPG tienden a presentarse juntas.
- En muestras de tumor de Wilms de pacientes que no han recibido quimioterapia, las variantes de SIX1 y SIX2 se relacionan con el subtipo blastémico de riesgo alto y con la presencia de blastema indiferenciado.
- En un estudio de 82 casos de tumor de Wilms con HF, se identificaron variantes en el punto caliente Q177R de SIX1 con mayor frecuencia en las muestras tumorales de recaída (11 cases; 13,4 %) que en las muestras del momento del diagnóstico (4 %). En 45 casos que contaban con muestras del momento del diagnóstico y de la recaída, se encontraron 6 casos con SIX1Q177R en el momento de la recaída, 3 de los cuales no tenían SIX1Q177R en el momento del diagnóstico. Este resultado indica que esta variante no es necesaria para la formación del tumor en algunas personas con tumor de Wilms.[
55 ]
- MLLT1. Alrededor del 4 % de los casos de tumor de Wilms tienen variantes en el dominio YEATS, altamente conservado, de MLLT1 (ENL), un gen involucrado en la elongación transcripcional producida por la RNA–polimerasa II durante el desarrollo temprano.[
19 ] Las proteínas MLLT1 alteradas exhiben una alteración en la unión a los extremos de histonas acetiladas. Los pacientes con tumores que exhiben alteraciones de MLLT1 desde edades tempranas tienen una prevalencia alta de restos nefrogénicos intralobulares precursores, lo que sustenta un modelo en el que se presentan variantes activadoras de MLLT1 durante el desarrollo renal temprano que producen tumor de Wilms. - TP53 (gen supresor de tumores). La mayoría de los casos de tumor de Wilms anaplásico exhiben variantes del gen supresor de tumores TP53.[
68 ,69 ,70 ] El TP53 quizás sea útil como marcador de pronóstico desfavorable.[68 ,69 ]En un estudio prospectivo de 118 pacientes con tumor de Wilms anaplásico difuso inscritos en el ensayo NWTS-5, se demostró que 57 pacientes (48 %) tenían variantes de TP53, 13 pacientes (11 %) tenían pérdida segmentaria del número de copias de TP53 sin variantes de este gen, y 48 pacientes (41 %) carecían de ambas anomalías (TP53 de tipo natural [wtTP53]). Todas las variantes de TP53 se detectaron mediante secuenciación sola. Los pacientes con enfermedad en estadio III o estadio IV y wtTP53 presentaron tasas de recaída y mortalidad significativamente más bajas que los pacientes con anomalías en TP53 (P = 0,00006 y P = 0,00007, respectivamente). El estado de TP53 no tuvo repercusión en los pacientes con tumores en estadio I o estadio II.[
71 ]- En un análisis detallado de un subconjunto de 39 pacientes con tumor de Wilms anaplásico difuso se observó que 7 pacientes (18 %) albergaban un wtTP53. En estos tumores con wtTP53 se demostró la expresión génica de la activación de la vía p53. En una revisión retrospectiva del resultado patológico de tumores con wtTP53 se observó un volumen muy bajo de anaplasia o ausencia de anaplasia en 6 de 7 tumores. Estos datos apoyan la función básica de la pérdida de TP53 en la formación de anaplasia en el tumor de Wilms, y respaldan la repercusión clínica significativa en pacientes con enfermedad anaplásica residual después de la cirugía.[
71 ]
- En un análisis detallado de un subconjunto de 39 pacientes con tumor de Wilms anaplásico difuso se observó que 7 pacientes (18 %) albergaban un wtTP53. En estos tumores con wtTP53 se demostró la expresión génica de la activación de la vía p53. En una revisión retrospectiva del resultado patológico de tumores con wtTP53 se observó un volumen muy bajo de anaplasia o ausencia de anaplasia en 6 de 7 tumores. Estos datos apoyan la función básica de la pérdida de TP53 en la formación de anaplasia en el tumor de Wilms, y respaldan la repercusión clínica significativa en pacientes con enfermedad anaplásica residual después de la cirugía.[
- FBXW7. Se identificó que el gen FBXW7, un componente de la ligasa de ubiquitina, es un gen supresor de tumores reconocido que presenta tasas bajas de alteraciones recurrentes en el tumor de Wilms y en otras neoplasias malignas. Las variantes de este gen se relacionaron con características histológicas tumorales de tipo epitelial.[
72 ]; [73 ][Nivel de evidencia C1] - TRIM28. El gen TRIM28 codifica una proteína con varios dominios que participa en la regulación de muchos procesos celulares y se considera un gen de predisposición al tumor de Wilms que se hereda de manera autosómica dominante. Las alteraciones en TRIM28 explican cerca del 8 % de los casos de tumor de Wilms familiar y 2 % de casos inespecíficos de tumor de Wilms.[
74 ,75 ,76 ,77 ]; [73 ][Nivel de evidencia C1]- Se ha observado una asociación fuerte entre las variantes de TRIM28 y el tumor de Wilms epitelial, la mayoría de las personas con una variante de TRIM28 presentan un tumor de Wilms con predominio de características histológicas epiteliales.[
74 ,75 ,76 ]; [73 ][Nivel de evidencia C1] - En una cohorte de 91 personas afectadas de 49 familias con árbol genealógico compatible con tumor de Wilms, se identificaron 33 personas que tenían variantes de predisposición al cáncer constitucionales, 21 de ellos presentaba una variante de TRIM28. Se observó un efecto fuerte del origen parental; los 10 casos evaluables presentaron variantes hereditarias transmitidas por vía materna.[
73 ][Nivel de evidencia C1] - La mayoría de los casos con variantes de TRIM28 exhibían variantes de cambio en el marco de lectura, terminadores o de corte y empalme en un alelo, en combinación con LOH en el otro alelo, lo que conlleva pérdida de la expresión de la proteína TRIM28 en el tumor. La tinción inmunohistoquímica para la pérdida de la expresión de la proteína TRIM28 se puede usar para identificar la mayoría de los pacientes que tienen variantes de TRIM28.[
77 ]
- Se ha observado una asociación fuerte entre las variantes de TRIM28 y el tumor de Wilms epitelial, la mayoría de las personas con una variante de TRIM28 presentan un tumor de Wilms con predominio de características histológicas epiteliales.[
- Síndrome de microdeleción de 9q22.3. Los pacientes con síndrome de microdeleción de 9q22.3 tienen riesgo elevado de presentar tumor de Wilms.[
78 ] La región cromosómica con deleción germinal abarca el gen PTCH1, que está alterado en el síndrome de Gorlin (síndrome del carcinoma nevoide basocelular asociado con osteosarcoma). El síndrome de microdeleción de 9q22.3 se caracteriza por las manifestaciones clínicas del síndrome de Gorlin, así como por un retraso del desarrollo o discapacidad intelectual, craniosinostosis metópica, hidrocefalia obstructiva, macrosomía prenatal y posnatal, y convulsiones. Se informó sobre 5 pacientes que presentaban tumor de Wilms en el marco de una microdeleción constitucional de 9q22.3.[78 ,79 ,80 ] - MYCN. Se han notificado alteraciones genómicas que afectan la red MYCN (por ejemplo, MYCN, MAX, MGA, NONO) en el 25 % al 30 % de los casos de tumor de Wilms.[
55 ] Las alteraciones genómicas específicas asociadas con la red MYCN son las siguientes:- La ganancia en el número de copias de MYCN se observó en cerca del 13 % de los casos de tumor de Wilms. La ganancia de MYCN fue más común en los casos anaplásicos (7 de 23 casos, 30 %) que en los casos no anaplásicos (11,2 %), y se relacionó con una supervivencia sin recaída (SSR) y supervivencia general más precarias, independientemente del tipo histológico.[
81 ] La duplicación en tándem de MYCN se notificó en 11 de 82 (13 %) muestras de tumor de Wilms con HF.[55 ] - La ganancia en el número de copias de MYCN de origen germinal se notificó en un caso de tumor de Wilms bilateral,[
81 ] y la duplicación germinal de MYCN también se notificó en un niño con nefroblastomatosis bilateral prenatal y antecedentes familiares de nefroblastoma.[82 ] - Se observaron variantes en el codón 44 (p.P44L) de MYCN en alrededor del 3 % al 4 % de los casos de tumor de Wilms en el momento del diagnóstico [
81 ,83 ] y en el 8,5 % de los casos en el momento de la recaída.[55 ] En un estudio de 810 casos de tumor de Wilms, 24 (3 %) exhibía variantes en el punto caliente P44L de MYCN. La SSR fue significativamente inferior (68,6 %) en los pacientes con variantes de P44L que en los pacientes con MYCN natural (87,1 %).[83 ] - La proteína de interacción con MYCN llamada MAX estaba alterada en el codón 60 (R60Q) en 7 de 782 casos de tumor de Wilms (0,9 %).[
83 ] La SSR fue inferior en los pacientes con una variante en el punto caliente R60Q de MAX que en los pacientes con MAX natural.
- La ganancia en el número de copias de MYCN se observó en cerca del 13 % de los casos de tumor de Wilms. La ganancia de MYCN fue más común en los casos anaplásicos (7 de 23 casos, 30 %) que en los casos no anaplásicos (11,2 %), y se relacionó con una supervivencia sin recaída (SSR) y supervivencia general más precarias, independientemente del tipo histológico.[
- CTR9. En 4 de 36 árboles genealógicos de tumor de Wilms familiar se encontraron variantes inactivadoras germinales de CTR9.[
11 ,84 ] El gen CTR9, ubicado en el cromosoma 11p15.3, es un componente clave del complejo del factor relacionado con la polimerasa de tipo 1 (PAF1c) que tiene múltiples funciones en la regulación de la RNA–polimerasa II y participa en la organogénesis embrionaria y la conservación de la pluripotencia de las células madre embrionarias. - REST. Se encontraron variantes inactivadoras germinales de REST (codificador del factor de transcripción de silenciamiento RE1) en 4 árboles genealógicos de tumor de Wilms familiar.[
10 ] El REST es un represor de la transcripción que afecta la diferenciación celular y el desarrollo embrionario. La mayoría de las variantes de REST se agrupan en la porción de REST que codifica el dominio de unión al DNA, y en el análisis funcional se observó que esas variantes afectan la represión transcripcional de REST. Cuando se hicieron pruebas para detectar variantes de REST, 9 de 519 personas con tumor de Wilms sin antecedentes familiares de la enfermedad obtuvieron un resultado positivo para una de estas variantes; algunos progenitores de estos pacientes también dieron positivo para una de estas variantes.[10 ] Estas observaciones permiten indicar que REST es un gen de predisposición al tumor de Wilms y que se asocia con casi un 2 % de estos tumores.
En la Figura 11 se resume el panorama genómico de una cohorte de pacientes con tumor de Wilms seleccionados debido a que presentaron recaída pese a exhibir HF.[

Alteraciones genómicas del tumor de Wilms en recaída
El tumor de Wilms en recaída conserva la mayoría de las alteraciones genómicas presentes en el momento del diagnóstico, aunque es posible que se presenten cambios en la prevalencia de alteraciones en genes específicos entre el momento del diagnóstico y de la recaída.[
- La prevalencia de ganancia de 1q en las muestras de tumores de Wilms en recaída (75 %) fue más alta que la observaba para los tumores en el momento del diagnóstico (47 %).[
55 ] El aumento de la prevalencia de la ganancia de 1q en el momento de la recaída es compatible con su asociación con un pronóstico adverso y progresión de la enfermedad. - Se identificaron variantes en el punto caliente SIX1Q177R con mayor frecuencia en las muestras tumorales de recaída (11 de 82 casos; 13,4 %) que en las muestras del momento del diagnóstico (4 %).[
55 ] En los 45 casos que contaban con muestras del momento del diagnóstico y recaída, 6 casos presentaron la variante en el punto caliente SIX1Q177R en el momento de la recaída, 3 de los cuales no tenían la variante SIX1Q177R en el momento del diagnóstico. Esto es compatible con que la variante SIX1Q177R no se considere un evento de carcinogénesis temprana en algunos casos.[55 ] - Se encontraron alteraciones genómicas en los genes asociados con la red MYCN el alrededor del 30 % de los casos de tumor de Wilms en recaída.[
55 ] Las alteraciones más comunes en la red MYCN fueron la duplicación en tándem de MYCN (13 %) y las variantes en el punto caliente P44L de MYCN (11 %).
Los tumores de Wilms recidivantes o resistentes al tratamiento de 56 pacientes pediátricos se sometieron a secuenciación tumoral en el ensayo Pediatric MATCH del National Cancer Institute–Children's Oncology Group (NCI-COG). Durante el proceso se encontraron alteraciones genómicas que se consideraron de interés para el tratamiento en los grupos del estudio MATCH en 6 de 56 tumores (10,7 %). En 2 de 56 tumores (3,6 %), se encontraron variantes de BRCA2.[
Alteraciones genómicas en adultos con tumor de Wilms
El tumor de Wilms en pacientes mayores de 16 años es raro con una tasa de incidencia de menos de 0,2 casos por millón de personas por año.[
En un estudio de 14 pacientes con diagnóstico de tumor de Wilms y edad mayor a 16 años (intervalo, 17–46 años; mediana de edad, 31 años), se evaluaron las variantes exónicas de 1425 genes relacionados con el cáncer.[
- Se encontró que 5 pacientes (36 %) albergaban variantes BRAF V600E. Si bien las variantes BRAF V600E son muy infrecuentes en el tumor de Wilms en la niñez, se encuentran en el 90 % de los adenomas metanéfricos de riñón, una afección que suele ser benigna y que solo se presenta en adultos.[
88 ] - Los 5 adultos con tumor de Wilms y variante BRAF V600E exhibían áreas con mejor diferenciación idénticas a un adenoma metanéfrico situadas de manera adyacente a áreas compatibles en apariencia con el tumor de Wilms epitelial.
- Entre los 5 casos con variantes BRAF V600E, 2 presentaban variantes del promotor de TERT además de variantes de BRAF.
- Se observaron variantes de ASXL1 en 4 de 14 casos, que incluyeron 1 de 5 casos con variantes BRAF V600E y 3 de 9 casos sin variantes BRAF V600E. Las variantes de ASXL1 no son comunes en el tumor de Wilms infantil (alrededor del 2 % de los casos).[
2 ] - Entre los 9 tumores que no tenían variantes de BRAF, algunos presentaban alteraciones genómicas asociadas con el tumor de Wilms de la niñez (por ejemplo, ganancia de 1q y variantes de WT1 [n = 2]).
En otro informe se describieron tumores renales con superposición histológica de adenoma metanéfrico y tumor de Wilms epitelial.[
Para obtener información sobre el tratamiento del carcinoma de células renales, consultar
Carcinoma de células renales
Características moleculares del carcinoma de células renales
Los carcinomas de riñón positivos para translocaciones se reconocen como un subtipo distintivo de carcinoma de células renales (RCC), que quizás sea el subtipo más común en niños y representa el 40 % al 50 % de los RCC infantiles.[
- ASPSCR en t(X;17)(p11.2;q25).
- PRCC en t(X;1)(p11.2;q21).
- SFPQ en t(X;1)(p11.2;p34).
- NONO en inv(X;p11.2;q12).
- CLTC en t(X;17)(p11;q23).
- VCP en t(x;9)(p11.23;p13.3).[
94 ]
En una investigación de una sola institución, los datos moleculares de 22 pacientes con RCC positivo para translocaciones se agruparon de acuerdo a los datos publicados con anterioridad. Los investigadores encontraron que ciertas variaciones en el número de copias se relacionaban con agresividad de la enfermedad en pacientes con RCC positivo para translocaciones. Los tumores con pérdida de 9p, ganancia de 17q o una carga alta desde el punto de vista genético de variaciones en el número de copias se relacionaron con una supervivencia deficiente en estos pacientes. En el estudio se incluyeron 3 pacientes pediátricos con un curso de enfermedad poco activo y se encontró que tenían cargas de número de copias más bajas, lo que se corresponde con un curso de la enfermedad menos agresivo en estos pacientes en comparación con los pacientes adultos.[
Otro subtipo de translocación menos común, t(6;11)(p21;q12), corresponde a una fusión del gen TFEB que induce la sobreexpresión de TFEB. Las translocaciones que afectan los genes TFE3 y TFEB inducen la sobreexpresión de estas proteínas que se pueden identificar mediante pruebas inmunohistoquímicas.[
La exposición previa a la quimioterapia es el único factor de riesgo conocido para la presentación de los RCC con translocación de Xp11. En un estudio, el intervalo posterior a la quimioterapia osciló entre 4 y 13 años. Todos los pacientes del informe recibieron un inhibidor de la DNA–topoisomerasa II o un alquilante.[
Hay polémica sobre el comportamiento biológico de los RCC con translocaciones en niños y adultos jóvenes. Si bien en algunas series se indicó que los pacientes que tienen un RCC con translocaciones exhiben un pronóstico más favorable cuando se tratan con cirugía sola pese a presentar el cáncer en un estadio más avanzado (III/IV), en un metanálisis se notificó que estos pacientes tienen desenlaces más precarios.[
El diagnóstico del RCC con una translocación de Xp11 se debe confirmar mediante un abordaje genético-molecular, en lugar de usar una prueba inmunohistoquímica sola para la identificación de TFE3, porque se han notificado casos que no exhiben esta translocación.
Hay un subgrupo infrecuente de casos de RCC que dan positivo para TFE3 pero no exhiben una translocación de TFE3, y en su lugar expresan una translocación de ALK. Estos casos conforman un nuevo subgrupo de RCC recientemente identificado que abarca el 15 % al 20 % de los RCC infantiles no clasificados. En 8 casos notificados en niños de 6 a 16 años, se observaron las siguientes características:[
- ALK se fusionó con VCL en la translocación t(2;10)(p23;q22) (n = 3). Todos los casos con la translocación de VCL se presentaron en niños con rasgo de células falciformes, mientras que ninguno de los casos con la translocación de TPM3 presentó este rasgo.
- ALK se fusionó con TPM3 (n = 3).
- ALK se fusionó con HOOK1 en 1p32 (n = 1).
- ALK se fusionó con TPM3 y formó la translocación t(1;2) (n = 1).
Para obtener información sobre el tratamiento del carcinoma de células renales, consultar
Tumores rabdoides de riñón
Características moleculares de los tumores rabdoides de riñón
Los tumores rabdoides, independientemente de su ubicación anatómica, exhiben una anomalía genética común: la pérdida de la función del gen SMARCB1 ubicado en el cromosoma 22q11.2 (>95 % de los tumores).[
Se han documentado variantes germinales de SMARCB1 en pacientes con uno o más tumores primarios de encéfalo o riñón, lo que es compatible con una predisposición genética a la formación de tumores rabdoides.[
En un estudio de 100 pacientes con tumores rabdoides de encéfalo, riñón o tejido blando, se encontró que 35 pacientes exhibían una anomalía germinal en SMARCB1. Estas anomalías fueron variantes de un solo nucleótido y del marco de lectura, deleciones intragénicas y duplicaciones, así como deleciones más grandes. En 9 casos se demostró la transmisión de progenitores a hijos de una copia alterada de SMARCB1. En 8 de los 9 casos, uno o más familiares contaban con un diagnóstico de tumor rabdoide o schwannoma. De las 8 familias, 2 contaban con varios niños afectados, lo que es compatible con mosaicismo gonadal.[
De manera infrecuente, los tumores rabdoides extracraneales también pueden albergar una inactivación de SMARCA4 en lugar de SMARCB1.[
Para obtener información sobre el tratamiento de los tumores rabdoides de riñón, consultar
Sarcoma de células claras de riñón
Características moleculares del sarcoma de células claras de riñón
Se sabe muy poco del origen molecular del sarcoma de células claras de riñón debido a que es un cáncer raro que carece de modelos experimentales. No obstante, se han descrito múltiples características moleculares del sarcoma de células claras de riñón, entre ellas, las siguientes:
- Se informó de duplicaciones internas en tándem en el exón 15 del gen BCOR (correpresor de BCL6) en el 90 % de los casos de sarcoma de células claras de riñón, y un subgrupo más pequeño alberga las fusiones génicas YWHAE::NUTM2B, YWHAE::NUTM2E o BCOR::CCNB3.[
123 ,124 ,125 ,126 ,127 ,128 ] Todas estas anomalías genéticas generan una firma transcripcional que se caracteriza por una expresión alta de mRNA de BCOR.[129 ] - La inmunorreactividad fuerte difusa para BCOR es muy sensible y específica para el diagnóstico del sarcoma de células claras de riñón. En una serie se evaluaron 79 neoplasias, incluso tumores de Wilms, nefromas mesoblásticos congénitos, sarcoma de células claras de riñón, tumores estromales metanéfricos, tumores rabdoides de riñón, tumor neuroectodérmico primitivo [TNEP] de riñón y fibrosarcomas epitelioides esclerosantes. Todas las muestras de sarcoma de células claras de riñón que se analizaron exhibieron marcación nuclear fuerte difusa para BCOR. La mayoría de las otras neoplasias renales infantiles fueron completamente negativas para BCOR.[
130 ]
Para obtener información sobre el tratamiento del sarcoma de células claras de riñón, consultar
Referencias:
- Coorens THH, Treger TD, Al-Saadi R, et al.: Embryonal precursors of Wilms tumor. Science 366 (6470): 1247-1251, 2019.
- Gadd S, Huff V, Walz AL, et al.: A Children's Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet 49 (10): 1487-1494, 2017.
- Wegert J, Wittmann S, Leuschner I, et al.: WTX inactivation is a frequent, but late event in Wilms tumors without apparent clinical impact. Genes Chromosomes Cancer 48 (12): 1102-11, 2009.
- Ruteshouser EC, Robinson SM, Huff V: Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer 47 (6): 461-70, 2008.
- Walz AL, Ooms A, Gadd S, et al.: Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 27 (2): 286-97, 2015.
- Wegert J, Ishaque N, Vardapour R, et al.: Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 27 (2): 298-311, 2015.
- Rakheja D, Chen KS, Liu Y, et al.: Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun 2: 4802, 2014.
- Torrezan GT, Ferreira EN, Nakahata AM, et al.: Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nat Commun 5: 4039, 2014.
- Dome JS, Huff V: Wilms Tumor Predisposition. In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp.
Available online . Last accessed August 16, 2022. - Mahamdallie SS, Hanks S, Karlin KL, et al.: Mutations in the transcriptional repressor REST predispose to Wilms tumor. Nat Genet 47 (12): 1471-4, 2015.
- Hanks S, Perdeaux ER, Seal S, et al.: Germline mutations in the PAF1 complex gene CTR9 predispose to Wilms tumour. Nat Commun 5: 4398, 2014.
- Huff V: Wilms tumor genetics. Am J Med Genet 79 (4): 260-7, 1998.
- Scott RH, Murray A, Baskcomb L, et al.: Stratification of Wilms tumor by genetic and epigenetic analysis. Oncotarget 3 (3): 327-35, 2012.
- Corbin M, de Reyniès A, Rickman DS, et al.: WNT/beta-catenin pathway activation in Wilms tumors: a unifying mechanism with multiple entries? Genes Chromosomes Cancer 48 (9): 816-27, 2009.
- Maiti S, Alam R, Amos CI, et al.: Frequent association of beta-catenin and WT1 mutations in Wilms tumors. Cancer Res 60 (22): 6288-92, 2000.
- Gadd S, Huff V, Huang CC, et al.: Clinically relevant subsets identified by gene expression patterns support a revised ontogenic model of Wilms tumor: a Children's Oncology Group Study. Neoplasia 14 (8): 742-56, 2012.
- Breslow NE, Beckwith JB, Perlman EJ, et al.: Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr Blood Cancer 47 (3): 260-7, 2006.
- Fukuzawa R, Heathcott RW, More HE, et al.: Sequential WT1 and CTNNB1 mutations and alterations of beta-catenin localisation in intralobar nephrogenic rests and associated Wilms tumours: two case studies. J Clin Pathol 60 (9): 1013-6, 2007.
- Perlman EJ, Gadd S, Arold ST, et al.: MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology Wilms tumours. Nat Commun 6: 10013, 2015.
- Diller L, Ghahremani M, Morgan J, et al.: Constitutional WT1 mutations in Wilms' tumor patients. J Clin Oncol 16 (11): 3634-40, 1998.
- Little SE, Hanks SP, King-Underwood L, et al.: Frequency and heritability of WT1 mutations in nonsyndromic Wilms' tumor patients: a UK Children's Cancer Study Group Study. J Clin Oncol 22 (20): 4140-6, 2004.
- Perlman EJ, Grundy PE, Anderson JR, et al.: WT1 mutation and 11P15 loss of heterozygosity predict relapse in very low-risk wilms tumors treated with surgery alone: a children's oncology group study. J Clin Oncol 29 (6): 698-703, 2011.
- Pelletier J, Bruening W, Kashtan CE, et al.: Germline mutations in the Wilms' tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 67 (2): 437-47, 1991.
- Barbosa AS, Hadjiathanasiou CG, Theodoridis C, et al.: The same mutation affecting the splicing of WT1 gene is present on Frasier syndrome patients with or without Wilms' tumor. Hum Mutat 13 (2): 146-53, 1999.
- Lipska BS, Ranchin B, Iatropoulos P, et al.: Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int 85 (5): 1169-78, 2014.
- Lehnhardt A, Karnatz C, Ahlenstiel-Grunow T, et al.: Clinical and molecular characterization of patients with heterozygous mutations in wilms tumor suppressor gene 1. Clin J Am Soc Nephrol 10 (5): 825-31, 2015.
- Scott RH, Stiller CA, Walker L, et al.: Syndromes and constitutional chromosomal abnormalities associated with Wilms tumour. J Med Genet 43 (9): 705-15, 2006.
- Marakhonov AV, Vasilyeva TA, Voskresenskaya AA, et al.: LMO2 gene deletions significantly worsen the prognosis of Wilms' tumor development in patients with WAGR syndrome. Hum Mol Genet 28 (19): 3323-3326, 2019.
- Green DM, Breslow NE, Beckwith JB, et al.: Screening of children with hemihypertrophy, aniridia, and Beckwith-Wiedemann syndrome in patients with Wilms tumor: a report from the National Wilms Tumor Study. Med Pediatr Oncol 21 (3): 188-92, 1993.
- Scott RH, Walker L, Olsen ØE, et al.: Surveillance for Wilms tumour in at-risk children: pragmatic recommendations for best practice. Arch Dis Child 91 (12): 995-9, 2006.
- Breslow NE, Norris R, Norkool PA, et al.: Characteristics and outcomes of children with the Wilms tumor-Aniridia syndrome: a report from the National Wilms Tumor Study Group. J Clin Oncol 21 (24): 4579-85, 2003.
- Scott RH, Douglas J, Baskcomb L, et al.: Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet 40 (11): 1329-34, 2008.
- Barbaux S, Niaudet P, Gubler MC, et al.: Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet 17 (4): 467-70, 1997.
- Koesters R, Ridder R, Kopp-Schneider A, et al.: Mutational activation of the beta-catenin proto-oncogene is a common event in the development of Wilms' tumors. Cancer Res 59 (16): 3880-2, 1999.
- Koesters R, Niggli F, von Knebel Doeberitz M, et al.: Nuclear accumulation of beta-catenin protein in Wilms' tumours. J Pathol 199 (1): 68-76, 2003.
- Major MB, Camp ND, Berndt JD, et al.: Wilms tumor suppressor WTX negatively regulates WNT/beta-catenin signaling. Science 316 (5827): 1043-6, 2007.
- Rivera MN, Kim WJ, Wells J, et al.: An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 315 (5812): 642-5, 2007.
- Fukuzawa R, Anaka MR, Weeks RJ, et al.: Canonical WNT signalling determines lineage specificity in Wilms tumour. Oncogene 28 (8): 1063-75, 2009.
- Jenkins ZA, van Kogelenberg M, Morgan T, et al.: Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nat Genet 41 (1): 95-100, 2009.
- Grohmann A, Tanneberger K, Alzner A, et al.: AMER1 regulates the distribution of the tumor suppressor APC between microtubules and the plasma membrane. J Cell Sci 120 (Pt 21): 3738-47, 2007.
- DeBaun MR, Siegel MJ, Choyke PL: Nephromegaly in infancy and early childhood: a risk factor for Wilms tumor in Beckwith-Wiedemann syndrome. J Pediatr 132 (3 Pt 1): 401-4, 1998.
- DeBaun MR, Tucker MA: Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr 132 (3 Pt 1): 398-400, 1998.
- Breslow N, Olshan A, Beckwith JB, et al.: Epidemiology of Wilms tumor. Med Pediatr Oncol 21 (3): 172-81, 1993.
- Satoh Y, Nakadate H, Nakagawachi T, et al.: Genetic and epigenetic alterations on the short arm of chromosome 11 are involved in a majority of sporadic Wilms' tumours. Br J Cancer 95 (4): 541-7, 2006.
- Algar EM, St Heaps L, Darmanian A, et al.: Paternally inherited submicroscopic duplication at 11p15.5 implicates insulin-like growth factor II in overgrowth and Wilms' tumorigenesis. Cancer Res 67 (5): 2360-5, 2007.
- Lennerz JK, Timmerman RJ, Grange DK, et al.: Addition of H19 'loss of methylation testing' for Beckwith-Wiedemann syndrome (BWS) increases the diagnostic yield. J Mol Diagn 12 (5): 576-88, 2010.
- Mussa A, Molinatto C, Baldassarre G, et al.: Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J Pediatr 176: 142-149.e1, 2016.
- Bliek J, Gicquel C, Maas S, et al.: Epigenotyping as a tool for the prediction of tumor risk and tumor type in patients with Beckwith-Wiedemann syndrome (BWS). J Pediatr 145 (6): 796-9, 2004.
- Rump P, Zeegers MP, van Essen AJ: Tumor risk in Beckwith-Wiedemann syndrome: A review and meta-analysis. Am J Med Genet A 136 (1): 95-104, 2005.
- Brioude F, Lacoste A, Netchine I, et al.: Beckwith-Wiedemann syndrome: growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Horm Res Paediatr 80 (6): 457-65, 2013.
- Bjornsson HT, Brown LJ, Fallin MD, et al.: Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst 99 (16): 1270-3, 2007.
- Fukuzawa R, Breslow NE, Morison IM, et al.: Epigenetic differences between Wilms' tumours in white and east-Asian children. Lancet 363 (9407): 446-51, 2004.
- Gratias EJ, Dome JS, Jennings LJ, et al.: Association of Chromosome 1q Gain With Inferior Survival in Favorable-Histology Wilms Tumor: A Report From the Children's Oncology Group. J Clin Oncol 34 (26): 3189-94, 2016.
- Chagtai T, Zill C, Dainese L, et al.: Gain of 1q As a Prognostic Biomarker in Wilms Tumors (WTs) Treated With Preoperative Chemotherapy in the International Society of Paediatric Oncology (SIOP) WT 2001 Trial: A SIOP Renal Tumours Biology Consortium Study. J Clin Oncol 34 (26): 3195-203, 2016.
- Gadd S, Huff V, Skol AD, et al.: Genetic changes associated with relapse in favorable histology Wilms tumor: A Children's Oncology Group AREN03B2 study. Cell Rep Med 3 (6): 100644, 2022.
- Grundy PE, Breslow NE, Li S, et al.: Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol 23 (29): 7312-21, 2005.
- Messahel B, Williams R, Ridolfi A, et al.: Allele loss at 16q defines poorer prognosis Wilms tumour irrespective of treatment approach in the UKW1-3 clinical trials: a Children's Cancer and Leukaemia Group (CCLG) Study. Eur J Cancer 45 (5): 819-26, 2009.
- Spreafico F, Gamba B, Mariani L, et al.: Loss of heterozygosity analysis at different chromosome regions in Wilms tumor confirms 1p allelic loss as a marker of worse prognosis: a study from the Italian Association of Pediatric Hematology and Oncology. J Urol 189 (1): 260-6, 2013.
- Gratias EJ, Jennings LJ, Anderson JR, et al.: Gain of 1q is associated with inferior event-free and overall survival in patients with favorable histology Wilms tumor: a report from the Children's Oncology Group. Cancer 119 (21): 3887-94, 2013.
- Hohenstein P, Pritchard-Jones K, Charlton J: The yin and yang of kidney development and Wilms' tumors. Genes Dev 29 (5): 467-82, 2015.
- Foulkes WD, Priest JR, Duchaine TF: DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer 14 (10): 662-72, 2014.
- Wu MK, Sabbaghian N, Xu B, et al.: Biallelic DICER1 mutations occur in Wilms tumours. J Pathol 230 (2): 154-64, 2013.
- Palculict TB, Ruteshouser EC, Fan Y, et al.: Identification of germline DICER1 mutations and loss of heterozygosity in familial Wilms tumour. J Med Genet 53 (6): 385-8, 2016.
- Astuti D, Morris MR, Cooper WN, et al.: Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet 44 (3): 277-84, 2012.
- Chang HM, Triboulet R, Thornton JE, et al.: A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature 497 (7448): 244-8, 2013.
- Hol JA, Kuiper RP, van Dijk F, et al.: Prevalence of (Epi)genetic Predisposing Factors in a 5-Year Unselected National Wilms Tumor Cohort: A Comprehensive Clinical and Genomic Characterization. J Clin Oncol 40 (17): 1892-1902, 2022.
- Alessandri JL, Cuillier F, Ramful D, et al.: Perlman syndrome: report, prenatal findings and review. Am J Med Genet A 146A (19): 2532-7, 2008.
- Bardeesy N, Falkoff D, Petruzzi MJ, et al.: Anaplastic Wilms' tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet 7 (1): 91-7, 1994.
- el Bahtimi R, Hazen-Martin DJ, Re GG, et al.: Immunophenotype, mRNA expression, and gene structure of p53 in Wilms' tumors. Mod Pathol 9 (3): 238-44, 1996.
- Wallkamm V, Dörlich R, Rahm K, et al.: Live imaging of Xwnt5A-ROR2 complexes. PLoS One 9 (10): e109428, 2014.
- Ooms AH, Gadd S, Gerhard DS, et al.: Significance of TP53 Mutation in Wilms Tumors with Diffuse Anaplasia: A Report from the Children's Oncology Group. Clin Cancer Res 22 (22): 5582-5591, 2016.
- Williams RD, Al-Saadi R, Chagtai T, et al.: Subtype-specific FBXW7 mutation and MYCN copy number gain in Wilms' tumor. Clin Cancer Res 16 (7): 2036-45, 2010.
- Mahamdallie S, Yost S, Poyastro-Pearson E, et al.: Identification of new Wilms tumour predisposition genes: an exome sequencing study. Lancet Child Adolesc Health 3 (5): 322-331, 2019.
- Armstrong AE, Gadd S, Huff V, et al.: A unique subset of low-risk Wilms tumors is characterized by loss of function of TRIM28 (KAP1), a gene critical in early renal development: A Children's Oncology Group study. PLoS One 13 (12): e0208936, 2018.
- Halliday BJ, Fukuzawa R, Markie DM, et al.: Germline mutations and somatic inactivation of TRIM28 in Wilms tumour. PLoS Genet 14 (6): e1007399, 2018.
- Diets IJ, Hoyer J, Ekici AB, et al.: TRIM28 haploinsufficiency predisposes to Wilms tumor. Int J Cancer 145 (4): 941-951, 2019.
- Hol JA, Diets IJ, de Krijger RR, et al.: TRIM28 variants and Wilms' tumour predisposition. J Pathol 254 (4): 494-504, 2021.
- Isidor B, Bourdeaut F, Lafon D, et al.: Wilms' tumor in patients with 9q22.3 microdeletion syndrome suggests a role for PTCH1 in nephroblastomas. Eur J Hum Genet 21 (7): 784-7, 2013.
- Garavelli L, Piemontese MR, Cavazza A, et al.: Multiple tumor types including leiomyoma and Wilms tumor in a patient with Gorlin syndrome due to 9q22.3 microdeletion encompassing the PTCH1 and FANC-C loci. Am J Med Genet A 161A (11): 2894-901, 2013.
- Cajaiba MM, Bale AE, Alvarez-Franco M, et al.: Rhabdomyosarcoma, Wilms tumor, and deletion of the patched gene in Gorlin syndrome. Nat Clin Pract Oncol 3 (10): 575-80, 2006.
- Williams RD, Chagtai T, Alcaide-German M, et al.: Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget 6 (9): 7232-43, 2015.
- Fievet A, Belaud-Rotureau MA, Dugay F, et al.: Involvement of germline DDX1-MYCN duplication in inherited nephroblastoma. Eur J Med Genet 56 (12): 643-7, 2013.
- Jiménez Martín O, Schlosser A, Furtwängler R, et al.: MYCN and MAX alterations in Wilms tumor and identification of novel N-MYC interaction partners as biomarker candidates. Cancer Cell Int 21 (1): 555, 2021.
- Martins AG, Pinto AT, Domingues R, et al.: Identification of a novel CTR9 germline mutation in a family with Wilms tumor. Eur J Med Genet 61 (5): 294-299, 2018.
- Parsons DW, Janeway KA, Patton DR, et al.: Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol 40 (20): 2224-2234, 2022.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute.
Available online . Last accessed February 25, 2025. - Argani P, Tickoo SK, Matoso A, et al.: Adult Wilms Tumor: Genetic Evidence of Origin of a Subset of Cases From Metanephric Adenoma. Am J Surg Pathol 46 (7): 988-999, 2022.
- Choueiri TK, Cheville J, Palescandolo E, et al.: BRAF mutations in metanephric adenoma of the kidney. Eur Urol 62 (5): 917-22, 2012.
- Wobker SE, Matoso A, Pratilas CA, et al.: Metanephric Adenoma-Epithelial Wilms Tumor Overlap Lesions: An Analysis of BRAF Status. Am J Surg Pathol 43 (9): 1157-1169, 2019.
- Geller JI, Dome JS: Local lymph node involvement does not predict poor outcome in pediatric renal cell carcinoma. Cancer 101 (7): 1575-83, 2004.
- van der Beek JN, Hol JA, Coulomb-l'Hermine A, et al.: Characteristics and outcome of pediatric renal cell carcinoma patients registered in the International Society of Pediatric Oncology (SIOP) 93-01, 2001 and UK-IMPORT database: A report of the SIOP-Renal Tumor Study Group. Int J Cancer 148 (11): 2724-2735, 2021.
- Geller JI, Ehrlich PF, Cost NG, et al.: Characterization of adolescent and pediatric renal cell carcinoma: A report from the Children's Oncology Group study AREN03B2. Cancer 121 (14): 2457-64, 2015.
- Ambalavanan M, Geller JI: Treatment of advanced pediatric renal cell carcinoma. Pediatr Blood Cancer 66 (8): e27766, 2019.
- Ge Y, Lin X, Zhang Q, et al.: Xp11.2 Translocation Renal Cell Carcinoma With TFE3 Rearrangement: Distinct Morphological Features and Prognosis With Different Fusion Partners. Front Oncol 11: 784993, 2021.
- Marcon J, DiNatale RG, Sanchez A, et al.: Comprehensive Genomic Analysis of Translocation Renal Cell Carcinoma Reveals Copy-Number Variations as Drivers of Disease Progression. Clin Cancer Res 26 (14): 3629-3640, 2020.
- Argani P, Hicks J, De Marzo AM, et al.: Xp11 translocation renal cell carcinoma (RCC): extended immunohistochemical profile emphasizing novel RCC markers. Am J Surg Pathol 34 (9): 1295-303, 2010.
- Argani P, Laé M, Ballard ET, et al.: Translocation carcinomas of the kidney after chemotherapy in childhood. J Clin Oncol 24 (10): 1529-34, 2006.
- Ramphal R, Pappo A, Zielenska M, et al.: Pediatric renal cell carcinoma: clinical, pathologic, and molecular abnormalities associated with the members of the mit transcription factor family. Am J Clin Pathol 126 (3): 349-64, 2006.
- Geller JI, Argani P, Adeniran A, et al.: Translocation renal cell carcinoma: lack of negative impact due to lymph node spread. Cancer 112 (7): 1607-16, 2008.
- Camparo P, Vasiliu V, Molinie V, et al.: Renal translocation carcinomas: clinicopathologic, immunohistochemical, and gene expression profiling analysis of 31 cases with a review of the literature. Am J Surg Pathol 32 (5): 656-70, 2008.
- Qiu Rao, Bing Guan, Zhou XJ: Xp11.2 Translocation renal cell carcinomas have a poorer prognosis than non-Xp11.2 translocation carcinomas in children and young adults: a meta-analysis. Int J Surg Pathol 18 (6): 458-64, 2010.
- Malouf GG, Camparo P, Oudard S, et al.: Targeted agents in metastatic Xp11 translocation/TFE3 gene fusion renal cell carcinoma (RCC): a report from the Juvenile RCC Network. Ann Oncol 21 (9): 1834-8, 2010.
- Rais-Bahrami S, Drabick JJ, De Marzo AM, et al.: Xp11 translocation renal cell carcinoma: delayed but massive and lethal metastases of a chemotherapy-associated secondary malignancy. Urology 70 (1): 178.e3-6, 2007.
- Thorner PS, Shago M, Marrano P, et al.: TFE3-positive renal cell carcinomas are not always Xp11 translocation carcinomas: Report of a case with a TPM3-ALK translocation. Pathol Res Pract 212 (10): 937-942, 2016.
- Cajaiba MM, Jennings LJ, Rohan SM, et al.: ALK-rearranged renal cell carcinomas in children. Genes Chromosomes Cancer 55 (5): 442-51, 2016.
- Smith NE, Deyrup AT, Mariño-Enriquez A, et al.: VCL-ALK renal cell carcinoma in children with sickle-cell trait: the eighth sickle-cell nephropathy? Am J Surg Pathol 38 (6): 858-63, 2014.
- Cajaiba MM, Jennings LJ, George D, et al.: Expanding the spectrum of ALK-rearranged renal cell carcinomas in children: Identification of a novel HOOK1-ALK fusion transcript. Genes Chromosomes Cancer 55 (10): 814-7, 2016.
- Versteege I, Sévenet N, Lange J, et al.: Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394 (6689): 203-6, 1998.
- Imbalzano AN, Jones SN: Snf5 tumor suppressor couples chromatin remodeling, checkpoint control, and chromosomal stability. Cancer Cell 7 (4): 294-5, 2005.
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011.
- Haruta M, Arai Y, Okita H, et al.: Frequent breakpoints of focal deletion and uniparental disomy in 22q11.1 or 11.2 segmental duplication region reveal distinct tumorigenesis in rhabdoid tumor of the kidney. Genes Chromosomes Cancer 60 (8): 546-558, 2021.
- Schneppenheim R, Frühwald MC, Gesk S, et al.: Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet 86 (2): 279-84, 2010.
- Hasselblatt M, Gesk S, Oyen F, et al.: Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 35 (6): 933-5, 2011.
- Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012.
- Biegel JA, Zhou JY, Rorke LB, et al.: Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59 (1): 74-9, 1999.
- Biegel JA: Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg Focus 20 (1): E11, 2006.
- Bourdeaut F, Lequin D, Brugières L, et al.: Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 17 (1): 31-8, 2011.
- Geller JI, Roth JJ, Biegel JA: Biology and Treatment of Rhabdoid Tumor. Crit Rev Oncog 20 (3-4): 199-216, 2015.
- Janson K, Nedzi LA, David O, et al.: Predisposition to atypical teratoid/rhabdoid tumor due to an inherited INI1 mutation. Pediatr Blood Cancer 47 (3): 279-84, 2006.
- Sévenet N, Sheridan E, Amram D, et al.: Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet 65 (5): 1342-8, 1999.
- Hasselblatt M, Nagel I, Oyen F, et al.: SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128 (3): 453-6, 2014.
- Andrianteranagna M, Cyrta J, Masliah-Planchon J, et al.: SMARCA4-deficient rhabdoid tumours show intermediate molecular features between SMARCB1-deficient rhabdoid tumours and small cell carcinomas of the ovary, hypercalcaemic type. J Pathol 255 (1): 1-15, 2021.
- Ueno-Yokohata H, Okita H, Nakasato K, et al.: Consistent in-frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat Genet 47 (8): 861-3, 2015.
- Argani P, Kao YC, Zhang L, et al.: Primary Renal Sarcomas With BCOR-CCNB3 Gene Fusion: A Report of 2 Cases Showing Histologic Overlap With Clear Cell Sarcoma of Kidney, Suggesting Further Link Between BCOR-related Sarcomas of the Kidney and Soft Tissues. Am J Surg Pathol 41 (12): 1702-1712, 2017.
- Karlsson J, Valind A, Gisselsson D: BCOR internal tandem duplication and YWHAE-NUTM2B/E fusion are mutually exclusive events in clear cell sarcoma of the kidney. Genes Chromosomes Cancer 55 (2): 120-3, 2016.
- Astolfi A, Melchionda F, Perotti D, et al.: Whole transcriptome sequencing identifies BCOR internal tandem duplication as a common feature of clear cell sarcoma of the kidney. Oncotarget 6 (38): 40934-9, 2015.
- Roy A, Kumar V, Zorman B, et al.: Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat Commun 6: 8891, 2015.
- Wong MK, Ng CCY, Kuick CH, et al.: Clear cell sarcomas of the kidney are characterised by BCOR gene abnormalities, including exon 15 internal tandem duplications and BCOR-CCNB3 gene fusion. Histopathology 72 (2): 320-329, 2018.
- Kao YC, Sung YS, Zhang L, et al.: Recurrent BCOR Internal Tandem Duplication and YWHAE-NUTM2B Fusions in Soft Tissue Undifferentiated Round Cell Sarcoma of Infancy: Overlapping Genetic Features With Clear Cell Sarcoma of Kidney. Am J Surg Pathol 40 (8): 1009-20, 2016.
- Argani P, Pawel B, Szabo S, et al.: Diffuse Strong BCOR Immunoreactivity Is a Sensitive and Specific Marker for Clear Cell Sarcoma of the Kidney (CCSK) in Pediatric Renal Neoplasia. Am J Surg Pathol 42 (8): 1128-1131, 2018.
Esta información no reemplaza el consejo de un médico. Ignite Healthwise, LLC, niega toda garantía y responsabilidad por el uso de esta información. El uso que usted haga de esta información implica que usted acepta los
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
Page Footer
Quiero...
Audiencia
Sitios seguros para miembros
Información sobre The Cigna Group
Aviso legal
Los planes individuales y familiares de seguro médico y dental están asegurados por Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc. y Cigna HealthCare of Texas, Inc. Los planes de beneficios de salud y de seguro de salud de grupo están asegurados o administrados por CHLIC, Connecticut General Life Insurance Company (CGLIC) o sus afiliadas (puedes ver
Todas las pólizas de seguros y los planes de beneficios de grupo contienen exclusiones y limitaciones. Para conocer la disponibilidad, los costos y detalles completos de la cobertura, comunícate con un agente autorizado o con un representante de ventas de Cigna. Este sitio web no está dirigido a los residentes de New Mexico.