Tratamiento de las manifestaciones de la enfermedad
Tratamiento de los tumores renales
Intervenciones quirúrgicas para los tumores renales
El tratamiento de la enfermedad de Von Hippel-Lindau (VHL) fue cambiando mucho a medida que los profesionales clínicos aprendían a equilibrar el riesgo de diseminación del cáncer al mismo tiempo que reducían la morbilidad renal. En algunas de las series quirúrgicas iniciales se usó la nefrectomía radical bilateral seguida de trasplante renal como tratamiento de tumores renales[1,2] Aunque es poco frecuente, algunos pacientes con enfermedad de VHL se someten a cirugías renales repetidas, lo que, con el tiempo, los vuelve dependientes de la terapia de reemplazo renal. Los trasplantes renales son inocuos en los pacientes con enfermedad de VHL, y los resultados de supervivencia del injerto son satisfactorios.[3] Sin embargo, la conservación de los riñones se ha convertido en una prioridad durante estos procedimientos. En la década de 1980 se introdujo la cirugía conservadora de nefronas para la enfermedad de VHL después de que varios grupos demostraron que este abordaje quirúrgico menos radical se asocia con un riesgo bajo de diseminación del cáncer.[4,5] En 1995, en una gran serie multiinstitucional se demostró que la cirugía conservadora de nefronas conlleva una excelente supervivencia específica del cáncer en pacientes con carcinoma de células renales (RCC).[6] Debido a múltiples informes de desenlaces excelentes, la cirugía conservadora de nefronas ahora se considera el estándar de atención quirúrgica para el RCC relacionado a la enfermedad de VHL, cuando es posible desde el punto de vista técnico. Con el tiempo, se han perfeccionado las técnicas de cirugía conservadora de nefronas para los pacientes con enfermedad de VHL a fin de minimizar el daño al parénquima normal adyacente. Por ejemplo, en la cirugía conservadora de nefronas tradicional, se extrae un margen amplio de tejido. Sin embargo, con un tipo posterior de cirugía conservadora de nefronas llamado enucleación, es posible conservar gran parte del parénquima normal adyacente.[7]
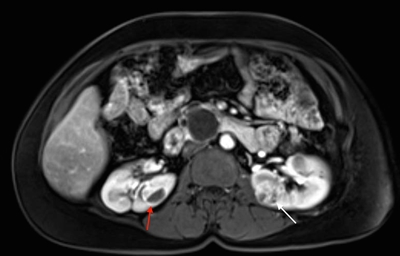
Los pacientes con enfermedad de VHL en ocasiones tienen docenas de tumores renales. Por lo tanto, la resección de toda la enfermedad renal evidente no siempre es posible. Con el fin de reducir al mínimo la morbilidad derivada de múltiples procedimientos quirúrgicos, la disminución del funcionamiento renal y el riesgo de progresión a distancia, se ha buscado un método que equilibre el tratamiento para evitar sobretratar o subtratar. El Instituto Nacional del Cáncer (NCI) evaluó un umbral específico de tamaño tumoral como desencadenante para llevar a cabo la intervención quirúrgica. En una evaluación de 52 pacientes con enfermedad de VHL o carcinoma renal papilar hereditario tratados cuando la lesión renal sólida más grande alcanzó los 3 cm, se demostró la ausencia de evidencia de metástasis a distancia o necesidad de terapia de reemplazo renal al cabo de una mediana de seguimiento de 60 meses.[8] En series retrospectivas posteriores se reforzó la importancia de este umbral de tamaño tumoral de 3 cm. Por ejemplo, en un estudio, ninguno de los 108 pacientes con enfermedad de VHL exhibió indicios de diseminación a distancia cuando el tamaño de los tumores tratados era de 3 cm o inferior.[9] En los pacientes con tumores que medían más de 3 cm, el 27,3 % (20 de 73) presentó recidivas a distancia.[9] Este umbral de tamaño tumoral de 3 cm se usa de manera generalizada en los Estados Unidos con el fin de definir el momento de la intervención quirúrgica para los pacientes con carcinomas renales de células claras (ccRCC) relacionados con la enfermedad de VHL. Sin embargo, algunos grupos de investigación internacionales han publicado datos que respaldan la vigilancia activa de los tumores renales hasta que alcanzan los 4 cm.[10,11,12,13] Cuando se opera a un paciente con enfermedad de VHL, es posible que la resección de tantos tumores renales como sea posible desde el punto de vista clínico retrase la necesidad de más intervenciones quirúrgicas.[14] La ecografía intraoperatoria es útil para identificar y extirpar las lesiones más pequeñas.[15]
Muchos pacientes con enfermedad de VHL continuamente están desarrollando nuevos RCC y es posible que necesiten más intervenciones. Las adherencias y las cicatrices perinéfricas hacen que los procedimientos quirúrgicos subsiguientes sean más difíciles. Si bien se podría considerar una nefrectomía radical, la cirugía conservadora de nefronas sigue siendo el abordaje preferido, cuando es posible. Aunque quizás la incidencia de complicaciones sea más alta con la cirugía conservadora de nefronas repetida y de rescate, es posible que esto permita mantener un funcionamiento renal excelente y desenlaces oncológicos favorables durante el seguimiento intermedio.[16,17] Es mejor que estas cirugías se lleven a cabo en centros especializados con gran experiencia en la atención de personas con formas hereditarias de cáncer de riñón.[18]
Nivel de evidencia: 3di
Procedimientos de ablación para tumores renales
Con las técnicas de ablación térmica se aplica calor o frío extremos en una masa para destruirla. La crioablación (CA) y la ablación por radiofrecuencia (ARF) se usaron por primera vez para el tratamiento de masas renales pequeñas a finales de la década de 1990.[19,20] En el caso de masas renales esporádicas, ambos procedimientos de ablación térmica logran la misma tasa de supervivencia sin recidiva, de cerca del 90 %, lo que hizo que la American Urologic Association recomendara que se tuvieran en cuenta para los pacientes de riesgo alto con masas renales pequeñas (≤4 cm).[21] Las aplicaciones clínicas de los procedimientos de ablación en pacientes con enfermedad de VHL no se han definido con claridad, y la cirugía continúa siendo la intervención más estudiada. Los procedimientos de ablación se usaron por primera vez para el tratamiento del RCC asociado a la enfermedad de VHL en un ensayo de fase II en el que se investigaron los efectos de la ablación en el momento de la resección de la lesión. En este estudio, se trataron 11 tumores, y se corroboró la eliminación del flujo sanguíneo a los tumores mediante ecografía intraoperatoria. En el análisis de patología definitivo se confirmó el efecto terapéutico en todos los tumores.[22] Desde entonces, algunos centros han empezado a utilizar con éxito los procedimientos de ablación térmica para el tratamiento primario y de rescate de pacientes con enfermedad de VHL.[23] En otros centros se encontró que procedimientos como la ARF exhiben una tasa alta de fracaso y se deben reservar para los pacientes con un funcionamiento renal marginal.[24] A pesar de la falta de datos a largo plazo, estos procedimientos se han empezado a usar cada vez más para el tratamiento del RCC en pacientes con enfermedad de VHL.[25,26] En un estudio de una sola institución se evaluaron las tendencias del tratamiento del RCC en 113 pacientes con enfermedad de VHL. Entre 2004 y 2009, el 43 % de los casos se trató con ARF.[26]
Es posible que la ablación térmica desempeñe una función cada vez más importante como terapia de rescate para las personas con un riesgo alto de morbilidad quirúrgica. La CA se evaluó como un tratamiento de rescate en una serie de 14 pacientes para evitar la morbilidad relacionada con la repetición de la cirugía conservadora de nefronas. Los resultados mostraron un cambio mínimo en el funcionamiento renal después del tratamiento con CA. Solo se sospechó recidiva en 4 de 33 tumores (12,1 %) al cabo de una mediana de seguimiento de 37 meses.[27] No obstante, la cirugía después de una ablación térmica es compleja y produce una tasa significativamente más alta de dificultades posoperatorias debido a las adherencias y la cicatrización, en especial, a lo largo del tracto por donde pasan las sondas de ablación.[28,29,30] Los profesionales clínicos deben tener en cuenta que la ablación térmica podría afectar el tratamiento futuro del RCC en personas más jóvenes, que tal vez necesiten otros tratamientos quirúrgicos durante su vida.[18,31]
En síntesis, las aplicaciones clínicas de los procedimientos de ablación en la enfermedad de VHL no están definidas de forma clara, y la cirugía sigue siendo la intervención más estudiada. La evidencia clínica disponible indica que los procedimientos ablativos solo se recomiendan para masas renales pequeñas (≤3 cm) con realce sólido en pacientes de edad avanzada con riesgo quirúrgico alto, en especial aquellos que se enfrentan a cirugía renal de rescate debido a una tasa alta de complicaciones. La edad temprana, un tamaño de tumor superior a 4 cm, los tumores hiliares y las lesiones quísticas se consideran contraindicaciones relativas para la ablación térmica.[32,33]
Nivel de evidencia: 3di
Tratamiento de los feocromocitomas
Vigilancia de los feocromocitomas
Los feocromocitomas a veces son una fuente significativa de morbilidad en los pacientes con enfermedad de VHL porque el exceso de catecolaminas puede provocar efectos cardiovasculares importantes.[34] Es posible, vigilar de manera inocua muchos feocromocitomas pequeños y asintomáticos (≤2 cm) porque tienen tasas de crecimiento y progresión muy lentas. Sin embargo, debido a que en ocasiones los feocromocitomas de personas con la enfermedad de VHL se vuelven malignos (5–10 % metastatizan), es esencial que se realice vigilancia muy estrecha con el fin de garantizar que haya tiempo suficiente para la intervención. Dado que la adrenalectomía parcial es la mejor opción quirúrgica para los feocromocitomas más pequeños, se debe considerar el tamaño y la ubicación anatómica de los mismos cuando se elige el método de vigilancia. Es posible que se necesiten consideraciones especiales para los pacientes que se someterán a futuras cirugías o a un parto. Cuando no se administra un tratamiento alfabloqueante preoperatorio en pacientes con un feocromocitoma, estos pacientes pueden experimentar una liberación masiva de catecolaminas, lo que a veces conlleva inestabilidad hemodinámica.
Intervenciones quirúrgicas para los feocromocitomas
La resección quirúrgica es una herramienta importante para el tratamiento de los feocromocitomas en personas con enfermedad de VHL. Es importante que se obtengan evaluaciones endocrinas detalladas en todos los pacientes y que se administre tratamiento alfabloqueante antes de la resección quirúrgica de los feocromocitomas. A menudo, los medicamentos se deben iniciar y ajustar poco a poco antes de la cirugía para prevenir complicaciones cardiovasculares potencialmente mortales. Para obtener más información en inglés, consultar la sección Preoperative management en Genetics of Endocrine and Neuroendocrine Neoplasias.
Los feocromocitomas en los pacientes con enfermedad de VHL quizás se traten de manera diferente a como se tratan en los pacientes con tumores esporádicos o que tienen otros síndromes de cáncer hereditario. Dado que los feocromocitomas son multifocales y bilaterales en casi el 50 % de los casos en personas con enfermedad de VHL, muchos pacientes se han sometido a una adrenalectomía bilateral y han necesitado reemplazo de corticoesteroides de por vida.[35] La morbilidad relacionada con el reemplazo hormonal de las glándulas suprarrenales y el desarrollo del síndrome de Cushing en personas con enfermedad de VHL ha aumentado el interés por la adrenalectomía parcial con conservación de la corteza. La glándula suprarrenal puede conservarse incluso después de gran movilización y resección tumoral puesto que esta glándula tiene mucha irrigación arterial colateral, además de drenaje venoso.[36] Se necesita conservar de un 15 % a un 30 % del volumen residual de la glándula para producir suficientes hormonas.[37] Usando los procedimientos modernos, la mayoría de las glándulas suprarrenales siguen produciendo cortisol después de una adrenalectomía parcial con conservación de corteza. Cuando queda una glándula suprarrenal solitaria, en una serie se demostró que solo 1 de 13 (8 %) pacientes necesitó reemplazo de corticoesteroides de por vida.[38]
La posibilidad de una adrenalectomía parcial donde quede cáncer residual es una preocupación para los pacientes con feocromocitoma maligno. Sin embargo, en la población con enfermedad de VHL, la tasa de neoplasia maligna para los feocromocitomas es baja (<5 %).[39] La tasa de recidiva local de los feocromocitomas después de una adrenalectomía parcial también es baja (0–33 %). Por lo tanto, cuando es posible y seguro desde el punto de vista oncológico, la mayoría de las directrices recomiendan una adrenalectomía parcial para el tratamiento de los feocromocitomas en pacientes con enfermedad de VHL.
Si se hace una adrenalectomía total, la vena suprarrenal se suele dividir temprano para limitar la liberación de catecolaminas durante la movilización de la glándula. Sin embargo, en una adrenalectomía parcial, la división de la vena suprarrenal puede provocar congestión venosa y compromiso de la glándula.[40] En un paciente con un bloqueo preoperatorio de catecolaminas eficaz, es posible que solo se necesite el pinzamiento de la vena suprarrenal durante la resección y luego, su liberación después de la extirpación del tumor. No está clara la cantidad óptima de parénquima normal adyacente que se debe extirpar. El abordaje quirúrgico inicial con adrenalectomía parcial en pacientes con tumores en la cola o la cabeza de la glándula suprarrenal incluye la extirpación de esta porción de la glándula. Sin embargo, en los pacientes con tumores en el cuerpo de la glándula suprarrenal, esta región se resecó junto con un borde delgado de parénquima normal. A medida que se han aclarado más los datos sobre el riesgo de neoplasia maligna y recidiva local de los feocromocitomas en pacientes con enfermedad de VHL, se ha descrito una técnica que implica una resección enucleativa de la pseudocápsula del tumor (que es similar a las técnicas de enucleación del tumor renal). Este abordaje permite conservar la mayor cantidad de tejido cortical y limitar el compromiso vascular de la glándula suprarrenal residual.[35] No obstante, las preocupaciones por una tasa más alta de recidiva local quizás limiten la aplicación de este abordaje.
Los abordajes quirúrgicos de resección abierta y laparoscópica son inocuos, pero se prefiere la extirpación por laparoscopia siempre que sea posible.[41,42] Los medios de exposición y el abordaje quirúrgico se basan en la ubicación anatómica del tumor. El acceso directo a la glándula suprarrenal y la región paraórtica se logra mediante un abordaje quirúrgico posterior, que es directo, inocuo y eficiente.[43] La exposición adecuada del tumor completo es importante para la extirpación total. Es posible usar asistencia robótica en casos seleccionados porque permite una vista tridimensional y magnificada del área anatómica del tumor.[44] Cuando una persona se ha sometido a varios procedimientos abdominales, a menudo no es factible un abordaje mínimamente invasivo debido a las adherencias. Por lo general, se recomienda la resección abierta para los pacientes con tumores suprarrenales grandes porque la laparoscopia es difícil de realizar dentro de un espacio confinado y aumenta el riesgo de complicaciones. Para obtener más información en inglés sobre los abordajes quirúrgicos para los feocromocitomas, consultar la sección Surgery en Genetics of Endocrine and Neuroendocrine Neoplasias.
Tratamiento de las manifestaciones pancreáticas
Los tumores relacionados con la enfermedad de VHL, como los tumores neuroendocrinos de páncreas, a veces se encuentran de manera fortuita al observar una imagen o al aplicar los protocolos de vigilancia de por vida.[45,46] Las características clínicas de las lesiones pancreáticas (es decir, quísticas vs. sólidas, sintomáticas vs. asintomáticas, tamaño) determinan si los pacientes son aptos para un tratamiento conservador con vigilancia por imágenes o si necesitan una intervención quirúrgica.[47]
Evaluación diagnóstica y pruebas con imágenes para las manifestaciones pancreáticas
Los quistes pancreáticos son benignos y casi nunca necesitan intervención. Los quistes pancreáticos en la enfermedad de VHL no muestran realce en las imágenes y no tienen potencial maligno, independientemente del tamaño. La enfermedad quística difusa del páncreas casi nunca afecta el funcionamiento endocrino. En pocas ocasiones, el quiste reemplaza el páncreas normal y a veces causa una pérdida del funcionamiento exocrino. Cuando se presenta distensión abdominal, cólicos, diarrea o dolor abdominal al ingerir comidas grasosas, es posible obtener estudios enzimáticos en la materia fecal para determinar si se indica la administración de suplementos exocrinos. Las lesiones pancreáticas sólidas o mixtas requieren evaluación y tratamiento especializados, ya que pueden ser cistoadenomas o tumores neuroendocrinos de páncreas. La mayoría de los tumores neuroendocrinos de páncreas no son funcionantes, pero se podría considerar la evaluación de laboratorio con marcadores bioquímicos, como la cromogranina A, durante la evaluación diagnóstica o el seguimiento. Las evaluaciones con imágenes por tomografía computarizada (TC) o resonancia magnética (IRM) con contraste son métodos excelentes para caracterizar las lesiones del páncreas. También se ha utilizado la tomografía por emisión de positrones (TEP) o TC con galio Ga 68-dotate para detectar tumores relacionados con la enfermedad de VHL.[48] Las medidas de desempeño varían según el tamaño de la lesión y el momento de la administración del contraste. Cuando las imágenes transversales no son concluyentes, las imágenes funcionales de medicina nuclear pueden ser útiles para ayudar a diagnosticar la enfermedad metastásica o para diferenciar los adenomas microquísticos sólidos de los tumores neuroendocrinos de páncreas sólidos.[49] La ecografía endoscópica es un método muy sensible. Este procedimiento a veces se ofrece a los pacientes que no pueden recibir un contraste intravenoso o cuando hay preocupación de que una lesión sea un adenoma seroso microquístico sólido, en lugar de un cáncer. Es posible obtener muestras tisulares durante un procedimiento endoscópico, pero casi nunca está indicado.
Vigilancia de las manifestaciones pancreáticas
Los cistoadenomas serosos no tienen potencial maligno y es seguro someterlos a observación. La obstrucción local del colédoco o el conducto pancreático es infrecuente con estas lesiones. Los tumores neuroendocrinos de páncreas sólidos tienen un potencial metastásico bajo, y es posible seguirlos con observación cuando son pequeños, localizados y asintomáticos. La duración y el tipo de imágenes del páncreas dependen del centro, pero los principios generales incluyen imágenes cada 1 a 2 años con el mismo método para permitir comparaciones significativas. Las lesiones pancreáticas con tiempo de duplicación lento y tamaño inferior a 3 cm en pacientes que no tienen variantes patogénica en el exón 3 exhiben los desenlaces más favorables.[50] En una revisión de 175 pacientes con enfermedad de VHL, aquellos con tumores de diámetro inferior a 1,2 cm no presentaron metástasis ni necesitaron cirugía.[51] Los pacientes con tumores más grandes (diámetro de 1,2-3,0 cm) y una variante de cambio de sentido en el gen VHL, en comparación con otros tipos de variantes, tuvieron más probabilidades de presentar metástasis o requerir una intervención quirúrgica. El tamaño del tumor, el tipo de variante y el exón afectado cumplirán en el futuro una función en la determinación del tipo de vigilancia en los pacientes con enfermedad de VHL.
Intervenciones quirúrgicas para las manifestaciones pancreáticas
Los quistes pancreáticos rara vez necesitan intervención quirúrgica, excepto cuando ejercen un efecto de masa. En estos casos raros es posible considerar una aspiración o decorticación. Las indicaciones quirúrgicas en los tumores neuroendocrinos de páncreas varían, pero las intervenciones se ofrecen para reducir el riesgo de diseminación. En una revisión de la historia natural de los tumores neuroendocrinos de páncreas se registró que estos tumores a veces presentan características de crecimiento no lineal.[52] Por lo general, los tumores neuroendocrinos de páncreas se extirpan si miden 3 cm o más (o ≥2 cm si el tumor se encuentra en la cabeza del páncreas).[53] La enucleación del tumor es inocua y eficaz si la lesión no se encuentra cerca del conducto pancreático. Si la enucleación de la lesión no es inocua, se lleva a cabo una pancreatectomía distal. Los tumores en la cabeza del páncreas de 2 cm o más, también se evalúan para resección, porque las lesiones más grandes en esta ubicación son más difíciles de enuclear. Si la lesión se encuentra demasiado cerca del conducto pancreático, se ofrece un procedimiento de Whipple. Para las situaciones poco frecuentes de lesiones multifocales grandes, se podría considerar una pancreatectomía total. Después de la cirugía, si los pacientes presentan disfunción exocrina, la administración de suplementos enzimáticos quizás mejore los síntomas digestivos y el estado nutricional.
Si se encuentran ganglios linfáticos comprometidos durante la cirugía, se deben extirpar. En las personas con tumores neuroendocrinos de páncreas localmente avanzados o metastásicos, es posible considerar la cirugía cuando sea posible hacer una citorreducción significativa. Además, las lesiones hepáticas metastásicas a menudo se tratan con procedimientos de ablación local o resección en algunos pacientes con enfermedad de VHL.
Tratamiento de los hemangioblastomas de retina
Intervenciones para los hemangioblastomas de retina
El tratamiento de los hemangioblastomas de retina incluye láser, terapia fotodinámica y vitrectomía. También se han probado terapias sistémicas y locales.
Fotocoagulación con láser.
La fotocoagulación con láser se usa de manera generalizada para tratar los hemangioblastomas de retina en pacientes con enfermedad de VHL. En una revisión retrospectiva de 304 hemangioblastomas de retina tratados en 100 ojos, se observó que la fotocoagulación con láser tuvo una tasa de control superior al 90 % y fue más eficaz en lesiones más pequeñas de hasta 1 disco de diámetro.[54]
Vitrectomía y retinectomía
En 21 pacientes con desprendimiento de retina grave se conservó la visión en grados variables cuando el tratamiento se hizo con vitrectomía a través de la parte plana con desprendimiento hialoideo posterior, disección de la membrana epirretiniana, e inyección de aceite de silicona o gas con retinectomía, fotocoagulación o crioterapia para extirpar el hemangioblastoma de retina.[55] La vitrectomía a través de la parte plana mejoró la función visual o conservó la visión en 23 pacientes con afección ocular avanzada asociada a la enfermedad de VHL.[56] Sin embargo, la progresión posoperatoria de dicha enfermedad quizás se aceleró en los casos tratados con retinotomía.
Terapia fotodinámica
La terapia fotodinámica redujo el edema macular en una serie de casos de 2 pacientes con hemangioblastomas bilaterales de retina.[57] Estos pacientes obtuvieron un beneficio mínimo, con poco o ningún aumento en la agudeza visual. En una segunda serie de 5 pacientes, entre ellos, 4 con enfermedad de VHL, se usó terapia fotodinámica en 6 ojos.[58] Este tratamiento produjo regresión o estabilización tumoral, así como la disminución de la cantidad de líquido subretiniano y exudados lipídicos en todos los pacientes. Sin embargo, la estabilización o mejora de la agudeza visual solo se presentó en el 50 % de los casos.
Tratamiento intravítreo
El tratamiento intravítreo con bevacizumab produjo estabilización de los hemangioblastomas capilares de retina durante más de 2 años en un informe de casos [59] y mejora de la visión en 1 de 5 ojos en otro informe de casos.[60] El ranibizumab intavítreo no produjo mejoría uniforme en una serie de casos en 5 pacientes.[61] El uso de bevacizumab sistémico produjo un beneficio marginal en informes de casos individuales.[62,63] El tratamiento con sunitinib produjo estabilización visual en 3 pacientes, pero también se asoció con toxicidad simultánea significativa.[64]
Terapia de protones
En un estudio de casos donde se usó la terapia de protones en 8 ojos de 8 pacientes, se observó resolución del edema macular en 7 de 8 pacientes. Todos los ojos tratados conservaron la visión después de una mediana de seguimiento de 84 meses.[65]
Tratamiento de los hemangioblastomas del sistema nervioso central
Vigilancia de los hemangioblastomas del sistema nervioso central
Muchas lesiones pequeñas se encuentran de manera fortuita durante la detección sistemática y es posible que los pacientes permanezcan asintomáticos durante un período largo. En un estudio de seguimiento por un corto plazo, del 35,5 % al 51 % de los hemangioblastomas del SNC permanecieron estables.[66]
En una serie de los Institutos Nacionales de la Salud, los investigadores observaron que el modelo de crecimiento de los hemangioblastomas del sistema nervioso central (SNC) varía, pero el más frecuente es el crecimiento saltatorio (72 %).[67] También se observaron otros patrones de crecimiento, como el crecimiento lineal (6 %) y el crecimiento exponencial (22 %). Se han evaluado los determinantes del crecimiento. La ubicación de las lesiones quizás repercuta en el crecimiento, porque las lesiones en el cerebelo y en el tronco encefálico crecen más rápido que las lesiones en la médula espinal o la cola de caballo.[67] Alrededor del 12 % de los pacientes con hemangioblastomas presentaron quistes peritumorales o intratumorales; el 6,4 % eran sintomáticos y necesitaron tratamiento. El aumento de la carga tumoral o del número de tumores detectados se relacionó con el sexo masculino, un tiempo de seguimiento más prolongado y el genotipo específico (todos los casos P < 0,01). Las deleciones germinales parciales se relacionaron con un número mayor de tumores por paciente que las variantes de cambio de sentido (P < 0,01). Se observaron otros hallazgos interesantes, como aumento del número de tumores por año en los pacientes más jóvenes y una tasa de crecimiento tumoral más rápida en los hombres (P < 0,01).
Otra serie pequeña (n = 52) tuvo como objetivo evaluar las tasas de crecimiento de los hemangioblastomas del SNC bajo vigilancia. Los investigadores encontraron que la presencia de síntomas era el único predictor independiente del crecimiento.[66] A medida que la mayoría de los hemangioblastomas del SNC crecen y se vuelven sintomáticos, la tasa de tratamiento también aumenta, con tasas de intervención a 1, 3 y 7 años que alcanzan el 11,5 %, el 50,1 % y el 73 %, respectivamente.
Intervenciones quirúrgicas para los hemangioblastomas del sistema nervioso central
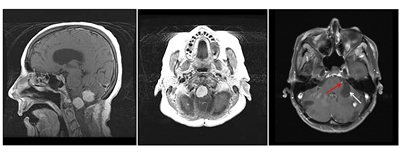
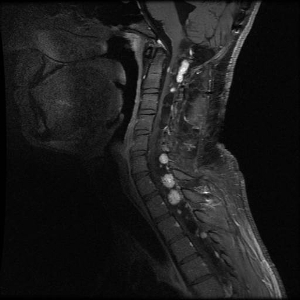
El abordaje de tratamiento estándar ha sido la resección quirúrgica de los hemangioblastomas de cerebelo o de médula espinal. Si bien la resección quirúrgica de los tumores por lo general se realiza antes de la aparición de los síntomas neurológicos,[68] esto varía según el centro y puede que se modifique de acuerdo a ciertos factores como edema, ubicación del tumor, hidrocefalia y tasa de crecimiento del tumor. A menudo, el acceso quirúrgico para las lesiones de la médula espinal es posterior y exige una laminectomía. Debido a que los pacientes suelen necesitar varias operaciones durante la vida, la pérdida de apoyo estructural a veces causa inestabilidad vertebral progresiva y es posible que exija estabilización o fusión.[69] En el caso de lesiones cerebelosas, el abordaje depende de la orientación lateral del tumor, en muchos casos es posible acceder a través de una incisión suboccipital en la línea media. La embolización preoperatoria a veces se hace para reducir la hemorragia, pero este abordaje depende de la preferencia del cirujano.[70]
Radioterapia para los hemangioblastomas del sistema nervioso central
Debido a que los pacientes a veces tienen tumores múltiples y requieren varios procedimientos quirúrgicos, la radioterapia externa se ha convertido en una alternativa cuando no es factible la cirugía de resección. La radiocirugía estereotáctica es un abordaje de uso generalizado para el tratamiento del hemangioblastoma.[71] En series retrospectivas se demostró que la radiocirugía redujo el tamaño de más del 50 % de las lesiones tratadas, con una tasa de complicaciones baja.[71] En un estudio prospectivo del NCI se evaluó el control local de las lesiones tratadas. Es posible que se necesiten series a largo plazo para evaluar la eficacia de la radiocirugía, porque algunos de los tumores exhiben un patrón de crecimiento saltatorio. En esta serie, el 33 % de los tumores asintomáticos tratados de menos de 1 centímetro progresaron durante el seguimiento. Debido a las preocupaciones del control local a largo plazo, los autores concluyeron que la radiocirugía estereotáctica se debe reservar para el tratamiento de los tumores que no se pueden someter a resección quirúrgica.[72] En otras series se observaron mejores desenlaces.[73] Sin embargo, no está claro si esto depende de la definición de control local, que varía entre los estudios.
Tratamiento de los tumores de saco endolinfático
Hay pocos datos sobre el tratamiento de los tumores de saco endolinfático, que en su mayoría provienen de series de casos en donde se describe el tratamiento quirúrgico de tumores esporádicos y tumores asociados a la enfermedad de VHL. Debido a que el compromiso audiovestibular no depende del tamaño del tumor y se puede presentar con tumores pequeños, por lo general se prefiere la intervención temprana. Es posible que la intervención temprana también reduzca al mínimo el riesgo de invasión de las estructuras circundantes y aumente la probabilidad de una resección completa. En un metaanálisis se evaluaron los desenlaces en 82 estudios donde se trataron 252 tumores.[74] Se observó un número limitado de pacientes y casi todos presentaron progresión clínica, lo que genera desconfianza sobre este abordaje. La cirugía ha sido el tratamiento principal para los tumores de saco endolinfático, y la resección completa es la mejor opción para evitar la recidiva. Se ha utilizado la embolización preoperatoria para minimizar el riesgo de morbilidad perioperatoria y hemorragia. El abordaje quirúrgico a menudo depende de la preferencia del cirujano, la ubicación del tumor y del estado auditivo inicial del paciente. La resección completa de la enfermedad no siempre es posible debido a la extensión del tumor y la invasión a las estructuras circundantes. Por lo tanto, las tasas de recidiva de los tumores de saco endolinfático después de la cirugía son altas (16 %). Si bien la radiación no suele ser una opción de tratamiento de primera línea, a menudo se usa como tratamiento adyuvante. Los datos sobre los abordajes de terapia sistémica para los tumores de saco endolinfático son limitados.
Terapia sistémica específica para la enfermedad de Von Hippel-Lindau localizada
Los pacientes con enfermedad de VHL a menudo requieren múltiples tratamientos locales para las manifestaciones de la enfermedad. Las intervenciones quirúrgicas repetidas contribuyen a la morbilidad y, a menudo, pueden causar daños irreversibles en los órganos afectados. Se puede presentar la pérdida permanente de funcionamiento en los siguientes órganos:
- Deterioro visual (retinas).
- Enfermedad renal crónica (riñones).
- Insuficiencia suprarrenal (glándulas suprarrenales).
- Diabetes y deficiencia exocrina pancreática (páncreas).
- Complicaciones neurológicas como alteraciones motoras o sensoriales (encéfalo y columna vertebral).
Los investigadores han buscado una terapia sistémica que reduzca o elimine la necesidad de intervenciones locales. La comprensión de las características biológicas de la enfermedad de VHL ha llevado al desarrollo de terapias dirigidas que interfieren con la cascada de señalización resultante asociada con la tumorogénesis.[75] Primero se desarrollaron fármacos eficaces dirigidos a la vía del factor inducible por hipoxia (HIF) de la enfermedad de VHL para tratar a los pacientes con ccRCC en estadio esporádico avanzado. Más tarde, se investigó la utilidad clínica de estos fármacos en personas con enfermedad de VHL que presentaban diversas manifestaciones relacionadas con la enfermedad.
En la investigación inicial sobre el tratamiento con tanespimicina (17-AAG), se destacó que la administración intravenosa de medicamentos tal vez no sea la preferida por algunos pacientes. La toxicidad modesta y la tolerabilidad deficiente relacionadas con 17-AAG pueden disuadir a los pacientes sanos (con otras opciones quirúrgicas) de usar este tratamiento.[76] Posteriormente, se evaluó una variedad de fármacos dirigidos a la vía del factor de crecimiento endotelial vascular (VEGF). La mayoría de estos fármacos demostraron actividad contra los tumores renales, pero solo una actividad modesta o ausente contra los hemangioblastomas del SNC, los tumores neuroendocrinos de páncreas y otras manifestaciones relacionadas con la enfermedad de VHL. Se administró un inhibidor de tirosina–cinasas de primera generación, el sunitinib, a pacientes con enfermedad de VHL durante un máximo de 4 ciclos (se administró durante 4 semanas, y luego se dieron 2 semanas de descanso). Este medicamento se evaluó por primera vez en una cohorte de 15 participantes con diversas manifestaciones relacionadas con la enfermedad de VHL; ninguno de los participantes necesitaba intervención médica inmediata.[77] Si bien los tumores no crecieron en estas personas (>90 % de los participantes), la toxicidad (incluso fatiga) condujo a reducciones frecuentes de la dosis de sunitinib en 10 de 15 participantes. La tasa de respuesta general (TRG) fue del 33 % (6 de 18 tumores) en los tumores renales localizados, lo que demuestra la posibilidad de que el tratamiento con sunitinib influya sobre la necesidad de intervención local. Desafortunadamente, no se observaron respuestas (según lo definido por los Response Evaluation Criteria in Solid Tumors [RECIST]) en los hemangioblastomas del SNC. En una revisión retrospectiva de 14 pacientes que habían recibido sunitinib, no se observaron respuestas en 11 pacientes con hemangioblastomas de cerebelo y 8 pacientes con hemangioblastomas de médula espinal.[78] En un estudio de fase II con 37 pacientes, se evaluó el vandetanib, un inhibidor de tirosina–cinasas con una especificidad amplia por varias dianas (incluso el receptor del factor de crecimiento endotelial vascular [VEGFR] y el receptor del factor de crecimiento epidérmico [EGFR]). En este estudio, el vandetanib se relacionó con actividad limitada y toxicidad, lo que requirió interrupciones frecuentes del tratamiento y reducciones de la dosis.[79] El pazopanib, un inhibidor de tirosina–cinasas de segunda generación con un perfil de toxicidad mejor que el del sunitinib, se estudió de manera similar en pacientes con enfermedad de VHL.[80] Una cohorte de 31 pacientes se trató con pazopanib durante 24 semanas con la opción de continuar recibiendo este tratamiento. Se observaron respuestas (de acuerdo a las definiciones de RECIST) en el 52 % de los tumores renales y el 53 % de las lesiones pancreáticas. En este estudio se incluyeron tumores neuroendocrinos de páncreas malignos y cistoadenomas serosos pancreáticos benignos. Sin embargo, hubo poca actividad contra los hemangioblastomas del SNC, y se observó una TRG del 4 %. El pazopanib también se toleró de manera precaria; solo el 23 % de los participantes optaron por continuar el tratamiento más allá del período inicial de 24 semanas.
Si bien la terapia anti-VEGF se administra de manera sistémica para la mayoría de las neoplasias relacionadas con la enfermedad de VHL, se puede administrar directamente en el ojo en casos de tumores de retina relacionados con esta enfermedad. Se evaluó la administración intravítrea del pegaptanib (terapia anti-VEGF) en 5 pacientes con hemangioblastomas de retina relacionados con la enfermedad de VHL.[81] Solo 2 pacientes pudieron completar el tratamiento con pegaptanib y no se observaron respuestas en los tumores primarios de los pacientes. Dos pacientes presentaron disminución del engrosamiento de la retina y un número reducido de exudados duros. Si bien la Administración de Alimentos y Medicamentos de los Estados Unidos aprobó el pegaptanib para el tratamiento de la degeneración macular, no está aprobado para el tratamiento de las lesiones de retina relacionadas con la enfermedad de VHL.
La investigación dirigida a las consecuencias del aumento regulado de HIF (debido a la inactivación del gen VHL) solo tuvo un éxito moderado. Por lo tanto, los esfuerzos recientes se han centrado en abordar las consecuencias directas de la pérdida de actividad del gen VHL. En muchos estudios se demostró que múltiples moléculas de HIF regulan de manera diferenciada la carcinogénesis, y que HIF2 es el mediador más crítico.[82] Este factor de transcripción se convirtió en una diana prometedora para el tratamiento, pero esto solo fue posible cuando se desarrolló una nueva clase de fármacos que pueden inhibir de manera selectiva el HIF2-α. Estos fármacos se diseñaron para encajar en el bolsillo de unión del dominio per-ARNT-SIM (PAS)-B de HIF2-α. Esto evitó la dimerización de HIF1-β con ARNT y mostró actividad en modelos de laboratorio.[83] En última instancia, esto condujo al desarrollo de belzutifán, un fármaco oral que se evaluó en un estudio internacional de fase II llevado a cabo en 11 centros con participación de 61 pacientes con RCC asociado a la enfermedad de VHL.[84] A diferencia de los fármacos dirigidos a la vía del VEGF, el belzutifán se toleró bien en los participantes y tuvo una tasa baja de toxicidad de grado alto. El acontecimiento adverso de grado 3 más común fue la anemia (8 % de los pacientes) y la mayoría de los pacientes siguieron tomando belzutifán después de la publicación de este artículo (mediana de período de seguimiento, 21,8 meses). Se observaron respuestas en el 49 % de los tumores renales, el 77 % de los tumores de páncreas y el 30 % de los tumores del SNC. Todos los tumores de retina (16/16) mejoraron después del tratamiento con belzutifán. La FDA aprobó el belzutifán para los hemangioblastomas de riñón, de páncreas y del SNC relacionados con la enfermedad de VHL que no requieren intervención quirúrgica inmediata, según las mediciones de actividad y tolerabilidad del ensayo. Se anticipa que otras investigaciones revelarán la eficacia y la toxicidad a largo plazo de este fármaco. El belzutifán se usa ahora como complemento del tratamiento quirúrgico en pacientes con tumores asociados a la enfermedad de VHL que se limitan a un órgano. No está claro cómo se utilizará el belzutifán en la práctica clínica y qué proveedores lo prescribirán a los pacientes.[76]
Tratamiento de la enfermedad de Von Hippel-Lindau durante el embarazo
En 2 estudios se examinó el efecto del embarazo en la progresión del hemangioblastoma en pacientes con enfermedad de Von Hippel-Lindau (VHL).[85,86] En un estudio retrospectivo de los Países bajos se examinaron los registros de 29 pacientes con enfermedad de VHL que quedaron embarazadas 48 veces (49 recién nacidos) entre 1966 y 2010 (40 % quedaron embarazadas antes de 1990). Se obtuvieron registros de las imágenes del 31 % de los embarazos. Los investigadores notificaron que en el 17 % de los embarazos se registraron complicaciones asociadas a la enfermedad de VHL, entre ellos, 3 pacientes con hemangioblastomas craneoespinales que presentaron un cambio significativo en la puntuación de progresión tumoral (P = 0,049) antes y después del embarazo.[85] Sin embargo, los hallazgos de este estudio contrastan con los de una investigación prospectiva pequeña.[86] Hasta que se cuente con un estudio prospectivo internacional de gran escala, estas investigaciones indican el uso de un abordaje conservador que incluya vigilancia médica durante el embarazo.
Referencias:
- Goldfarb DA, Neumann HP, Penn I, et al.: Results of renal transplantation in patients with renal cell carcinoma and von Hippel-Lindau disease. Transplantation 64 (12): 1726-9, 1997.
- Fetner CD, Barilla DE, Scott T, et al.: Bilateral renal cell carcinoma in von Hippel-Lindau syndrome: treatment with staged bilateral nephrectomy and hemodialysis. J Urol 117 (4): 534-6, 1977.
- Antony MB, Rompré-Brodeur A, Chaurasia A, et al.: Outcomes of and indications for renal transplantation in patients with von Hippel Lindau disease. Urol Oncol 41 (12): 487.e1-487.e6, 2023.
- Pearson JC, Weiss J, Tanagho EA: A plea for conservation of kidney in renal adenocarcinoma associated with von Hippel-Lindau disease. J Urol 124 (6): 910-2, 1980.
- Loughlin KR, Gittes RF: Urological management of patients with von Hippel-Lindau's disease. J Urol 136 (4): 789-91, 1986.
- Steinbach F, Novick AC, Zincke H, et al.: Treatment of renal cell carcinoma in von Hippel-Lindau disease: a multicenter study. J Urol 153 (6): 1812-6, 1995.
- Walther MM, Thompson N, Linehan W: Enucleation procedures in patients with multiple hereditary renal tumors. World J Urol 13 (4): 248-50, 1995.
- Walther MM, Choyke PL, Glenn G, et al.: Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol 161 (5): 1475-9, 1999.
- Duffey BG, Choyke PL, Glenn G, et al.: The relationship between renal tumor size and metastases in patients with von Hippel-Lindau disease. J Urol 172 (1): 63-5, 2004.
- Peng X, Chen J, Wang J, et al.: Natural history of renal tumours in von Hippel-Lindau disease: a large retrospective study of Chinese patients. J Med Genet 56 (6): 380-387, 2019.
- Zhang J, Pan JH, Dong BJ, et al.: Active surveillance of renal masses in von Hippel-Lindau disease: growth rates and clinical outcome over a median follow-up period of 56 months. Fam Cancer 11 (2): 209-14, 2012.
- Jilg CA, Neumann HP, Gläsker S, et al.: Growth kinetics in von Hippel-Lindau-associated renal cell carcinoma. Urol Int 88 (1): 71-8, 2012.
- Neumann HP, Bender BU, Berger DP, et al.: Prevalence, morphology and biology of renal cell carcinoma in von Hippel-Lindau disease compared to sporadic renal cell carcinoma. J Urol 160 (4): 1248-54, 1998.
- Fadahunsi AT, Sanford T, Linehan WM, et al.: Feasibility and outcomes of partial nephrectomy for resection of at least 20 tumors in a single renal unit. J Urol 185 (1): 49-53, 2011.
- Choyke PL, Pavlovich CP, Daryanani KD, et al.: Intraoperative ultrasound during renal parenchymal sparing surgery for hereditary renal cancers: a 10-year experience. J Urol 165 (2): 397-400, 2001.
- Bratslavsky G, Liu JJ, Johnson AD, et al.: Salvage partial nephrectomy for hereditary renal cancer: feasibility and outcomes. J Urol 179 (1): 67-70, 2008.
- Johnson A, Sudarshan S, Liu J, et al.: Feasibility and outcomes of repeat partial nephrectomy. J Urol 180 (1): 89-93; discussion 93, 2008.
- Shuch B, Linehan WM, Bratslavsky G: Repeat partial nephrectomy: surgical, functional and oncological outcomes. Curr Opin Urol 21 (5): 368-75, 2011.
- Gill IS, Novick AC, Soble JJ, et al.: Laparoscopic renal cryoablation: initial clinical series. Urology 52 (4): 543-51, 1998.
- McGovern FJ, Wood BJ, Goldberg SN, et al.: Radio frequency ablation of renal cell carcinoma via image guided needle electrodes. J Urol 161 (2): 599-600, 1999.
- Campbell SC, Novick AC, Belldegrun A, et al.: Guideline for management of the clinical T1 renal mass. J Urol 182 (4): 1271-9, 2009.
- Walther MC, Shawker TH, Libutti SK, et al.: A phase 2 study of radio frequency interstitial tissue ablation of localized renal tumors. J Urol 163 (5): 1424-7, 2000.
- Shingleton WB, Sewell PE: Percutaneous renal cryoablation of renal tumors in patients with von Hippel-Lindau disease. J Urol 167 (3): 1268-70, 2002.
- Park BK, Kim CK: Percutaneous radio frequency ablation of renal tumors in patients with von Hippel-Lindau disease: preliminary results. J Urol 183 (5): 1703-7, 2010.
- Chan VW, Lenton J, Smith J, et al.: Multimodal image-guided ablation on management of renal cancer in Von-Hippel-Lindau syndrome patients from 2004 to 2021 at a specialist centre: A longitudinal observational study. Eur J Surg Oncol 48 (3): 672-679, 2022.
- Joly D, Méjean A, Corréas JM, et al.: Progress in nephron sparing therapy for renal cell carcinoma and von Hippel-Lindau disease. J Urol 185 (6): 2056-60, 2011.
- Yang B, Autorino R, Remer EM, et al.: Probe ablation as salvage therapy for renal tumors in von Hippel-Lindau patients: the Cleveland Clinic experience with 3 years follow-up. Urol Oncol 31 (5): 686-92, 2013.
- Nguyen CT, Lane BR, Kaouk JH, et al.: Surgical salvage of renal cell carcinoma recurrence after thermal ablative therapy. J Urol 180 (1): 104-9; discussion 109, 2008.
- Karam JA, Wood CG, Compton ZR, et al.: Salvage surgery after energy ablation for renal masses. BJU Int 115 (1): 74-80, 2015.
- Kowalczyk KJ, Hooper HB, Linehan WM, et al.: Partial nephrectomy after previous radio frequency ablation: the National Cancer Institute experience. J Urol 182 (5): 2158-63, 2009.
- Shuch B, Singer EA, Bratslavsky G: The surgical approach to multifocal renal cancers: hereditary syndromes, ipsilateral multifocality, and bilateral tumors. Urol Clin North Am 39 (2): 133-48, v, 2012.
- Dominguez-Escrig JL, Sahadevan K, Johnson P: Cryoablation for small renal masses. Adv Urol : 479495, 2008.
- Aron M, Gill IS: Minimally invasive nephron-sparing surgery (MINSS) for renal tumours. Part II: probe ablative therapy. Eur Urol 51 (2): 348-57, 2007.
- Sanford T, Gomella PT, Siddiqui R, et al.: Long term outcomes for patients with von Hippel-Lindau and Pheochromocytoma: defining the role of active surveillance. Urol Oncol 39 (2): 134.e1-134.e8, 2021.
- Benhammou JN, Boris RS, Pacak K, et al.: Functional and oncologic outcomes of partial adrenalectomy for pheochromocytoma in patients with von Hippel-Lindau syndrome after at least 5 years of followup. J Urol 184 (5): 1855-9, 2010.
- Baghai M, Thompson GB, Young WF, et al.: Pheochromocytomas and paragangliomas in von Hippel-Lindau disease: a role for laparoscopic and cortical-sparing surgery. Arch Surg 137 (6): 682-8; discussion 688-9, 2002.
- Brauckhoff M, Gimm O, Thanh PN, et al.: Critical size of residual adrenal tissue and recovery from impaired early postoperative adrenocortical function after subtotal bilateral adrenalectomy. Surgery 134 (6): 1020-7; discussion 1027-8, 2003.
- Walther MM, Keiser HR, Choyke PL, et al.: Management of hereditary pheochromocytoma in von Hippel-Lindau kindreds with partial adrenalectomy. J Urol 161 (2): 395-8, 1999.
- Fallon SC, Feig D, Lopez ME, et al.: The utility of cortical-sparing adrenalectomy in pheochromocytomas associated with genetic syndromes. J Pediatr Surg 48 (6): 1422-5, 2013.
- Roukounakis N, Dimas S, Kafetzis I, et al.: Is preservation of the adrenal vein mandatory in laparoscopic adrenal-sparing surgery? JSLS 11 (2): 215-8, 2007 Apr-Jun.
- Timmers HJ, Gimenez-Roqueplo AP, Mannelli M, et al.: Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer 16 (2): 391-400, 2009.
- Vargas HI, Kavoussi LR, Bartlett DL, et al.: Laparoscopic adrenalectomy: a new standard of care. Urology 49 (5): 673-8, 1997.
- Perrier ND, Kennamer DL, Bao R, et al.: Posterior retroperitoneoscopic adrenalectomy: preferred technique for removal of benign tumors and isolated metastases. Ann Surg 248 (4): 666-74, 2008.
- Dickson PV, Jimenez C, Chisholm GB, et al.: Posterior retroperitoneoscopic adrenalectomy: a contemporary American experience. J Am Coll Surg 212 (4): 659-65; discussion 665-7, 2011.
- Binderup ML, Bisgaard ML, Harbud V, et al.: Von Hippel-Lindau disease (vHL). National clinical guideline for diagnosis and surveillance in Denmark. 3rd edition. Dan Med J 60 (12): B4763, 2013.
- Kruizinga RC, Sluiter WJ, de Vries EG, et al.: Calculating optimal surveillance for detection of von Hippel-Lindau-related manifestations. Endocr Relat Cancer 21 (1): 63-71, 2014.
- Keutgen XM, Hammel P, Choyke PL, et al.: Evaluation and management of pancreatic lesions in patients with von Hippel-Lindau disease. Nat Rev Clin Oncol 13 (9): 537-49, 2016.
- Shell J, Tirosh A, Millo C, et al.: The utility of 68Gallium-DOTATATE PET/CT in the detection of von Hippel-Lindau disease associated tumors. Eur J Radiol 112: 130-135, 2019.
- Sadowski SM, Weisbrod AB, Ellis R, et al.: Prospective evaluation of the clinical utility of 18-fluorodeoxyglucose PET CT scanning in patients with von hippel-lindau-associated pancreatic lesions. J Am Coll Surg 218 (5): 997-1003, 2014.
- Blansfield JA, Choyke L, Morita SY, et al.: Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs). Surgery 142 (6): 814-8; discussion 818.e1-2, 2007.
- Tirosh A, Sadowski SM, Linehan WM, et al.: Association of VHL Genotype With Pancreatic Neuroendocrine Tumor Phenotype in Patients With von Hippel-Lindau Disease. JAMA Oncol 4 (1): 124-126, 2018.
- Weisbrod AB, Kitano M, Thomas F, et al.: Assessment of tumor growth in pancreatic neuroendocrine tumors in von Hippel Lindau syndrome. J Am Coll Surg 218 (2): 163-9, 2014.
- Libutti SK, Choyke PL, Bartlett DL, et al.: Pancreatic neuroendocrine tumors associated with von Hippel Lindau disease: diagnostic and management recommendations. Surgery 124 (6): 1153-9, 1998.
- Krivosic V, Kamami-Levy C, Jacob J, et al.: Laser photocoagulation for peripheral retinal capillary hemangioblastoma in von Hippel-Lindau disease. Ophthalmol Retina 1 (1): 59-67, 2017. Also available online. Last accessed October 22, 2024.
- Gaudric A, Krivosic V, Duguid G, et al.: Vitreoretinal surgery for severe retinal capillary hemangiomas in von hippel-lindau disease. Ophthalmology 118 (1): 142-9, 2011.
- Krzystolik K, Stopa M, Kuprjanowicz L, et al.: PARS PLANA VITRECTOMY IN ADVANCED CASES OF VON HIPPEL-LINDAU EYE DISEASE. Retina 36 (2): 325-34, 2016.
- Papastefanou VP, Pilli S, Stinghe A, et al.: Photodynamic therapy for retinal capillary hemangioma. Eye (Lond) 27 (3): 438-42, 2013.
- Sachdeva R, Dadgostar H, Kaiser PK, et al.: Verteporfin photodynamic therapy of six eyes with retinal capillary haemangioma. Acta Ophthalmol 88 (8): e334-40, 2010.
- Ach T, Thiemeyer D, Hoeh AE, et al.: Intravitreal bevacizumab for retinal capillary haemangioma: longterm results. Acta Ophthalmol 88 (4): e137-8, 2010.
- Slim E, Antoun J, Kourie HR, et al.: Intravitreal bevacizumab for retinal capillary hemangioblastoma: A case series and literature review. Can J Ophthalmol 49 (5): 450-7, 2014.
- Wong WT, Liang KJ, Hammel K, et al.: Intravitreal ranibizumab therapy for retinal capillary hemangioblastoma related to von Hippel-Lindau disease. Ophthalmology 115 (11): 1957-64, 2008.
- von Buelow M, Pape S, Hoerauf H: Systemic bevacizumab treatment of a juxtapapillary retinal haemangioma. Acta Ophthalmol Scand 85 (1): 114-6, 2007.
- Wackernagel W, Lackner EM, Pilz S, et al.: von Hippel-Lindau disease: treatment of retinal haemangioblastomas by targeted therapy with systemic bevacizumab. Acta Ophthalmol 88 (7): e271-2, 2010.
- Knickelbein JE, Jacobs-El N, Wong WT, et al.: Systemic Sunitinib Malate Treatment for Advanced Juxtapapillary Retinal Hemangioblastomas Associated with von Hippel-Lindau Disease. Ophthalmol Retina 1 (3): 181-187, 2017 May-Jun.
- Seibel I, Cordini D, Hager A, et al.: Long-term results after proton beam therapy for retinal papillary capillary hemangioma. Am J Ophthalmol 158 (2): 381-6, 2014.
- Byun J, Yoo HJ, Kim JH, et al.: Growth rate and fate of untreated hemangioblastomas: clinical assessment of the experience of a single institution. J Neurooncol 144 (1): 147-154, 2019.
- Lonser RR, Butman JA, Huntoon K, et al.: Prospective natural history study of central nervous system hemangioblastomas in von Hippel-Lindau disease. J Neurosurg 120 (5): 1055-62, 2014.
- Harati A, Satopää J, Mahler L, et al.: Early microsurgical treatment for spinal hemangioblastomas improves outcome in patients with von Hippel-Lindau disease. Surg Neurol Int 3: 6, 2012.
- Asthagiri AR, Mehta GU, Butman JA, et al.: Long-term stability after multilevel cervical laminectomy for spinal cord tumor resection in von Hippel-Lindau disease. J Neurosurg Spine 14 (4): 444-52, 2011.
- Das JM, Kesavapisharady K, Sadasivam S, et al.: Microsurgical Treatment of Sporadic and von Hippel-Lindau Disease Associated Spinal Hemangioblastomas: A Single-Institution Experience. Asian Spine J 11 (4): 548-555, 2017.
- Kano H, Shuto T, Iwai Y, et al.: Stereotactic radiosurgery for intracranial hemangioblastomas: a retrospective international outcome study. J Neurosurg 122 (6): 1469-78, 2015.
- Asthagiri AR, Mehta GU, Zach L, et al.: Prospective evaluation of radiosurgery for hemangioblastomas in von Hippel-Lindau disease. Neuro Oncol 12 (1): 80-6, 2010.
- Liebenow B, Tatter A, Dezarn WA, et al.: Gamma Knife Stereotactic Radiosurgery favorably changes the clinical course of hemangioblastoma growth in von Hippel-Lindau and sporadic patients. J Neurooncol 142 (3): 471-478, 2019.
- Tang JD, Grady AJ, Nickel CJ, et al.: Systematic Review of Endolymphatic Sac Tumor Treatment and Outcomes. Otolaryngol Head Neck Surg 168 (3): 282-290, 2023.
- Linehan WM, Pinto PA, Bratslavsky G, et al.: Hereditary kidney cancer: unique opportunity for disease-based therapy. Cancer 115 (10 Suppl): 2252-61, 2009.
- Shuch B: HIF2 Inhibition for von-Hippel Lindau Associated Kidney Cancer: Will Urology Lead or Follow? Urol Oncol 39 (5): 277-280, 2021.
- Jonasch E, McCutcheon IE, Waguespack SG, et al.: Pilot trial of sunitinib therapy in patients with von Hippel-Lindau disease. Ann Oncol 22 (12): 2661-6, 2011.
- Roma A, Maruzzo M, Basso U, et al.: First-Line sunitinib in patients with renal cell carcinoma (RCC) in von Hippel-Lindau (VHL) disease: clinical outcome and patterns of radiological response. Fam Cancer 14 (2): 309-16, 2015.
- Stamatakis L, Shuch B, Singer EA, et al.: Phase II trial of vandetanib in Von Hippel-Lindau-associated renal cell carcinoma. J Clin Oncol 31 (15 Suppl): 4584, 2013. Also available online. Last accessed October 22, 2024.
- Jonasch E, McCutcheon IE, Gombos DS, et al.: Pazopanib in patients with von Hippel-Lindau disease: a single-arm, single-centre, phase 2 trial. Lancet Oncol 19 (10): 1351-1359, 2018.
- Dahr SS, Cusick M, Rodriguez-Coleman H, et al.: Intravitreal anti-vascular endothelial growth factor therapy with pegaptanib for advanced von Hippel-Lindau disease of the retina. Retina 27 (2): 150-8, 2007.
- Kondo K, Kim WY, Lechpammer M, et al.: Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol 1 (3): E83, 2003.
- Courtney KD, Infante JR, Lam ET, et al.: Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2α Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J Clin Oncol 36 (9): 867-874, 2018.
- Jonasch E, Donskov F, Iliopoulos O, et al.: Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N Engl J Med 385 (22): 2036-2046, 2021.
- Frantzen C, Kruizinga RC, van Asselt SJ, et al.: Pregnancy-related hemangioblastoma progression and complications in von Hippel-Lindau disease. Neurology 79 (8): 793-6, 2012.
- Ye DY, Bakhtian KD, Asthagiri AR, et al.: Effect of pregnancy on hemangioblastoma development and progression in von Hippel-Lindau disease. J Neurosurg 117 (5): 818-24, 2012.