Genetics of Prostate Cancer (PDQ®): Genetics - Health Professional Information [NCI]
Germline Genetics for Prostate Cancer
Prostate cancer is highly heritable. More than half of an individual's prostate cancer risk is inherited from one's parents.[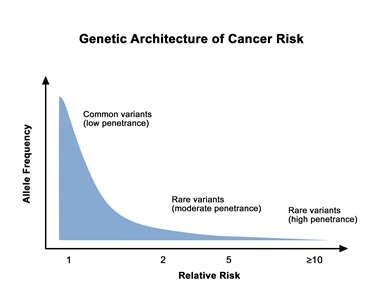
Genetic architecture of cancer risk. This graph depicts the general finding of a low relative risk associated with common, low-penetrance genetic variants, such as single-nucleotide polymorphisms identified in genome-wide association studies, and a higher relative risk associated with rare, high-penetrance genetic variants, such as mutations in the BRCA1/ BRCA2 genes associated with hereditary breast and ovarian cancer and the mismatch repair genes associated with Lynch syndrome.
Clinically Relevant Genes for Prostate Cancer
BRCA1andBRCA2
Studies of male carriers of BRCA1 and BRCA2 pathogenic variants demonstrate that these individuals have a higher risk of prostate cancer and other cancers.[
BRCA–associated prostate cancer risk
The risk of prostate cancer in carriers of BRCA pathogenic variants has been studied in various settings.
In an effort to clarify the relationship between BRCA pathogenic variants and prostate cancer risk, findings from a systematic review and meta-analysis are summarized in
| Population | Number of Studies | Fixed-Effect Pooled Prostate Cancer RR (95% CI) | Random-Effect Pooled Prostate Cancer RR (95% CI) | I2 |
|---|---|---|---|---|
| CI = confidence interval; RR = relative risk. | ||||
| a Adapted from Nyberg et al. | ||||
| BRCA1 | ||||
| All | 20 | 1.57 (1.30–1.91) | 1.69 (1.30–2.20) | 30% |
| Unselected for age, aggressive prostate cancer, or prostate cancer family history | 15 | 1.43 (1.71–1.75) | 1.47 (1.13–1.91) | 25% |
| Unselected for age, aggressive prostate cancer, or prostate cancer family historyand did not use historical controls | 13 | 1.32 (1.07–1.64) | 1.33 (1.05–1.69) | 8% |
| Prostate cancer diagnosed <65 y | 4 | 2.21 (1.47–3.30) | 2.19 (1.21–3.98) | 57% |
| Prostate cancer diagnosed >65 y | 3 | 1.18 (0.83–1.70) | 1.43 (0.71–2.87) | 65% |
| BRCA2 | ||||
| All | 21 | 5.24 (4.63–5.49) | 3.94 (2.79–5.56) | 83% |
| Unselected for age, aggressive prostate cancer, or prostate cancer family history | 15 | 3.87 (3.34–4.47) | 3.33 (2.57–4.33) | 58% |
| Prostate cancer diagnosed <65 y | 5 | 6.37 (4.81–8.43) | 5.28 (3.10–9.00) | 63% |
| Prostate cancer diagnosed >65 y | 3 | 3.74 (2.82–4.96) | 3.74 (2.82–4.96) | 0% |
Prevalence ofBRCAfounder pathogenic variants in men with prostate cancer
Ashkenazi Jewish population
Several studies in Israel and in North America have analyzed the frequency of BRCAfounder pathogenic variants among Ashkenazi Jewish (AJ) men with prostate cancer.[
| Population | Number of Studies | Fixed-Effect Pooled Prostate Cancer RR (95% CI) | Random-Effect Pooled Prostate Cancer RR (95% CI) | I2 |
|---|---|---|---|---|
| CI = confidence interval; RR = relative risk. | ||||
| a Adapted from Nyberg et al. | ||||
| BRCA1 | ||||
| All | 3 | 1.12 (0.55–2.31) | 1.12 (0.55–2.31) | 0% |
| BRCA2 | ||||
| All | 6 | 2.08 (1.38–3.12) | 2.08 (1.38–3.12) | 0% |
This systematic review and meta-analysis provide further evidence that prostate cancer occurs more often in Ashkenazi Jewish BRCA founder variant carriers and suggests that prostate cancer risk may be greater in men with BRCA2 6174delT founder pathogenic variants than in men with BRCA1 85delAG or BRCA1 5382insC founder pathogenic variants.
Other populations
The association between prostate cancer and pathogenic variants in BRCA1 and BRCA2 has also been studied in other populations.
| Population | Number of Studies | Fixed-Effect Pooled Prostate Cancer RR (95% CI) | Random-Effect Pooled Prostate Cancer RR (95% CI) | I2 |
|---|---|---|---|---|
| CI = confidence interval; RR = relative risk. | ||||
| a Adapted from Nyberg et al. | ||||
| BRCA1 | ||||
| Non-Ashkenazi European Ancestry | 8 | 1.30 (1.03–1.64) | 1.30 (0.95–1.79) | 30% |
| African Ancestry | 1 | 1.11 (0.09–13.61) | 1.11 (0.09–13.61) | - |
| Asian Ancestry | 1 | 2.27 (0.92–5.59) | 2.27 (0.92–5.59) | - |
| BRCA2 | ||||
| Non-Ashkenazi European Ancestry | 7 | 4.07 (3.45–4.80) | 3.69 (2.71–5.04) | 66% |
| African Ancestry | 1 | 10.30 (1.28–82.73) | 10.30 (1.28–82.73) | - |
| Asian Ancestry | 1 | 5.65 (3.49–9.15) | 5.65 (3.49–9.15) | - |
Prostate cancer aggressiveness in carriers ofBRCApathogenic variants
A systematic review and meta-analysis found that BRCA1 and BRCA2 showed differences in prostate cancer aggressiveness.[
- BRCA1: RR, 1.98 (1.35–2.90; I² = 0%).
- BRCA2: RR, 6.08 (3.44–10.8; I² = 82%).
Men harboring pathogenic variants in the United Kingdom and Ireland were prospectively followed for prostate cancer diagnoses (BRCA1 [n = 16/376] and BRCA2 [n = 26/447]; median follow-up, 5.9 y and 5.3 y, respectively).[
- BRCA1 Gleason score less than 6; standardized incidence ratio (SIR), 3.50 (95% CI, 1.67–7.35) and Gleason score greater than 7; SIR, 1.80 (95% CI, 0.89–3.65).
- BRCA2 Gleason score less than 6; SIR, 3.03 (95% CI, 1.24–7.44) and Gleason score greater than 7; SIR, 5.07 (95% CI, 3.20–8.02).
This study was followed by a large, retrospective, international study of men diagnosed with prostate cancer who had pathogenic variants in BRCA1 (n = 3,453) and BRCA2 (n = 3,051).[
These studies suggest that prostate cancer in BRCA carriers is associated with aggressive disease features including a high Gleason score, and a high tumor stage and/or grade at diagnosis. This is a finding that warrants consideration when patients undergo cancer risk assessment and genetic counseling.[
BRCA1/BRCA2and survival outcomes
Analyses of prostate cancer cases in families with known BRCA1 or BRCA2 pathogenic variants have been examined for survival. A meta-analysis that examined BRCA1/BRCA2 prostate cancer risk, BRCA1/BRCA2 frequency in patients with prostate cancer, and prostate cancer mortality found that BRCA1/BRCA2 carriers who were diagnosed with prostate cancer had decreased cancer-specific survival (HR, 2.53; 95% CI, 1.98–3.22; P < .0001) when compared with noncarriers.[
HOXB13
Key points
HOXB13 was the first gene found to be associated with hereditary prostate cancer. The HOXB13 G84E variant has been extensively studied because of its association with prostate cancer risk.
- Overall risk of prostate cancer with the G84E variant ranges from 3- to 5-fold, with a higher risk of early-onset prostate cancer with the G84E variant of up to 10-fold.
- Penetrance for carriers of the G84E variant is an approximate 60% lifetime risk of prostate cancer by age 80 years.
- There is no clear association of the G84E variant with aggressive prostate cancer or other cancers.
- Preliminary studies suggest additional variants in HOXB13 may be relevant for prostate cancer risk in diverse populations.
Background
Linkage to 17q21-22 was initially reported by the UM-PCGP from 175 pedigrees of families with hereditary prostate cancer.[
- Men with a positive family history of prostate cancer, 2.2% versus negative, 0.8% (OR, 2.8; 95% CI, 1.6–5.1; P = 1.2 × 10-4).
- Men younger than 55 years at diagnosis, 2.2% versus older than 55 years, 0.8% (OR, 2.7; 95% CI, 1.6–4.7; P = 1.1 × 10-4).
- Men with a positive family history of prostate cancer and younger than 55 years at diagnosis, 3.1% versus a negative family history of prostate cancer and age at diagnosis older than 55 years, 0.6% (OR, 5.1; 95% CI, 2.4–12.2; P = 2.0 × 10-6).
- Men with a positive family history of prostate cancer and older than 55 years at diagnosis, 1.2%.
- Controls, 0.1% to 0.2%.[
19 ]
The clinical utility of genetic testing for the HOXB13 G84E variant is evolving.[
Validation and confirmatory studies
A validation study from the International Consortium of Prostate Cancer Genetics confirmed HOXB13 as a susceptibility gene for prostate cancer risk.[
Additional studies have emerged that better define the carrier frequency and prostate cancer risk associated with the HOXB13 G84E pathogenic variant.[
Risk of prostate cancer by HOXB13 G84E pathogenic variant status has been reported to vary by age of onset, family history, and geographical region. A validation study in an independent cohort of 9,988 cases and 61,994 controls from six studies of men of European ancestry, including 4,537 cases and 54,444 controls from Iceland whose genotypes were largely imputed, reported an OR of 7.06 (95% CI, 4.62–10.78; P = 1.5 × 10−19) for prostate cancer risk by G84E carrier status.[
Another meta-analysis that included 11 case-control studies also reported higher risk estimates for prostate cancer in HOXB13 G84E carriers (OR, 4.51; 95% CI, 3.28–6.20; P < .00001) and found a stronger association between HOXB13 G84E and early-onset disease (OR, 9.73; 95% CI, 6.57–14.39; P < .00001).[
However, a 2018 publication of a study combining multiple prostate cancer cases and controls of Nordic origin along with functional analysis reported that simultaneous presence of HOXB13 (G84E) and CIP2A (R229Q) predisposes men to an increased risk of prostate cancer (OR, 21.1; P = .000024).[
HOXB13pathogenic variants in diverse populations
A study of Chinese men with and without prostate cancer failed to identify the HOXB13 G84E pathogenic variant; however, there was an excess of a novel variant, G135E, in cases compared with controls.[
Two studies confirmed the association between the HOXB13 X285K variant and increased prostate cancer risk in African American men after this variant was identified in Martinique.[
Penetrance
Penetrance estimates for prostate cancer development in carriers of the HOXB13 G84E pathogenic variant are also being reported. One study from Sweden estimated a 33% lifetime risk of prostate cancer among G84E carriers.[
Biology
HOXB13 plays a role in prostate cancer development and interacts with the androgen receptor; however, the mechanism by which it contributes to the pathogenesis of prostate cancer remains unknown. This is the first gene identified to account for a fraction of hereditary prostate cancer, particularly early-onset prostate cancer. The clinical utility and implications for genetic counseling regarding HOXB13 G84E or other pathogenic variants have yet to be defined.
DNA mismatch repair genes (Lynch syndrome)
Five genes are implicated in mismatch repair (MMR), namely MLH1, MSH2, MSH6, PMS2, and EPCAM. Germline pathogenic variants in these five genes have been associated with Lynch syndrome, which manifests by cases of nonpolyposis colorectal cancer and a constellation of other cancers in families, including endometrial, ovarian, duodenal cancers, and transitional cell cancers of the ureter and renal pelvis. For more information about other cancers that are associated with Lynch syndrome, see the
One study that included two familial cancer registries found an increased cumulative incidence and risk of prostate cancer among 198 independent families with MMR gene pathogenic variants and Lynch syndrome.[
A systematic review and meta-analysis that included 23 studies (6 studies with molecular characterization and 18 risk studies, of which 12 studies quantified risk for prostate cancer) reported an association of prostate cancer with Lynch syndrome.[
A study from three sites participating in the Colon Cancer Family Registry examined 32 cases of prostate cancer (mean age at diagnosis, 62 y; standard deviation, 8 y) in men with a documented MMR gene pathogenic variant (23 MSH2 carriers, 5 MLH1 carriers, and 4 MSH6 carriers).[
Although the risk of prostate cancer appears to be elevated in families with Lynch syndrome, strategies for germline testing for MMR gene pathogenic variants in index prostate cancer patients remain to be determined.
A study of 1,133 primary prostate adenocarcinomas and 43 neuroendocrine prostate cancers (NEPC) conducted screening by MSH2 immunohistochemistry with confirmation by NGS.[
EPCAM testing has been included in some multigene panels likely due to EPCAM variants silencing MSH2. Specific large genomic rearrangement variants at the 3' end of EPCAM (which lies near the MSH2 gene) induce methylation of the MSH2 promoter, resulting in MSH2 protein loss.[
ATM
Ataxia telangiectasia (AT) is an autosomal recessive disorder characterized by neurological deterioration, telangiectasias, immunodeficiency states, and hypersensitivity to ionizing radiation. It is estimated that 1% of the general population may be heterozygous carriers of ATM pathogenic variants.[
CHEK2
CHEK2 has also been investigated for a potential association with prostate cancer risk. For more information on other cancers associated with CHEK2 pathogenic variants, see the
TP53
TP53 has also been investigated for a potential association with prostate cancer risk. For more information about other cancers associated with TP53 pathogenic variants, see the
Germline TP53 pathogenic variants have also been identified in men with prostate cancer who have undergone tumor testing. A prospective case series of 42 men with either localized, biochemically recurrent, or metastatic prostate cancer unselected for cancer family history or age at diagnosis undergoing tumor-only somatic testing found that 2 of 42 men (5%) were found to have a suspected TP53 germline pathogenic variant.[
Further evidence supports an association between prostate cancer and germline TP53 pathogenic variants.[
NBN
NBN, which is also known as NBS1, has been investigated due to a potential association with prostate cancer risk, with the literature constantly evolving. Studies mostly from Polish populations reported that the NBN 657del5 variant is associated with prostate cancer risk (OR, 2.5; P < .001), mortality (HR, 1.6; P = .001), and familial prostate cancer (OR, 4.6; P < .0001).[
Multigene testing studies in prostate cancer
Prevalence of pathogenic variants with prostate cancer risk on multigene panel testing
The following section gives information about additional genes that may be on hereditary prostate cancer panel tests.
One retrospective case series of 692 men with metastatic prostate cancer unselected for cancer family history or age at diagnosis assessed the incidence of germline pathogenic variants in 16 DNA repair genes. Pathogenic variants were identified in 11.8% (82 of 692), a rate higher than in men with localized prostate cancer (4.6%, P < .001), suggesting that genetic aberrations are more commonly observed in men with aggressive forms of disease.[
A case-control study in a Japanese population of 7,636 men with prostate cancer and 12,366 men without prostate cancer evaluated pathogenic variants in eight genes (BRCA1, BRCA2, CHEK2, ATM, NBN, PALB2, HOXB13, and BRIP1) for an association with prostate cancer.[
Germline pathogenic variants associated with metastatic prostate cancer
The metastatic prostate cancer setting is also contributing insights into the germline pathogenic variant spectrum of prostate cancer. Clinical sequencing of 150 metastatic tumors from men with castrate-resistant prostate cancer identified alterations in genes involved in DNA repair in 23% of men.[
| Study | Cohort | Germline Results for Prostate Cancer | Comments | ||
|---|---|---|---|---|---|
| mCRPC = metastatic castration-resistant prostate cancer. | |||||
| a Potential overlap of cohorts. | |||||
| Robinson et al. (2015)a[ |
Whole-exomeand transcriptome sequencing of bone or soft tissue tumor biopsies from a cohort of 150 men with mCRPC | 8% had germline pathogenic variants: | |||
| —BRCA2: 9/150 (6.0%) | |||||
| —ATM: 2/150 (1.3%) | |||||
| —BRCA1: 1/150 (0.7%) | |||||
| Pritchard et al. (2016)a[ |
692 men with metastatic prostate cancer, unselected for family history; analysis focused on 20 genes involved in maintaining DNA integrity and associated withautosomal dominantcancer–predisposing syndromes | 82/692 (11.8%) had germline pathogenic variants: | Frequency of germline pathogenic variants in DNA repair genes among men with metastatic prostate cancer significantly exceeded the prevalence of 4.6% among 499 men with localized prostate cancer in the Cancer Genome Atlas (P < .001) | ||
| —BRCA2: 37/692 (5.3%) | |||||
| —ATM: 11/692 (1.6%) | |||||
| —BRCA1: 6/692 (0.9%) | |||||
| Schrader et al. (2016)[ |
1,566 patients undergoing tumor profiling (341 genes) with matched normal DNA at a single institution; 97 cases of prostate cancer included | 10/97 (10.3%) had germline pathogenic variants: | |||
| —BRCA2: 6/97 (6.2%) | |||||
| —BRCA1: 1/97 (1.0%) | |||||
| —MSH6: 1/97 (1.0%) | |||||
| —MUTYH: 1/97 (1.0%) | |||||
| —PMS2: 1/97 (1.0%) | |||||
Common Risk Variants and Polygenic Risk Scores for Prostate Cancer
The most prevalent prostate cancer risk variants in the human genome were discovered in genome-wide association studies (GWAS). GWAS evaluate the millions of common single nucleotide polymorphisms (SNPs) in the human population (typically >5% prevalence) and ask if each variant is enriched in individuals with a given disease. With great statistical rigor, GWAS have revealed over 250 prostate cancer risk variants. Each single SNP confers a very modest prostate cancer risk. However, when compounded, these SNPs comprise a substantial portion of inherited prostate cancer risk. Research continues to translate these discoveries into clinical practice, with use in tools like polygenic risk scores (PRS).
GWAS and SNPs
- GWAS can identify inherited genetic variants that influence a specific phenotype, such as risk of a particular disease.
- For complex diseases, such as prostate cancer, risk of developing the disease is the product of multiple genetic and environmental factors; each individual factor contributes relatively little to overall risk.
- To date, GWAS have discovered more than 250 common genetic variants associated with prostate cancer risk.
- Individuals can be genotyped for all known prostate cancer risk markers relatively easily; but, to date, studies have not demonstrated that this information substantially refines risk estimates from commonly used variables, such as family history.
- The clinical relevance of variants identified from GWAS remains unclear.
Although the statistical evidence for an association between genetic variation at these loci and prostate cancer risk is overwhelming, the clinical relevance of the variants and the mechanism(s) by which they lead to increased risk are unclear and will require further characterization. Additionally, these loci are associated with very modest risk estimates and explain only a fraction of overall inherited risk. However, when combined into a PRS, these confirmed genetic risk variants may prove to be useful for prostate cancer risk stratification and to identify men for targeted screening and early detection. Further work will include genome-wide analysis of rarer alleles catalogued via sequencing efforts. Disease-associated alleles with frequencies of less than 1% in the population may prove to be more highly penetrant and clinically useful. In addition, further work is needed to describe the landscape of genetic risk in non-European populations. Finally, until the individual and collective influences of genetic risk alleles are evaluated prospectively, their clinical utility will remain difficult to fully assess.
Beginning in 2006, multiple genome-wide studies seeking associations with prostate cancer risk converged on the same chromosomal locus, 8q24.[
Since prostate cancer risk loci have been discovered at 8q24, more than 250 variants have been identified at other chromosomal risk loci. These chromosomal risk loci were detected by multistage GWAS, which were comprised of thousands of cases and controls and were validated in independent cohorts.[
Most prostate cancer GWAS data generated to date have been derived from populations of European descent. This shortcoming is profound, considering that linkage disequilibrium structure, SNV frequencies, and incidence of disease differ across ancestral groups. To provide meaningful genetic data to all patients, well-designed, adequately powered GWAS must be aimed at specific ethnic groups.[
The African American population is of particular interest because American men with West African ancestry are at higher risk of prostate cancer than any other group. A handful of studies have sought to determine whether GWAS findings in men of European ancestry are applicable to men of African ancestry.[
Statistically well-powered GWAS have also been launched to examine inherited cancer risk in Japanese and Chinese populations. Investigators discovered that these populations share many risk regions observed in African American men.[
Polygenic risk scores for prostate cancer
Current GWAS findings account for an estimated 58% of heritable prostate cancer risk. Another 6% of familial prostate cancer risk is attributed to rare genetic variants.[
In a 2023 study, PRS were created for a multi-ethnic cohort of over 150,000 prostate cancer cases and over 750,000 controls.[
As GWAS elucidate these networks, it is hoped that new therapies and chemopreventive strategies will follow.[
Germline SNPs associated with prostate cancer aggressiveness
Prostate cancer is biologically and clinically heterogeneous. Many tumors are indolent and are successfully managed with observation alone. Other tumors are quite aggressive and prove deadly. Several variables are used to determine prostate cancer aggressiveness at the time of diagnosis, such as Gleason score and PSA, but these are imperfect. Additional markers are needed because sound treatment decisions depend on accurate prognostic information. Germline genetic variants are attractive markers because they are present, easily detectable, and static throughout life.
Findings regarding inherited risk of aggressive disease are considered preliminary. Further work is needed to validate findings and assess these associations prospectively.
References:
- Mucci LA, Hjelmborg JB, Harris JR, et al.: Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315 (1): 68-76, 2016.
- Thompson D, Easton DF; Breast Cancer Linkage Consortium: Cancer Incidence in BRCA1 mutation carriers. J Natl Cancer Inst 94 (18): 1358-65, 2002.
- Nyberg T, Tischkowitz M, Antoniou AC: BRCA1 and BRCA2 pathogenic variants and prostate cancer risk: systematic review and meta-analysis. Br J Cancer 126 (7): 1067-1081, 2022.
- Mersch J, Jackson MA, Park M, et al.: Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 121 (2): 269-75, 2015.
- Nastiuk KL, Mansukhani M, Terry MB, et al.: Common mutations in BRCA1 and BRCA2 do not contribute to early prostate cancer in Jewish men. Prostate 40 (3): 172-7, 1999.
- Vazina A, Baniel J, Yaacobi Y, et al.: The rate of the founder Jewish mutations in BRCA1 and BRCA2 in prostate cancer patients in Israel. Br J Cancer 83 (4): 463-6, 2000.
- Lehrer S, Fodor F, Stock RG, et al.: Absence of 185delAG mutation of the BRCA1 gene and 6174delT mutation of the BRCA2 gene in Ashkenazi Jewish men with prostate cancer. Br J Cancer 78 (6): 771-3, 1998.
- Struewing JP, Abeliovich D, Peretz T, et al.: The carrier frequency of the BRCA1 185delAG mutation is approximately 1 percent in Ashkenazi Jewish individuals. Nat Genet 11 (2): 198-200, 1995.
- Oddoux C, Struewing JP, Clayton CM, et al.: The carrier frequency of the BRCA2 6174delT mutation among Ashkenazi Jewish individuals is approximately 1%. Nat Genet 14 (2): 188-90, 1996.
- Roa BB, Boyd AA, Volcik K, et al.: Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat Genet 14 (2): 185-7, 1996.
- Struewing JP, Hartge P, Wacholder S, et al.: The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med 336 (20): 1401-8, 1997.
- Nyberg T, Frost D, Barrowdale D, et al.: Prostate Cancer Risks for Male BRCA1 and BRCA2 Mutation Carriers: A Prospective Cohort Study. Eur Urol 77 (1): 24-35, 2020.
- Patel VL, Busch EL, Friebel TM, et al.: Association of Genomic Domains in BRCA1 and BRCA2 with Prostate Cancer Risk and Aggressiveness. Cancer Res 80 (3): 624-638, 2020.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2.2024. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023.
Available online with free registration. Last accessed September 18, 2024. - Taylor RA, Fraser M, Livingstone J, et al.: Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat Commun 8: 13671, 2017.
- Oh M, Alkhushaym N, Fallatah S, et al.: The association of BRCA1 and BRCA2 mutations with prostate cancer risk, frequency, and mortality: A meta-analysis. Prostate 79 (8): 880-895, 2019.
- Lange EM, Gillanders EM, Davis CC, et al.: Genome-wide scan for prostate cancer susceptibility genes using families from the University of Michigan prostate cancer genetics project finds evidence for linkage on chromosome 17 near BRCA1. Prostate 57 (4): 326-34, 2003.
- Lange EM, Robbins CM, Gillanders EM, et al.: Fine-mapping the putative chromosome 17q21-22 prostate cancer susceptibility gene to a 10 cM region based on linkage analysis. Hum Genet 121 (1): 49-55, 2007.
- Ewing CM, Ray AM, Lange EM, et al.: Germline mutations in HOXB13 and prostate-cancer risk. N Engl J Med 366 (2): 141-9, 2012.
- Schroeck FR, Zuhlke KA, Siddiqui J, et al.: Testing for the recurrent HOXB13 G84E germline mutation in men with clinical indications for prostate biopsy. J Urol 189 (3): 849-53, 2013.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer Early Detection. Version 2.2023. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023.
Available online with free registration. Last accessed November 30, 2023. - Xu J, Lange EM, Lu L, et al.: HOXB13 is a susceptibility gene for prostate cancer: results from the International Consortium for Prostate Cancer Genetics (ICPCG). Hum Genet 132 (1): 5-14, 2013.
- Chen Z, Greenwood C, Isaacs WB, et al.: The G84E mutation of HOXB13 is associated with increased risk for prostate cancer: results from the REDUCE trial. Carcinogenesis 34 (6): 1260-4, 2013.
- Shang Z, Zhu S, Zhang H, et al.: Germline homeobox B13 (HOXB13) G84E mutation and prostate cancer risk in European descendants: a meta-analysis of 24,213 cases and 73, 631 controls. Eur Urol 64 (1): 173-6, 2013.
- Handorf E, Crumpler N, Gross L, et al.: Prevalence of the HOXB13 G84E mutation among unaffected men with a family history of prostate cancer. J Genet Couns 23 (3): 371-6, 2014.
- Laitinen VH, Wahlfors T, Saaristo L, et al.: HOXB13 G84E mutation in Finland: population-based analysis of prostate, breast, and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev 22 (3): 452-60, 2013.
- Witte JS, Mefford J, Plummer SJ, et al.: HOXB13 mutation and prostate cancer: studies of siblings and aggressive disease. Cancer Epidemiol Biomarkers Prev 22 (4): 675-80, 2013.
- Beebe-Dimmer JL, Hathcock M, Yee C, et al.: The HOXB13 G84E Mutation Is Associated with an Increased Risk for Prostate Cancer and Other Malignancies. Cancer Epidemiol Biomarkers Prev 24 (9): 1366-72, 2015.
- Gudmundsson J, Sulem P, Gudbjartsson DF, et al.: A study based on whole-genome sequencing yields a rare variant at 8q24 associated with prostate cancer. Nat Genet 44 (12): 1326-9, 2012.
- Huang H, Cai B: G84E mutation in HOXB13 is firmly associated with prostate cancer risk: a meta-analysis. Tumour Biol 35 (2): 1177-82, 2014.
- Cai Q, Wang X, Li X, et al.: Germline HOXB13 p.Gly84Glu mutation and cancer susceptibility: a pooled analysis of 25 epidemiological studies with 145,257 participates. Oncotarget 6 (39): 42312-21, 2015.
- Stott-Miller M, Karyadi DM, Smith T, et al.: HOXB13 mutations in a population-based, case-control study of prostate cancer. Prostate 73 (6): 634-41, 2013.
- Alanee S, Shah S, Vijai J, et al.: Prevalence of HOXB13 mutation in a population of Ashkenazi Jewish men treated for prostate cancer. Fam Cancer 12 (4): 597-600, 2013.
- Kote-Jarai Z, Mikropoulos C, Leongamornlert DA, et al.: Prevalence of the HOXB13 G84E germline mutation in British men and correlation with prostate cancer risk, tumour characteristics and clinical outcomes. Ann Oncol 26 (4): 756-61, 2015.
- Sipeky C, Gao P, Zhang Q, et al.: Synergistic Interaction of HOXB13 and CIP2A Predisposes to Aggressive Prostate Cancer. Clin Cancer Res 24 (24): 6265-6276, 2018.
- Lin X, Qu L, Chen Z, et al.: A novel germline mutation in HOXB13 is associated with prostate cancer risk in Chinese men. Prostate 73 (2): 169-75, 2013.
- Momozawa Y, Iwasaki Y, Hirata M, et al.: Germline Pathogenic Variants in 7636 Japanese Patients With Prostate Cancer and 12 366 Controls. J Natl Cancer Inst 112 (4): 369-376, 2020.
- Marlin R, Créoff M, Merle S, et al.: Mutation HOXB13 c.853delT in Martinican prostate cancer patients. Prostate 80 (6): 463-470, 2020.
- Na R, Wei J, Sample CJ, et al.: The HOXB13 variant X285K is associated with clinical significance and early age at diagnosis in African American prostate cancer patients. Br J Cancer 126 (5): 791-796, 2022.
- Darst BF, Hughley R, Pfennig A, et al.: A Rare Germline HOXB13 Variant Contributes to Risk of Prostate Cancer in Men of African Ancestry. Eur Urol 81 (5): 458-462, 2022.
- Karlsson R, Aly M, Clements M, et al.: A population-based assessment of germline HOXB13 G84E mutation and prostate cancer risk. Eur Urol 65 (1): 169-76, 2014.
- MacInnis RJ, Severi G, Baglietto L, et al.: Population-based estimate of prostate cancer risk for carriers of the HOXB13 missense mutation G84E. PLoS One 8 (2): e54727, 2013.
- Nyberg T, Govindasami K, Leslie G, et al.: Homeobox B13 G84E Mutation and Prostate Cancer Risk. Eur Urol 75 (5): 834-845, 2019.
- Soravia C, van der Klift H, Bründler MA, et al.: Prostate cancer is part of the hereditary non-polyposis colorectal cancer (HNPCC) tumor spectrum. Am J Med Genet 121A (2): 159-62, 2003.
- Haraldsdottir S, Hampel H, Wei L, et al.: Prostate cancer incidence in males with Lynch syndrome. Genet Med 16 (7): 553-7, 2014.
- Grindedal EM, Møller P, Eeles R, et al.: Germ-line mutations in mismatch repair genes associated with prostate cancer. Cancer Epidemiol Biomarkers Prev 18 (9): 2460-7, 2009.
- Langeberg WJ, Kwon EM, Koopmeiners JS, et al.: Population-based study of the association of variants in mismatch repair genes with prostate cancer risk and outcomes. Cancer Epidemiol Biomarkers Prev 19 (1): 258-64, 2010.
- Bauer CM, Ray AM, Halstead-Nussloch BA, et al.: Hereditary prostate cancer as a feature of Lynch syndrome. Fam Cancer 10 (1): 37-42, 2011.
- Dominguez-Valentin M, Joost P, Therkildsen C, et al.: Frequent mismatch-repair defects link prostate cancer to Lynch syndrome. BMC Urol 16: 15, 2016.
- Raymond VM, Mukherjee B, Wang F, et al.: Elevated risk of prostate cancer among men with Lynch syndrome. J Clin Oncol 31 (14): 1713-8, 2013.
- Ryan S, Jenkins MA, Win AK: Risk of prostate cancer in Lynch syndrome: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev 23 (3): 437-49, 2014.
- Rosty C, Walsh MD, Lindor NM, et al.: High prevalence of mismatch repair deficiency in prostate cancers diagnosed in mismatch repair gene mutation carriers from the colon cancer family registry. Fam Cancer 13 (4): 573-82, 2014.
- Dominguez-Valentin M, Sampson JR, Seppälä TT, et al.: Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med 22 (1): 15-25, 2020.
- Guedes LB, Antonarakis ES, Schweizer MT, et al.: MSH2 Loss in Primary Prostate Cancer. Clin Cancer Res 23 (22): 6863-6874, 2017.
- Kovacs ME, Papp J, Szentirmay Z, et al.: Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat 30 (2): 197-203, 2009.
- Pritchard CC, Mateo J, Walsh MF, et al.: Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med 375 (5): 443-53, 2016.
- Savitsky K, Bar-Shira A, Gilad S, et al.: A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268 (5218): 1749-53, 1995.
- Dombernowsky SL, Weischer M, Allin KH, et al.: Risk of cancer by ATM missense mutations in the general population. J Clin Oncol 26 (18): 3057-62, 2008.
- Angèle S, Falconer A, Edwards SM, et al.: ATM polymorphisms as risk factors for prostate cancer development. Br J Cancer 91 (4): 783-7, 2004.
- Meyer A, Wilhelm B, Dörk T, et al.: ATM missense variant P1054R predisposes to prostate cancer. Radiother Oncol 83 (3): 283-8, 2007.
- Schumacher FR, Al Olama AA, Berndt SI, et al.: Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet 50 (7): 928-936, 2018.
- Karlsson Q, Brook MN, Dadaev T, et al.: Rare Germline Variants in ATM Predispose to Prostate Cancer: A PRACTICAL Consortium Study. Eur Urol Oncol 4 (4): 570-579, 2021.
- Wang Y, Dai B, Ye D: CHEK2 mutation and risk of prostate cancer: a systematic review and meta-analysis. Int J Clin Exp Med 8 (9): 15708-15, 2015.
- Conti DV, Wang K, Sheng X, et al.: Two Novel Susceptibility Loci for Prostate Cancer in Men of African Ancestry. J Natl Cancer Inst 109 (8): , 2017.
- Mai PL, Best AF, Peters JA, et al.: Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 122 (23): 3673-3681, 2016.
- Ruijs MW, Verhoef S, Rookus MA, et al.: TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet 47 (6): 421-8, 2010.
- Bougeard G, Renaux-Petel M, Flaman JM, et al.: Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol 33 (21): 2345-52, 2015.
- Cheng HH, Klemfuss N, Montgomery B, et al.: A Pilot Study of Clinical Targeted Next Generation Sequencing for Prostate Cancer: Consequences for Treatment and Genetic Counseling. Prostate 76 (14): 1303-11, 2016.
- Stacey SN, Sulem P, Jonasdottir A, et al.: A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat Genet 43 (11): 1098-103, 2011.
- Mittal RD, George GP, Mishra J, et al.: Role of functional polymorphisms of P53 and P73 genes with the risk of prostate cancer in a case-control study from Northern India. Arch Med Res 42 (2): 122-7, 2011.
- Xu B, Xu Z, Cheng G, et al.: Association between polymorphisms of TP53 and MDM2 and prostate cancer risk in southern Chinese. Cancer Genet Cytogenet 202 (2): 76-81, 2010.
- Maxwell KN, Cheng HH, Powers J, et al.: Inherited TP53 Variants and Risk of Prostate Cancer. Eur Urol 81 (3): 243-250, 2022.
- Rusak B, Kluźniak W, Wokołorczykv D, et al.: Inherited NBN Mutations and Prostate Cancer Risk and Survival. Cancer Res Treat 51 (3): 1180-1187, 2019.
- Wokołorczyk D, Kluźniak W, Huzarski T, et al.: Mutations in ATM, NBN and BRCA2 predispose to aggressive prostate cancer in Poland. Int J Cancer 147 (10): 2793-2800, 2020.
- Giri VN, Hegarty SE, Hyatt C, et al.: Germline genetic testing for inherited prostate cancer in practice: Implications for genetic testing, precision therapy, and cascade testing. Prostate 79 (4): 333-339, 2019.
- Nicolosi P, Ledet E, Yang S, et al.: Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol 5 (4): 523-528, 2019.
- Lang SH, Swift SL, White H, et al.: A systematic review of the prevalence of DNA damage response gene mutations in prostate cancer. Int J Oncol 55 (3): 597-616, 2019.
- Robinson D, Van Allen EM, Wu YM, et al.: Integrative clinical genomics of advanced prostate cancer. Cell 161 (5): 1215-28, 2015.
- Mandelker D, Zhang L, Kemel Y, et al.: Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. JAMA 318 (9): 825-835, 2017.
- Schrader KA, Cheng DT, Joseph V, et al.: Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol 2 (1): 104-11, 2016.
- Amundadottir LT, Sulem P, Gudmundsson J, et al.: A common variant associated with prostate cancer in European and African populations. Nat Genet 38 (6): 652-8, 2006.
- Schumacher FR, Feigelson HS, Cox DG, et al.: A common 8q24 variant in prostate and breast cancer from a large nested case-control study. Cancer Res 67 (7): 2951-6, 2007.
- Suuriniemi M, Agalliu I, Schaid DJ, et al.: Confirmation of a positive association between prostate cancer risk and a locus at chromosome 8q24. Cancer Epidemiol Biomarkers Prev 16 (4): 809-14, 2007.
- Wang L, McDonnell SK, Slusser JP, et al.: Two common chromosome 8q24 variants are associated with increased risk for prostate cancer. Cancer Res 67 (7): 2944-50, 2007.
- Yeager M, Orr N, Hayes RB, et al.: Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 39 (5): 645-9, 2007.
- Zheng SL, Sun J, Cheng Y, et al.: Association between two unlinked loci at 8q24 and prostate cancer risk among European Americans. J Natl Cancer Inst 99 (20): 1525-33, 2007.
- Savage SA, Greene MH: The evidence for prostate cancer risk loci at 8q24 grows stronger. J Natl Cancer Inst 99 (20): 1499-501, 2007.
- Salinas CA, Kwon E, Carlson CS, et al.: Multiple independent genetic variants in the 8q24 region are associated with prostate cancer risk. Cancer Epidemiol Biomarkers Prev 17 (5): 1203-13, 2008.
- Zheng SL, Hsing AW, Sun J, et al.: Association of 17 prostate cancer susceptibility loci with prostate cancer risk in Chinese men. Prostate 70 (4): 425-32, 2010.
- Zeegers MP, Khan HS, Schouten LJ, et al.: Genetic marker polymorphisms on chromosome 8q24 and prostate cancer in the Dutch population: DG8S737 may not be the causative variant. Eur J Hum Genet 19 (1): 118-20, 2011.
- Gudmundsson J, Sulem P, Manolescu A, et al.: Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet 39 (5): 631-7, 2007.
- Haiman CA, Patterson N, Freedman ML, et al.: Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet 39 (5): 638-44, 2007.
- Yeager M, Chatterjee N, Ciampa J, et al.: Identification of a new prostate cancer susceptibility locus on chromosome 8q24. Nat Genet 41 (10): 1055-7, 2009.
- Al Olama AA, Kote-Jarai Z, Giles GG, et al.: Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat Genet 41 (10): 1058-60, 2009.
- Matejcic M, Saunders EJ, Dadaev T, et al.: Germline variation at 8q24 and prostate cancer risk in men of European ancestry. Nat Commun 9 (1): 4616, 2018.
- Conti DV, Darst BF, Moss LC, et al.: Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat Genet 53 (1): 65-75, 2021.
- Cook MB, Wang Z, Yeboah ED, et al.: A genome-wide association study of prostate cancer in West African men. Hum Genet 133 (5): 509-21, 2014.
- Haiman CA, Chen GK, Blot WJ, et al.: Characterizing genetic risk at known prostate cancer susceptibility loci in African Americans. PLoS Genet 7 (5): e1001387, 2011.
- Han Y, Signorello LB, Strom SS, et al.: Generalizability of established prostate cancer risk variants in men of African ancestry. Int J Cancer 136 (5): 1210-7, 2015.
- Han Y, Rand KA, Hazelett DJ, et al.: Prostate Cancer Susceptibility in Men of African Ancestry at 8q24. J Natl Cancer Inst 108 (7): , 2016.
- Takata R, Akamatsu S, Kubo M, et al.: Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nat Genet 42 (9): 751-4, 2010.
- Akamatsu S, Takata R, Haiman CA, et al.: Common variants at 11q12, 10q26 and 3p11.2 are associated with prostate cancer susceptibility in Japanese. Nat Genet 44 (4): 426-9, S1, 2012.
- Xu J, Mo Z, Ye D, et al.: Genome-wide association study in Chinese men identifies two new prostate cancer risk loci at 9q31.2 and 19q13.4. Nat Genet 44 (11): 1231-5, 2012.
- Takata R, Takahashi A, Fujita M, et al.: 12 new susceptibility loci for prostate cancer identified by genome-wide association study in Japanese population. Nat Commun 10 (1): 4422, 2019.
- Benafif S, Kote-Jarai Z, Eeles RA, et al.: A Review of Prostate Cancer Genome-Wide Association Studies (GWAS). Cancer Epidemiol Biomarkers Prev 27 (8): 845-857, 2018.
- Wang A, Shen J, Rodriguez AA, et al.: Characterizing prostate cancer risk through multi-ancestry genome-wide discovery of 187 novel risk variants. Nat Genet 55 (12): 2065-2074, 2023.
- Chou A, Darst BF, Wilkens LR, et al.: Association of Prostate-Specific Antigen Levels with Prostate Cancer Risk in a Multiethnic Population: Stability Over Time and Comparison with Polygenic Risk Score. Cancer Epidemiol Biomarkers Prev 31 (12): 2199-2207, 2022.
- Kachuri L, Hoffmann TJ, Jiang Y, et al.: Genetically adjusted PSA levels for prostate cancer screening. Nat Med 29 (6): 1412-1423, 2023.
- Nyberg T, Brook MN, Ficorella L, et al.: CanRisk-Prostate: A Comprehensive, Externally Validated Risk Model for the Prediction of Future Prostate Cancer. J Clin Oncol 41 (5): 1092-1104, 2023.
- Dite GS, Spaeth E, Murphy NM, et al.: Development and validation of a simple prostate cancer risk prediction model based on age, family history, and polygenic risk. Prostate 83 (10): 962-969, 2023.
- Black MH, Li S, LaDuca H, et al.: Validation of a prostate cancer polygenic risk score. Prostate 80 (15): 1314-1321, 2020.
- Yoon BW, Shin HT, Seo JH: Risk Allele Frequency Analysis and Risk Prediction of Single-Nucleotide Polymorphisms for Prostate Cancer. Genes (Basel) 13 (11): , 2022.
- Huntley C, Torr B, Sud A, et al.: Utility of polygenic risk scores in UK cancer screening: a modelling analysis. Lancet Oncol 24 (6): 658-668, 2023.
- Karunamuni RA, Huynh-Le MP, Fan CC, et al.: Performance of African-ancestry-specific polygenic hazard score varies according to local ancestry in 8q24. Prostate Cancer Prostatic Dis 25 (2): 229-237, 2022.
- Chen F, Madduri RK, Rodriguez AA, et al.: Evidence of Novel Susceptibility Variants for Prostate Cancer and a Multiancestry Polygenic Risk Score Associated with Aggressive Disease in Men of African Ancestry. Eur Urol 84 (1): 13-21, 2023.
- Zhang W, Nicholson T, Zhang K: Deciphering the Polygenic Basis of Racial Disparities in Prostate Cancer By an Integrative Analysis of Genomic and Transcriptomic Data. Cancer Prev Res (Phila) 15 (3): 161-171, 2022.
- Ruan X, Huang D, Huang J, et al.: Application of European-specific polygenic risk scores for predicting prostate cancer risk in different ancestry populations. Prostate 83 (1): 30-38, 2023.
- Darst BF, Shen J, Madduri RK, et al.: Evaluating approaches for constructing polygenic risk scores for prostate cancer in men of African and European ancestry. Am J Hum Genet 110 (7): 1200-1206, 2023.
- Siltari A, Lönnerbro R, Pang K, et al.: How Well do Polygenic Risk Scores Identify Men at High Risk for Prostate Cancer? Systematic Review and Meta-Analysis. Clin Genitourin Cancer 21 (2): 316.e1-316.e11, 2023.
- Chen F, Darst BF, Madduri RK, et al.: Validation of a multi-ancestry polygenic risk score and age-specific risks of prostate cancer: A meta-analysis within diverse populations. Elife 11: , 2022.
- Ruan X, Huang D, Huang J, et al.: Genetic risk assessment of lethal prostate cancer using polygenic risk score and hereditary cancer susceptibility genes. J Transl Med 21 (1): 446, 2023.
- Freedman ML, Monteiro AN, Gayther SA, et al.: Principles for the post-GWAS functional characterization of cancer risk loci. Nat Genet 43 (6): 513-8, 2011.
- Pomerantz MM, Beckwith CA, Regan MM, et al.: Evaluation of the 8q24 prostate cancer risk locus and MYC expression. Cancer Res 69 (13): 5568-74, 2009.
- Jia L, Landan G, Pomerantz M, et al.: Functional enhancers at the gene-poor 8q24 cancer-linked locus. PLoS Genet 5 (8): e1000597, 2009.
- Ahmadiyeh N, Pomerantz MM, Grisanzio C, et al.: 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci U S A 107 (21): 9742-6, 2010.
- Sotelo J, Esposito D, Duhagon MA, et al.: Long-range enhancers on 8q24 regulate c-Myc. Proc Natl Acad Sci U S A 107 (7): 3001-5, 2010.
- Meyer KB, Maia AT, O'Reilly M, et al.: A functional variant at a prostate cancer predisposition locus at 8q24 is associated with PVT1 expression. PLoS Genet 7 (7): e1002165, 2011.
- Spisák S, Lawrenson K, Fu Y, et al.: CAUSEL: an epigenome- and genome-editing pipeline for establishing function of noncoding GWAS variants. Nat Med 21 (11): 1357-63, 2015.
- Hazelett DJ, Rhie SK, Gaddis M, et al.: Comprehensive functional annotation of 77 prostate cancer risk loci. PLoS Genet 10 (1): e1004102, 2014.
- Jiang J, Cui W, Vongsangnak W, et al.: Post genome-wide association studies functional characterization of prostate cancer risk loci. BMC Genomics 14 (Suppl 8): S9, 2013.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
Page Footer
Quiero...
Audiencia
Sitios seguros para miembros
Información sobre The Cigna Group
Aviso legal
Los planes individuales y familiares de seguro médico y dental están asegurados por Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc. y Cigna HealthCare of Texas, Inc. Los planes de beneficios de salud y de seguro de salud de grupo están asegurados o administrados por CHLIC, Connecticut General Life Insurance Company (CGLIC) o sus afiliadas (puedes ver
Todas las pólizas de seguros y los planes de beneficios de grupo contienen exclusiones y limitaciones. Para conocer la disponibilidad, los costos y detalles completos de la cobertura, comunícate con un agente autorizado o con un representante de ventas de Cigna. Este sitio web no está dirigido a los residentes de New Mexico.