Síndrome de Birt-Hogg-Dubé (PDQ®) información para profesionales de salud [NCI]
Manifestaciones clínicas
Las 3 características principales del síndrome de Birt-Hogg-Dubé (BHD) son los fibrofoliculomas o tricodiscomas, los quistes pulmonares y el neumotórax espontáneo, y los tumores renales.[
Lesiones cutáneas
Las personas con síndrome de BHD suelen presentar múltiples pápulas pequeñas en forma de cúpula, del mismo color de la piel, en la cara, el cuello y la parte superior del tronco. Estas manifestaciones dermatológicas características se llaman fibrofoliculomas o tricodiscomas (hamartoma del folículo piloso).[
Desde el punto de vista histológico, los fibrofoliculomas o tricodiscomas se caracterizan por múltiples hebras epiteliales anastomosantes que emanan de un folículo central. A veces el estroma está formado por tejido conjuntivo denso o rico en mucina y encapsula el componente epitelial.[
Quistes pulmonares y neumotórax espontáneo
En tomografía computarizada (TC) se observa que hay quistes pulmonares en el 85 % al 87 % de los pacientes con síndrome de BHD.[
En un estudio de 198 pacientes con BHD, los casos de neumotórax espontáneo fueron comparables entre hombres (20 %) y mujeres (29 %). Los primero neumotórax se presentaron entre los 22 y los 75 años.[
El cuadro clínico del neumotórax espontáneo varía desde ausencia de síntomas hasta disnea y dolor torácico. Los hallazgos clínicos incluyen taquipnea o murmullo respiratorio disminuido o ausente. Es posible que la investigación radiográfica exija el uso de una TC torácica de alta resolución para confirmar el diagnóstico porque la radiografía del tórax quizás no sea lo suficientemente sensible como para detectar un neumotórax loculado. Hasta el 75 % de los pacientes con antecedentes de neumotórax espontáneo presentarán un segundo caso de neumotórax espontáneo. Las diferencias en la notificación de la recidiva del neumotórax espontáneo quizás sean un reflejo de la eficacia de las diferentes modalidades de tratamiento.
Los hallazgos histológicos de las lesiones pleuropulmonares relacionadas con el síndrome de BHD incluyen bullas y quistes pleurales y subpleurales de pared delgada, quistes de aire intraparenquimatosos, vesículas pleurales y cambios compatibles con neumotórax espontáneo, además de cambios enfisematosos subyacentes en el parénquima pulmonar adyacente a las bullas.[
Tumores renales
Del 25 % al 35 % de las personas afectadas por el síndrome de BHD presentarán tumores renales,[
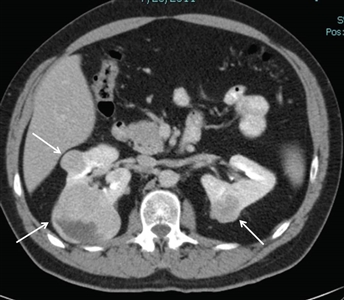
Los tumores más comunes son un híbrido de los tipos histológicos celulares oncocitoma y cromófobo, los llamados tumores híbridos oncocíticos, el carcinoma de células renales cromófobo y el oncocitoma renal. Solo el oncocitoma renal se considera un tumor benigno.[
Entre 70 pacientes con síndrome de BHD, tumores renales y una variante patogénica de FLCN analizados por los Institutos Nacionales de la Salud e identificados mediante revisión de la literatura, se notificó que 5 (7 %) fallecieron debido a RCC metastásico.[
Otras manifestaciones
Se notificaron oncocitomas de parótida multifocales bilaterales [
Cabe señalar que también se encontraron variantes germinales de FLCN en pacientes sin hallazgos cutáneos, pero con sospecha de síndrome de BHD debido a manifestaciones renales y pulmonares específicas.[
Además, se han notificado lipomas, angiolipomas,[
Aunque las observaciones epidemiológicas iniciales permitieron establecer un vínculo entre el síndrome de BHD y un aumento del riesgo de pólipos en el colon, en estudios epidemiológicos posteriores no se confirmó esta asociación.[
Referencias:
- Toro JR, Wei MH, Glenn GM, et al.: BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 45 (6): 321-31, 2008.
- Schmidt LS, Nickerson ML, Warren MB, et al.: Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet 76 (6): 1023-33, 2005.
- Schmidt LS, Linehan WM: Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol 12 (10): 558-69, 2015.
- Toro JR, Glenn G, Duray P, et al.: Birt-Hogg-Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol 135 (10): 1195-202, 1999.
- Graham RB, Nolasco M, Peterlin B, et al.: Nonsense mutations in folliculin presenting as isolated familial spontaneous pneumothorax in adults. Am J Respir Crit Care Med 172 (1): 39-44, 2005.
- Painter JN, Tapanainen H, Somer M, et al.: A 4-bp deletion in the Birt-Hogg-Dubé gene (FLCN) causes dominantly inherited spontaneous pneumothorax. Am J Hum Genet 76 (3): 522-7, 2005.
- Vernooij M, Claessens T, Luijten M, et al.: Birt-Hogg-Dubé syndrome and the skin. Fam Cancer 12 (3): 381-5, 2013.
- Toro JR, Pautler SE, Stewart L, et al.: Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dubé syndrome. Am J Respir Crit Care Med 175 (10): 1044-53, 2007.
- Matsumoto K, Lim D, Pharoah PD, et al.: A systematic review assessing the existence of pneumothorax-only variants of FLCN. Implications for lifelong surveillance of renal tumours. Eur J Hum Genet 29 (11): 1595-1600, 2021.
- Zbar B, Alvord WG, Glenn G, et al.: Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomarkers Prev 11 (4): 393-400, 2002.
- Pavlovich CP, Walther MM, Eyler RA, et al.: Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol 26 (12): 1542-52, 2002.
- Pavlovich CP, Grubb RL, Hurley K, et al.: Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J Urol 173 (5): 1482-6, 2005.
- Shuch B, Vourganti S, Ricketts CJ, et al.: Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 32 (5): 431-7, 2014.
- Sattler EC, Reithmair M, Steinlein OK: Kidney cancer characteristics and genotype-phenotype-correlations in Birt-Hogg-Dubé syndrome. PLoS One 13 (12): e0209504, 2018.
- Howlader N, Noone AM, Krapcho M, et al.: SEER Cancer Statistics Review (CSR) 1975-2017. Bethesda, Md: National Cancer Institute, 2020.
Available online . Last accessed February 7, 2025. - Vocke CD, Yang Y, Pavlovich CP, et al.: High frequency of somatic frameshift BHD gene mutations in Birt-Hogg-Dubé-associated renal tumors. J Natl Cancer Inst 97 (12): 931-5, 2005.
- da Silva NF, Gentle D, Hesson LB, et al.: Analysis of the Birt-Hogg-Dubé (BHD) tumour suppressor gene in sporadic renal cell carcinoma and colorectal cancer. J Med Genet 40 (11): 820-4, 2003.
- Khoo SK, Kahnoski K, Sugimura J, et al.: Inactivation of BHD in sporadic renal tumors. Cancer Res 63 (15): 4583-7, 2003.
- Liu V, Kwan T, Page EH: Parotid oncocytoma in the Birt-Hogg-Dubé syndrome. J Am Acad Dermatol 43 (6): 1120-2, 2000.
- Vinit J, Friedel J, Bielefeld P, et al.: [Birt-Hogg-Dubé syndrome and multiple recurrent tumors]. Rev Med Interne 32 (3): e40-2, 2011.
- Maffé A, Toschi B, Circo G, et al.: Constitutional FLCN mutations in patients with suspected Birt-Hogg-Dubé syndrome ascertained for non-cutaneous manifestations. Clin Genet 79 (4): 345-54, 2011.
- Chung JY, Ramos-Caro FA, Beers B, et al.: Multiple lipomas, angiolipomas, and parathyroid adenomas in a patient with Birt-Hogg-Dube syndrome. Int J Dermatol 35 (5): 365-7, 1996.
- Vincent A, Farley M, Chan E, et al.: Birt-Hogg-Dubé syndrome: two patients with neural tissue tumors. J Am Acad Dermatol 49 (4): 717-9, 2003.
- Drummond C, Grigoris I, Dutta B: Birt-Hogg-Dubé syndrome and multinodular goitre. Australas J Dermatol 43 (4): 301-4, 2002.
- Welsch MJ, Krunic A, Medenica MM: Birt-Hogg-Dubé Syndrome. Int J Dermatol 44 (8): 668-73, 2005.
- Tomassetti S, Carloni A, Chilosi M, et al.: Pulmonary features of Birt-Hogg-Dubé syndrome: cystic lesions and pulmonary histiocytoma. Respir Med 105 (5): 768-74, 2011.
- Walter P, Kirchhof B, Korge B, et al.: Flecked chorioretinopathy associated with Birt-Hogg-Dubé syndrome. Graefes Arch Clin Exp Ophthalmol 235 (6): 359-61, 1997.
- Nahorski MS, Lim DH, Martin L, et al.: Investigation of the Birt-Hogg-Dube tumour suppressor gene (FLCN) in familial and sporadic colorectal cancer. J Med Genet 47 (6): 385-90, 2010.
- van de Beek I, Glykofridis IE, Wolthuis RMF, et al.: No evidence for increased prevalence of colorectal carcinoma in 399 Dutch patients with Birt-Hogg-Dubé syndrome. Br J Cancer 122 (4): 590-594, 2020.
Esta información no reemplaza el consejo de un médico. Ignite Healthwise, LLC, niega toda garantía y responsabilidad por el uso de esta información. El uso que usted haga de esta información implica que usted acepta los
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
Page Footer
Quiero...
Audiencia
Sitios seguros para miembros
Información sobre The Cigna Group
Aviso legal
Los planes individuales y familiares de seguro médico y dental están asegurados por Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc. y Cigna HealthCare of Texas, Inc. Los planes de beneficios de salud y de seguro de salud de grupo están asegurados o administrados por CHLIC, Connecticut General Life Insurance Company (CGLIC) o sus afiliadas (puedes ver
Todas las pólizas de seguros y los planes de beneficios de grupo contienen exclusiones y limitaciones. Para conocer la disponibilidad, los costos y detalles completos de la cobertura, comunícate con un agente autorizado o con un representante de ventas de Cigna. Este sitio web no está dirigido a los residentes de New Mexico.