Comprar planes
Comprar tu propia cobertura
Planes a través de tu empleador
Conoce sobre los beneficios médicos, dentales, de farmacia y voluntarios que tu empleador puede ofrecer.
Aprender
¿Vives o trabajas fuera del país?
Tratamiento de la leucemia mieloide aguda y otras neoplasias mieloides malignas infantiles (PDQ®) : Tratamiento - información para profesionales de salud [NCI]
Esta información es producida y suministrada por el Instituto Nacional del Cáncer (NCI, por sus siglas en inglés). La información en este tema puede haber cambiado desde que se escribió. Para la información más actual, comuníquese con el Instituto Nacional del Cáncer a través del Internet en la página web http://cancer.gov o llame al 1-800-4-CANCER.
Información general sobre la leucemia mieloide aguda infantil
Se han logrado mejoras notables en la supervivencia de niños y adolescentes con cáncer.[
Características de las leucemias mieloides y otras neoplasias mieloides malignas en los niños
Cerca de un 20 % de las leucemias infantiles son de origen mieloide y corresponden a un grupo de neoplasias malignas hematopoyéticas.[
Las características generales de las leucemias mieloides y otras neoplasias mieloides malignas se describen a continuación:
- Leucemia mieloide aguda (LMA). La LMA se define como un trastorno clonal causado por la transformación maligna de un producto de la médula ósea, células madre o progenitoras que se renuevan solas, lo que conduce a una acumulación de células mieloides inmaduras no funcionales. Dichas circunstancias llevan a un aumento de la acumulación de estas células mieloides malignas en la médula ósea y otros órganos. Para considerarse una leucemia aguda, por lo general la médula ósea debe contener más de un 20 % de blastocitos leucémicos inmaduros, con algunas excepciones según se describe en las secciones siguientes. Para obtener más información, consultar las secciones Aspectos generales de las opciones de tratamiento de la leucemia mieloide aguda infantil y Tratamiento de la leucemia mieloide aguda infantil.
- Mielopoyesis anormal transitoria (MAT). La MAT también se llama trastorno mieloproliferativo transitorio o leucemia transitoria. La MAT que se observa en lactantes con síndrome de Down representa una expansión clonal de mieloblastos que es difícil de distinguir de una LMA. Es importante destacar que la MAT remite de manera espontánea en la mayoría de los casos durante los 3 primeros meses de vida. La MAT se presenta en un 4 % a un 10 % de los lactantes con síndrome de Down.[
3 ,4 ,5 ]En presencia de trisomía 21, los blastocitos de la MAT a menudo exhiben características de diferenciación megacariocítica y mutaciones específicas que afectan el gen GATA1.[
6 ,7 ] La MAT a veces se presenta en lactantes con fenotipo normal y mosaicismo genético de trisomía 21 en la médula ósea. Aunque la MAT por lo general no se caracteriza por anomalías citogenéticas diferentes a la trisomía 21, la presencia de otras manifestaciones citogenéticas quizá pronostique un aumento de riesgo de LMA posterior.[8 ] Cerca de un 20 % de los lactantes con MAT vinculada al síndrome de Down con el tiempo presentan LMA; la mayoría de los casos se diagnostican durante los primeros 3 años de vida.[7 ,8 ]La muerte prematura por complicaciones relacionadas con la MAT se presenta en un 10 % a un 20 % de los lactantes afectados.[
8 ,9 ] Los lactantes con organomegalia progresiva, derrames viscerales, recuento elevado de blastocitos (>100 000 células/μl) y hallazgos de laboratorio de insuficiencia hepática progresiva tienen un riesgo particularmente alto de mortalidad prematura.[8 ,9 ] Para obtener más información, consultar la sección Proliferaciones mieloides relacionadas con el síndrome de Down. - Síndrome mielodisplásico (SMD). El SMD en niños representa un grupo heterogéneo de trastornos que se caracteriza por hematopoyesis ineficaz, alteración en la maduración de los progenitores mieloides con características morfológicas displásicas y citopenias. Aunque no se conoce la causa subyacente de los SMD en los niños, a menudo se relacionan con síndromes de insuficiencia medular. La mayoría de los pacientes con SMD tiene una médula ósea hipercelular sin aumento del número de blastocitos leucémicos; sin embargo, algunos pacientes tienen una médula ósea muy hipocelular que dificulta la distinción entre una anemia aplásica grave y un SMD.[
10 ,11 ]La presencia de una anomalía cariotípica en una médula hipocelular es congruente con un SMD y se debe anticipar una transformación a LMA. Debido a que es frecuente que un SMD evolucione a una LMA, los pacientes con SMD por lo general se derivan para trasplante de células madre antes de que ocurra la transformación a LMA. Para obtener más información, consultar la sección Síndromes mielodisplásicos.
- Leucemia mielomonocítica juvenil (LMMJ). La LMMJ representa el síndrome mieloproliferativo más frecuente en niños pequeños. La mediana de edad del inicio de la LMMJ es de 1,8 años.
La LMMJ se manifiesta de forma característica con hepatoesplenomegalia, linfadenopatía, fiebre y erupción cutánea, junto con un recuento elevado de glóbulos blancos (GB) y un aumento de monocitos circulantes.[
12 ] Además, los pacientes a menudo tienen concentraciones altas de hemoglobina F, hipersensibilidad de las células leucémicas al factor estimulante de las colonias de granulocitos y macrófagos (GM-CSF), monosomía 7 y mutaciones en las células leucémicas en un gen que participa en la vía de señalización RAS (por ejemplo, NF1, KRAS, NRAS, PTPN11 o CBL).[12 ,13 ,14 ] Para obtener más información, consultar la sección Leucemia mielomonocítica juvenil. - Leucemia mielógena crónica (LMC). La LMC es, en esencia, una enfermedad de adultos; sin embargo, es la forma más común de trastorno mieloproliferativo infantil; representa cerca de 10 % de las leucemias mieloides infantiles.[
2 ] Aunque se han notificado casos de LMC en niños muy pequeños, la mayoría de los pacientes tienen 6 años o más.La LMC es una panmielopatía clonal que afecta todos los linajes de células hematopoyéticas. Aunque el recuento de glóbulos blancos (GB) puede estar muy elevado, la médula ósea no exhibe un número alto de blastocitos leucémicos durante la fase crónica de esta enfermedad. La causa de la LMC es la presencia del cromosoma Filadelfia, una translocación entre los cromosomas 9 y 22 (es decir, t(9;22)) que produce la fusión de los genes BCR y ABL1. Para obtener más información, consultar la sección Leucemia mielógena crónica.
Otros síndromes mieloproliferativos crónicos, como la policitemia vera y la trombocitosis esencial, son muy infrecuentes en los niños.
Afecciones relacionadas con las neoplasias mieloides malignas
Ciertas anomalías genéticas (síndromes de predisposición al cáncer) se relacionan con la formación de LMA. Hay una tasa alta de coincidencia de LMA en gemelos monocigóticos; sin embargo, no se cree que se relacione con un riesgo genético sino con la circulación compartida y la incapacidad de uno de los gemelos de rechazar las células leucémicas del otro gemelo durante el desarrollo fetal.[
La aparición de LMA también se ha relacionado con diversos síndromes hereditarios o familiares que, en la clasificación de las neoplasias mieloides y la leucemia aguda de la Organización Mundial de la Salud (OMS) de 2016, se reconocen como una categoría única. También hay varias afecciones adquiridas que aumentan el riesgo de presentar LMA. Estas afecciones hereditarias y adquiridas pueden provocar leucemogénesis a través de mecanismos que incluyen desequilibrio o inestabilidad cromosómica, defectos en la reparación del DNA, anomalías en la activación del receptor de citocinas o de la vía de señalización celular, y cambios en la síntesis de proteínas.[
Síndromes hereditarios
- Desequilibrios cromosómicos:
- Síndrome de Down.
- Monosomía familiar 7.
- Síndromes de inestabilidad cromosómica:
- Anemia de Fanconi.
- Disqueratosis congénita.
- Síndrome de Bloom.
- Síndromes de crecimiento y defectos de las vías de señalización de supervivencia celular:
- Neurofibromatosis de tipo 1 (en particular, predisposición a la LMMJ).
- Síndrome de Noonan (en particular, predisposición a la LMMJ).
- Neutropenia congénita grave (síndrome de Kostmann, mutaciones en HAX1) y neutropenia cíclica (mutaciones en ELANE).
- Síndrome de Shwachman-Diamond.
- Anemia de Diamond-Blackfan.
- Trombocitopenia amegacariocítica congénita (mutaciones en MPL).
- Síndrome de la línea germinal de CBL (en particular, predisposición a la LMMJ).
- Síndrome de Li-Fraumeni (mutaciones en TP53).
- Trastornos trombocitopénicos y plaquetarios hereditarios con predisposición germinal a neoplasias mieloides (mutaciones en RUNX1, ANKRD26 y ETV6).
- Deficiencia de GATA2 (mutaciones en GATA2).
Síndromes adquiridos
- Anemia aplásica grave.
- Hemoglobinuria paroxística nocturna.
- Trombocitopenia amegacariocítica.
- Monosomía 7 adquirida.
En el sistema de clasificación de la OMS de 2016 se categorizaron las neoplasias mieloides con predisposición germinal de la siguiente manera:
- Neoplasias mieloides con predisposición germinal sin trastorno preexistente ni disfunción orgánica.[
22 ]- Leucemia mieloide aguda con mutaciones de la línea germinal en CEBPA.
- Neoplasias mieloides con mutaciones de la línea germinal en DDX41.
- Neoplasias mieloides con predisposición germinal y trastornos plaquetarios preexistentes.[
22 ]- Neoplasias mieloides con mutaciones de la línea germinal en RUNX1.
- Neoplasias mieloides con mutaciones de la línea germinal en ANKRD26.
- Neoplasias mieloides con mutaciones de la línea germinal en ETV6.
- Neoplasias mieloides con predisposición germinal y otra disfunción orgánica.[
22 ]- Neoplasias mieloides con mutaciones de la línea germinal en GATA2.
- Neoplasias mieloides vinculadas a síndromes de insuficiencia medular (incluso anemia de Fanconi, anemia de Diamond-Blackfan y síndrome de Shwachman-Diamond).
- Neoplasias mieloides relacionadas con trastornos biológicos de los telómeros (incluso disqueratosis congénita).
- Leucemia mielomonocítica juvenil vinculada a neurofibromatosis, síndrome de Noonan o trastornos similares al síndrome de Noonan (mutaciones de la línea germinal en CBL).
- Neoplasias mieloides relacionadas con el síndrome de Down.
También está en estudio la susceptibilidad genética a la LMA no sindrómica. Por ejemplo, la homocigosis de un polimorfismo específico de IKZF1 se ha relacionado con un aumento de riesgo de LMA infantil.[
Referencias:
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Smith MA, Ries LA, Gurney JG, et al.: Leukemia. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 17-34. Also available online. Last accessed August 11, 2022.
- Roberts I, Alford K, Hall G, et al.: GATA1-mutant clones are frequent and often unsuspected in babies with Down syndrome: identification of a population at risk of leukemia. Blood 122 (24): 3908-17, 2013.
- Zipursky A: Transient leukaemia--a benign form of leukaemia in newborn infants with trisomy 21. Br J Haematol 120 (6): 930-8, 2003.
- Gamis AS, Smith FO: Transient myeloproliferative disorder in children with Down syndrome: clarity to this enigmatic disorder. Br J Haematol 159 (3): 277-87, 2012.
- Hitzler JK, Cheung J, Li Y, et al.: GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood 101 (11): 4301-4, 2003.
- Mundschau G, Gurbuxani S, Gamis AS, et al.: Mutagenesis of GATA1 is an initiating event in Down syndrome leukemogenesis. Blood 101 (11): 4298-300, 2003.
- Massey GV, Zipursky A, Chang MN, et al.: A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481. Blood 107 (12): 4606-13, 2006.
- Gamis AS, Alonzo TA, Gerbing RB, et al.: Natural history of transient myeloproliferative disorder clinically diagnosed in Down syndrome neonates: a report from the Children's Oncology Group Study A2971. Blood 118 (26): 6752-9; quiz 6996, 2011.
- Hasle H, Niemeyer CM: Advances in the prognostication and management of advanced MDS in children. Br J Haematol 154 (2): 185-95, 2011.
- Schwartz JR, Ma J, Lamprecht T, et al.: The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun 8 (1): 1557, 2017.
- Niemeyer CM, Arico M, Basso G, et al.: Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89 (10): 3534-43, 1997.
- Loh ML: Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol 152 (6): 677-87, 2011.
- Stieglitz E, Taylor-Weiner AN, Chang TY, et al.: The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47 (11): 1326-33, 2015.
- Zuelzer WW, Cox DE: Genetic aspects of leukemia. Semin Hematol 6 (3): 228-49, 1969.
- Miller RW: Persons with exceptionally high risk of leukemia. Cancer Res 27 (12): 2420-3, 1967.
- Inskip PD, Harvey EB, Boice JD, et al.: Incidence of childhood cancer in twins. Cancer Causes Control 2 (5): 315-24, 1991.
- Kurita S, Kamei Y, Ota K: Genetic studies on familial leukemia. Cancer 34 (4): 1098-101, 1974.
- Greaves M: Pre-natal origins of childhood leukemia. Rev Clin Exp Hematol 7 (3): 233-45, 2003.
- Puumala SE, Ross JA, Aplenc R, et al.: Epidemiology of childhood acute myeloid leukemia. Pediatr Blood Cancer 60 (5): 728-33, 2013.
- West AH, Godley LA, Churpek JE: Familial myelodysplastic syndrome/acute leukemia syndromes: a review and utility for translational investigations. Ann N Y Acad Sci 1310: 111-8, 2014.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Ross JA, Linabery AM, Blommer CN, et al.: Genetic variants modify susceptibility to leukemia in infants: a Children's Oncology Group report. Pediatr Blood Cancer 60 (1): 31-4, 2013.
Clasificación de las neoplasias mieloides malignas infantiles
Sistema de clasificación para la leucemia mieloide aguda infantil del grupo French-American-British
El primer sistema de clasificación integral (morfológica e histoquímica) de la leucemia mieloide aguda (LMA) lo formuló el French-American-British (FAB) Cooperative Group.[
Los subtipos principales de LMA son los siguientes:
- M0: leucemia mieloblástica aguda sin diferenciación.[
6 ,7 ] La LMA M0, que también se conoce como LMA con diferenciación mínima, no expresa mieloperoxidasa (MPO) en la microscopía óptica, pero a veces exhibe gránulos característicos en la microscopía electrónica. La LMA M0 se puede definir por la expresión de marcadores de conglomerados determinantes (CD) como el CD13, CD33 y CD117 (c-KIT) en ausencia de diferenciación linfoide. - M1: leucemia mieloblástica aguda con diferenciación mínima, pero que expresa MPO detectable mediante análisis inmunohistoquímicos o citometría de flujo.
- M2: leucemia mieloblástica aguda con diferenciación.
- M3: leucemia promielocítica aguda (LPA) de tipo hipergranular. Para obtener más información, consultar la sección Leucemia promielocítica aguda.
- M3v: leucemia promielocítica aguda, variante microgranular. El citoplasma de los promielocitos muestra una granularidad fina y núcleos que a menudo están plegados. La M3v tiene las mismas repercusiones clínicas, citogenéticas y terapéuticas que la FAB M3.
- M4: leucemia mielomonocítica aguda (LMMA).
- M4Eo: leucemia mielomonocítica aguda con eosinofilia (eosinófilos anormales con gránulos basofílicos displásicos).
- M5: leucemia monocítica aguda (LMoA).
- M5a: leucemia monocítica aguda sin diferenciación (monoblástica).
- M5b: leucemia monocítica aguda con diferenciación.
- M6: leucemia eritroide aguda (LEA).
- M6a: eritroleucemia.
- M6b: leucemia eritroide pura (el componente de mieloblastos no es aparente).
- M6c: presencia de mieloblastos y proeritroblastos.
- M7: leucemia megacariocítica aguda (LMCA).
Otros subtipos de LMA muy infrecuentes son la leucemia eosinofílica aguda y la leucemia basofílica aguda.
La clasificación FAB se reemplazó con la clasificación de la OMS descrita a continuación, pero sigue siendo importante porque constituye la base de la subcategoría de LMA sin otra indicación (LMA, SAI) que se usa en la OMS.
Sistema de clasificación de la Organización Mundial de la Salud para la leucemia mieloide aguda infantil
En 2001, la Organización Mundial de la Salud (OMS) propuso un sistema de clasificación nuevo en el que se incorporó información citogenética de utilidad diagnóstica y que se correlaciona de forma más confiable con los desenlaces. En esta clasificación, los pacientes con t(8;21), inv(16), t(15;17) o translocaciones de KMT2A (MLL), que en conjunto eran casi la mitad de los casos de LMA infantil, se clasificaron como LMA con anomalías citogenéticas recidivantes. Este sistema de clasificación también disminuyó de 30 a 20 % el requisito del porcentaje de blastocitos leucémicos en la médula ósea necesarios para diagnosticar una LMA; además, se aclaró que los pacientes con anomalías citogenéticas recurrentes no necesitan cumplir los requisitos mínimos de blastocitos para considerar que tienen una LMA.[
En 2008, la OMS aumentó el número de anomalías citogenéticas vinculadas con la clasificación de la LMA y, por primera vez, incluyó mutaciones génicas específicas (CEBPA y NPM) en su sistema de clasificación.[
Clasificación de la Organización Mundial de la Salud de 2016 para la leucemia mieloide aguda y las neoplasias relacionadas
- Leucemia mieloide aguda con anomalías genéticas recurrentes:
- Leucemia mieloide aguda con t(8;21)(q22;q22) con fusión génica RUNX1::RUNX1T1.
- Leucemia mieloide aguda con inv(16)(p13.1;q22) o t(16;16)(p13.1;q22) y fusión génica CBFB::MYH11.
- Leucemia promielocítica aguda con fusión génica PML::RARA.
- Leucemia mieloide aguda con t(9;11)(p21.3;q23.3) y fusión génica MLLT3::KMT2A.
- Leucemia mieloide aguda con t(6;9)(p23;q34.1) y fusión génica DEK::NUP214.
- Leucemia mieloide aguda con inv(3)(q21.3;q26.2) o t(3;3)(q21.3;q26.2) y alteración en GATA2 o MECOMGATA2, MECOM.
- Leucemia mieloide aguda (megacarioblástica) con t(1;22)(p13.3;q13.3) y fusión génica RBM15::MKL1.
- Leucemia mieloide aguda con fusión génica BCR::ABL1 (entidad provisional).
- Leucemia mieloide aguda con mutación en NPM1.
- Leucemia mieloide aguda con mutaciones bialélicas en CEBPA.
- Leucemia mieloide aguda con mutación en RUNX1 (entidad provisional).
- Leucemia mieloide aguda con características relacionadas con mielodisplasia.
- Neoplasias mieloides relacionadas con el tratamiento.
- Leucemia mieloide aguda, sin otra indicación:
- Leucemia mieloide aguda con diferenciación mínima.
- Leucemia mieloide aguda sin maduración.
- Leucemia mieloide aguda con maduración.
- Leucemia mielomonocítica aguda.
- Leucemia monoblástica o monocítica aguda.
- Leucemia eritroide pura.
- Leucemia megacarioblástica aguda.
- Leucemia basofílica aguda.
- Panmielosis aguda con mielofibrosis.
- Sarcoma mieloide.
- Proliferaciones mieloides relacionadas con el síndrome de Down:
- Mielopoyesis anormal transitoria (MAT).
- Leucemia mieloide relacionada con el síndrome de Down.
Clasificación de la Organización Mundial de la Salud de 2016 para las leucemias agudas o de linaje ambiguo
El grupo de las leucemias agudas que tienen características de LMA y leucemia linfoblástica aguda (LLA), que se conoce como leucemias agudas de linaje ambiguo en el sistema de clasificación de la OMS, se resume en el Cuadro 1.[
| Afección | Definición |
|---|---|
| SAI = sin otra indicación. | |
| a Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.[ |
|
| Leucemia aguda indiferenciada | Leucemia aguda que no expresa ningún marcador que se considere específico para el linaje linfoide ni el linaje mieloide |
| Leucemia aguda de fenotipo mixto con t(9;22)(q34;q11.2) y fusión génicaBCR::ABL1 | Leucemia aguda que cumple con los criterios diagnósticos de la leucemia aguda de fenotipo mixto en la que los blastocitos también expresan la translocación (9;22) o un reordenamientoBCR::ABL1 |
| Leucemia aguda de fenotipo mixto con t(v;11q23) y reordenamiento deKMT2A(MLL) | Leucemia aguda que cumple con los criterios diagnósticos de la leucemia aguda de fenotipo mixto en la que los blastocitos también expresan una translocación que afecta el genKMT2A |
| Leucemia aguda de fenotipo mixto, B o mieloide, SAI | Leucemia aguda que cumple con los criterios diagnósticos para asignar un linaje B y un linaje mieloide, en la que los blastocitos carecen de anomalías genéticas que incluyan la fusión génicaBCR::ABL1o alteraciones enKMT2A |
| Leucemia aguda de fenotipo mixto, T o mieloide, SAI | Leucemia aguda que cumple con los criterios diagnósticos para asignar un linaje T y un linaje mieloide, en la que los blastocitos carecen de anomalías genéticas que incluyan la fusión génicaBCR::ABL1oKMT2A |
| Leucemia aguda de fenotipo mixto, B o mieloide, SAI (tipos poco frecuentes) | Leucemia aguda que cumple con los criterios diagnósticos para asignar un linaje B y un linaje T |
| Otras leucemias de linaje ambiguo | Leucemia o linfoma linfoblásticos de células citolíticas naturales |
| Linaje | Criterios |
|---|---|
| a Adaptado de Arber et al.[ |
|
| b Fuerte se define como igual o más brillante que las células B o T normales en la muestra. | |
| Linaje mieloide | Mieloperoxidasa (pruebas de citometría de flujo, inmunohistoquímica o citoquímica);o diferenciación monocítica (por lo menos dos de los siguientes aspectos: prueba citoquímica de esterasa inespecífica, CD11c, CD14, CD64, lisozima) |
| Linaje T | Fuerteb CD3 citoplasmático fuerte (con anticuerpos contra la cadena ε de CD3);o CD3 de superficie |
| Linaje B | Fuerteb CD19 fuerte y expresión fuerte de por lo menos una de las siguientes moléculas: CD79a, CD22 citoplasmático o CD10;o CD19 débil y expresión fuerte de por lo menos dos de las siguientes moléculas: CD79a, CD22 citoplasmático o CD10 |
Es posible que se observen leucemias de fenotipos mixtos en distintas presentaciones; por ejemplo, las siguientes:
- Leucemias bilineales en las que hay dos poblaciones de células diferentes; a menudo, una linfoide y otra mieloide.
- Leucemias bifenotípicas en las que los blastocitos exhiben a la vez características de linaje linfoide y mieloide.
Los casos bifenotípicos representan la mayoría de las leucemias de fenotipo mixto.[
Clasificación de la Organización Mundial de la Salud sobre los hallazgos en la médula ósea y la sangre periférica de los síndromes mielodisplásicos
La clasificación FAB de los síndromes mielodisplásicos (SMD) no era completamente apropiada para niños.[
- Mielodisplasia.
- Tipos de citopenia.
- Anomalías cromosómicas específicas.
- Porcentaje de mieloblastos.
La OMS publicó en 2008 un esquema modificado de clasificación para los SMD y los trastornos mieloproliferativos (TMP) que incluyó subsecciones dedicadas a los SMD y TMP infantiles.[
Cabe destacar que es difícil diferenciar un SMD de otras causas reactivas de displasia o citopenias que se parecen. En general, la presencia de más del 10 % de displasia en un linaje celular es un criterio diagnóstico de SMD; sin embargo, en las directrices de la OMS de 2016 se advierte que las causas reactivas, más que las clonales, quizá presenten más del 10 % de displasia y se deben excluir; en particular, cuando la displasia es mínima o afecta un solo linaje.[
Para determinar el riesgo de progresión a LMA y el desenlace de los pacientes adultos con SMD, se usa el International Prognostic Scoring System. Cuando este sistema se aplicó a los niños con SMD o leucemia mielomonocítica juvenil (LMMJ), solo un recuento de blastocitos de menos del 5 % y un recuento de plaquetas de más de 100 × 109 /l se relacionaron con mejor supervivencia para el SMD; un recuento de plaquetas de más de 40 × 109 /l pronosticó un mejor desenlace de la LMMJ.[
Los SMD infantiles se agrupan en diferentes categorías, cada una con características clínicas y biológicas específicas, de la siguiente manera:[
- Síndrome mielodisplásico que surge a partir de un síndrome de insuficiencia medular hereditario, por ejemplo, la anemia de Fanconi, la neutropenia congénita grave y el síndrome de Shwachman-Diamond.
- Síndrome mielodisplásico que surge a partir de una anemia aplásica grave.
- Síndrome mielodisplásico secundario que surge a partir de una agresión citotóxica, como dosis altas de quimioterapia alquilante.
- Síndrome mielodisplásico primario que incluye casos de SMD que no se enumeraron antes, teniendo en cuenta que algunos de los casos caracterizados como SMD primarios también se vinculan a síndromes de predisposición.
La caracterización genómica de los SMD primarios infantiles permitió identificar subgrupos específicos definidos mediante alteraciones en ciertos genes. Por ejemplo, las mutaciones de la línea germinal en GATA2,[
| Tipo de síndrome mielodisplásico | Médula ósea | Sangre periférica | |
|---|---|---|---|
| a Adaptado de Arber et al.[ |
|||
| b Se destaca que los casos con pancitopenia se clasificarán como SMD-SC. | |||
| c Cuando la médula tiene <5 % de mieloblastos, pero la sangre periférica tiene 2–4 % de mieloblastos, el diagnóstico es SMD-EB-1. | |||
| d El diagnóstico de SMD-EB-2 se establece si se cumple alguno de los siguientes criterios: médula con 10–19 % de blastocitos, sangre periférica con 5–19 % de blastocitos o cuerpos de Auer. | |||
| e Anomalías cromosómicas recurrentes en el SMD: desequilibradas: +8, -7 o del(7q), -5 o del(5q), del(20q), -Y, i(17q) o t(17p), -13 o del(13q), del(11q), del(12p) o t(12p), del(9q), idic(X)(q13); equilibradas: t(11;16)(q23;p13.3), t(3;21)(q26.2;q22.1), t(1;3)(p36.3;q21.2), t(2;11)(p21;q23), inv(3)(q21q26.2), t(6;9)(p23;q34). En la clasificación de la OMS se indica que se deberá considerar la presencia de estas anomalías cromosómicas si hay citopenias persistentes de origen indeterminado con el fin de respaldar un diagnóstico provisional de SMD cuando no se observan características morfológicas. | |||
| f Los criterios diagnósticos del SMD infantil (citopenia refractaria infantil-anotación provisional) son los siguientes: 1) citopenia persistente en 1 a 3 líneas celulares con <5 % de blastocitos en la médula ósea, <2 % de blastocitos en sangre periférica, ausencia de sideroblastos en anillo y 2) se deben encontrar cambios displásicos en 1–3 linajes. | |||
| SMD con displasia de un solo linaje | Displasia de un linaje: ≥10 % de un linaje mieloide | 1–2 citopeniasb | |
| <5 % de blastocitos | Blastocitos <1 %c | ||
| <15 % de sideroblastos en anillo | |||
| SMD con sideroblastos en anillo (SMD-SA) | Displasia eritroide sola | ||
| <5 % de blastocitos | Sin blastocitos | ||
| ≥15 % de sideroblastos en anillo | |||
| SMD con displasia de linajes múltiples | Displasia en ≥10 % de las células en ≥2 linajes mieloides | 1–3 citopenias | |
| <5 % de blastocitos | Blastocitos (ninguno o <1 %)c | ||
| ±15 % de sideroblastos en anillo | |||
| Sin bastones de Auer | Sin bastones de Auer | ||
| <1×109 monocitos/l | |||
| SMD con exceso de blastocitos-1 (SMD-EB-1) | Displasia de un solo linaje o de linajes múltiples | Citopenias | |
| <5–9 % de blastocitosc | <5 % de blastocitosc | ||
| Sin bastones de Auer | Sin bastones de Auer | ||
| <1×109 monocitos/l | |||
| SMD con exceso de blastocitos-2 (SMD-EB-2) | Displasia de un solo linaje o de linajes múltiples | Citopenias | |
| 10–19 % de blastocitosd | 5–19 % de blastocitosd | ||
| ± bastones de Auerd | ± bastones de Auerd | ||
| <1×109 monocitos/l | |||
| SMD relacionado con del(5q) aislada | Megacariocitos normales o aumentados (núcleos hipolobulados) | Anemia | |
| <5 % de blastocitos | Blastocitos (ninguno o <1 %) | ||
| Sin bastones de Auer | Recuento de plaquetas normal o aumentado | ||
| del(5q) aislada | |||
| SMD sin clasificar (SMD-SC) | Displasia de <10 % de las células en ≥1 linaje mieloide | Citopenias | |
| Anomalías citogenéticas relacionadas con un diagnóstico de SMDe | <1 % de blastocitosc | ||
| <5 % de blastocitos | |||
| Entidad provisional: Citopenia refractaria infantilf | Para obtener más información, consultar el Cuadro 4. | ||
| | Linaje eritroide | Linaje mieloide | Linaje megacariocítico |
|---|---|---|---|
| a Adaptado de Baumann et al.[ |
|||
| b Es posible que se necesite una trepanación o biopsia de médula ósea porque la médula ósea en la citopenia refractaria infantil a menudo es hipocelular. | |||
| c Las características incluyen lobulación anómala del núcleo, células multinucleadas y puentes nucleares. | |||
| d Presencia de células con anomalía pseudo–Pelger-Huet, citoplasma hipogranular o agranular, formas enbanda gigantes. | |||
| e Los megacariocitos tienen tamaño variable y, a menudo, núcleos redondos o separados; la ausencia de megacariocitos no excluye el diagnóstico de citopenia refractaria infantil. | |||
| Aspirado de médula óseab | Displasia o cambios megaloblastoides en ≥10 % de los precursores eritroidesc | Displasia en ≥10 % de los precursores granulocíticos o neutrófilos | Micromegacariocitos y otras características displásicase |
| <5 % de blastocitosd | |||
| Biopsia de médula ósea | Hay precursores eritroides | No hay criterios adicionales | Micromegacariocitos y otras características displásicase |
| Aumento de proeritroblastos | Prueba inmunohistoquímica positiva para CD61 y CD41 | ||
| Aumento del número de mitosis | |||
| Sangre periférica | Displasia en ≥10 % de los neutrófilos | ||
| <2 % de blastocitos | |||
Evaluación histoquímica, inmunofenotípica y molecular de la leucemia mieloide aguda infantil
Evaluación histoquímica
El tratamiento de los niños con leucemia mieloide aguda (LMA) varía de forma significativa del tratamiento para los niños con leucemia linfoblástica aguda (LLA). En consecuencia, es fundamental diferenciar la LMA de la LLA. Las tinciones histoquímicas especiales que se aplican a las muestras de médula ósea de los niños con leucemia aguda sirven para confirmar el diagnóstico. Las tinciones que se usan más a menudo son la mieloperoxidasa, el ácido peryódico de Schiff, el negro Sudán B y la esterasa. En la mayoría de los casos, el patrón de tinción con estas técnicas histoquímicas permitirá diferenciar la LMA de la leucemia mielomonocítica aguda (LMMA) y de la LLA (consultar el Cuadro 5). La inmunofenotipificación por citometría de flujo reemplazó la mayoría de las tinciones histoquímicas.
| | M0 | LMA, LPA (M1-M3) | LMMA (M4) | LMoA (M5) | LEA (M6) | LMCA (M7) | LLA | |
|---|---|---|---|---|---|---|---|---|
| LEA = leucemia eritroide aguda; LLA = leucemia linfoblástica aguda; LMA = leucemia mieloide aguda; LMCA = leucemia megacariocítica aguda; LMMA = leucemia mielomonocítica aguda; LMoA = leucemia monocítica aguda; LPA = leucemia promielocítica aguda; PAS = ácido peryódico de Schiff. | ||||||||
| a Para obtener más información sobre el sistema de clasificación morfológica e histoquímica de la LMA, consultar la sección Clasificación de la leucemia mieloide aguda infantil del French-American-British. | ||||||||
| b Estas reacciones se inhiben con fluoruro. | ||||||||
| Mieloperoxidasa | - | + | + | - | - | - | - | |
| Esterasas inespecíficas | ||||||||
| Cloroacetato | - | + | + | ± | - | - | - | |
| Acetato de alfanaftol | - | - | +b | +b | - | ±b | - | |
| Negro Sudán B | - | + | + | - | - | - | - | |
| PAS | - | - | ± | ± | + | - | + | |
Evaluación inmunofenotípica
El uso de anticuerpos monoclonales para determinar los antígenos de superficie celular de la LMA ayuda a reforzar el diagnóstico histológico. En el momento del proceso diagnóstico inicial de la leucemia, se deben emplear varios anticuerpos monoclonales específicos de cada linaje que detectan los antígenos celulares de la LMA, junto con un conjunto de marcadores específicos del linaje de los linfocitos T y B que ayuden a distinguir la LMA de la LLA y las leucemias agudas de linaje ambiguo. La expresión de diversas proteínas de conglomerados determinantes (CD), consideradas como relativamente específicas de cada linaje de la LMA son CD33, CD13, CD14, CDw41 (o antiglicoproteína plaquetaria IIb/IIIa), CD15, CD11B, CD36 y antiglicoforina A. Los antígenos relacionados con el linaje de linfocitos B CD10, CD19, CD20, CD22 y CD24 se encuentran en un 10 % a un 20 % de los casos de LMA, pero a menudo no expresan inmunoglobulina monoclonal de superficie ni las cadenas pesadas de inmunoglobulina citoplasmática; de manera parecida, se encuentran antígenos relacionados con el linaje de linfocitos T CD2, CD3, CD5 y CD7 en un 20 % a un 40 % de los casos de LMA.[
La inmunofenotipificación también puede ser útil para distinguir los siguientes subtipos de LMA según la clasificación French-American-British (FAB):
- Las pruebas que identifican la presencia del antígeno HLA relacionado con D (HLA-DR) a veces son útiles para identificar la leucemia promielocítica aguda (LPA). En general, el HLA-DR se expresa en un 75 % a un 80 % de las células de LMA, pero, pocas veces en las células de LPA.[
39 ,40 ] Además, la LPA se caracteriza por una expresión fuerte de CD33 y expresión de CD117 (c-KIT) en la mayoría de los casos, además de expresión heterogénea de CD13 y, a menudo, expresión baja o ausente de CD34, CD11a y CD18.[39 ,40 ] La variante microgranular de LPA M3v por lo común expresa CD34 y CD2.[39 ,41 ] - Las pruebas para identificar la glicoproteína Ib, la glicoproteína IIb/IIIa o la expresión del antígeno del factor VIII son útiles para el diagnóstico de la M7 (leucemia megacariocítica).
- La expresión de glucoforina es útil para el diagnóstico de la M6 (leucemia eritroide).
Menos del 5 % de los casos de leucemia aguda infantil tiene linaje ambiguo, con características tanto de linaje mieloide como linfoide.[
Evaluación molecular
Características moleculares de la leucemia mieloide aguda
Una caracterización molecular integral de la leucemia mieloide aguda (LMA) en niños y adultos indicó que la LMA es una enfermedad que exhibe coincidencias y diferencias en todos los grupos de edades.[
- La LMA infantil, a diferencia de la de adultos, es por lo general una enfermedad de alteraciones cromosómicas recurrentes. Consultar el Cuadro 6 para obtener una lista de fusiones génicas frecuentes.[
44 ,46 ] Dentro del grupo de edad pediátrica, algunas fusiones génicas ocurren en especial en niños menores de 5 años (por ejemplo, en los genes NUP98 y KMT2A y la fusión génica CBFA2T3::GLIS2), mientras que otras ocurren por lo general en niños de 5 años o más (por ejemplo, fusiones génicas RUNX1::RUNX1T1, CBFB::MYH11 y NPM1::RARA). - Los pacientes pediátricos con LMA presentan tasas bajas de mutación, en la mayoría de los casos exhiben menos de un cambio somático por megabase en las regiones de codificación de proteínas.[
45 ] Esta tasa de mutación es algo más baja que la que se observa en la LMA de adultos y es mucho más baja que la tasa de mutación de los cánceres que responden a los inhibidores de puntos de control (por ejemplo, el melanoma).[45 ] - El patrón de mutaciones génicas difiere entre los casos de LMA infantil y en adultos. Por ejemplo, las mutaciones en IDH1, IDH2, TP53, RUNX1 y DNMT3A son más comunes en la LMA en adultos que en la infantil, mientras que las mutaciones en NRAS y WT1 son significativamente más comunes en la LMA infantil.[
44 ,45 ]
En los niños con LMA se hacen análisis genéticos de los blastocitos de la leucemia (mediante métodos citogenéticos convencionales y métodos moleculares) porque las anomalías cromosómicas y moleculares son marcadores diagnósticos y pronósticos importantes.[
La detección de anomalías moleculares también ayuda a estratificar el riesgo y asignar el tratamiento. Por ejemplo, las mutaciones en NPM y CEBPA se relacionan con desenlaces favorables mientras que determinadas mutaciones en FLT3 acarrean riesgo alto de recaída; es posible que identificar estas últimas mutaciones permita usar terapia dirigida.[
En la revisión de 2016 de la clasificación de las neoplasias mieloides y la leucemia aguda de la Organización Mundial de la Salud (OMS), se enfatiza que las translocaciones cromosómicas recurrentes de la LMA infantil tal vez sean únicas o tengan una prevalencia diferente de la LMA en adultos.[
| Producto de la fusión génica | Translocación cromosómica | Prevalencia en la LMA infantil (%) |
|---|---|---|
| a Translocación cromosómica críptica; LMA: leucemia mieloide aguda. | ||
| Translocación deKMT2A(MLL) | 11q23.3 | 25,0 |
| NUP98::NSD1a | t(5;11)(q35.3;p15.5) | 7,0 |
| CBFA2T3::GLIS2a | inv(16)(p13.3;q24.3) | 3,0 |
| NUP98::KDM5Aa | t(11;12)(p15.5;p13.5) | 3,0 |
| DEK::NUP214 | t(6;9)(p22.3;q34.1) | 1,7 |
| RBM15(OTT)::MKL1 (MAL) | t(1;22)(p13.3;q13.1) | 0,8 |
| MNX1::ETV6 | t(7;12)(q36.3;p13.2) | 0,8 |
| KAT6A::CREBBP | t(8;16)(p11.2;p13.3) | 0,5 |
| RUNX1::RUNX1T1 | t(8;21)(q22;q22) | 13–14 |
| CBFB::MYH11 | inv(16)(p13.1;q22) o t(16;16)(p13.1;q22) | 4–9 |
| PML::RARA | t(15;17)(q24;q21) | 6–11 |
El panorama genómico de los casos de LMA infantil puede cambiar desde el momento del diagnóstico hasta la recidiva. Las mutaciones detectables en el momento del diagnóstico a veces no se encuentran en el momento de la recaída y, a la inversa, aparecen mutaciones nuevas en ese momento. Un hallazgo clave en un estudio de 20 casos para los que se disponía de datos de secuenciación en el momento del diagnóstico y de la recaída fue que la frecuencia de una variante alélica en el momento del diagnóstico se correlacionó de manera sólida con persistencia de las mutaciones en el momento de la recaída.[
A continuación, se presenta una descripción breve de las anomalías citogenéticas y moleculares recurrentes específicas. Las anomalías se enumeran según aquellas en uso clínico que permiten identificar a los pacientes con un pronóstico favorable o desfavorable, seguidas de otras anomalías. Cuando se considera relevante se incluye la nomenclatura de la revisión de 2016 de la clasificación de las neoplasias mieloides y la leucemia aguda de la OMS.
Anomalías genéticas relacionadas con un pronóstico favorable
Las anomalías genéticas relacionadas con un pronóstico favorable son las siguientes:
- La LMA con factor de unión nuclear (CBF) incluye casos con las fusiones génicas RUNX1::RUNX1T1 y CBFB::MYH11 que alteran la actividad del CBF conformado por RUNX1 y CBFB. Estas son entidades específicas en la revisión de 2016 de la clasificación de las neoplasias mieloides y la leucemia aguda de la OMS.
- LMA con fusiones génicas RUNX1::RUNX1T1 (t(8;21)(q22;q22.1)). En las leucemias con t(8;21), el gen RUNX1 (AML1) del cromosoma 21 se fusiona con el gen RUNX1T1 (ETO) del cromosoma 8. La translocación t(8;21) se relaciona con el subtipo FAB M2 y con los sarcomas granulocíticos. Los adultos que tienen LMA con t(8;21) tienen un pronóstico más favorable que los adultos que tienen otros tipos de LMA.[
47 ] Los niños que tienen LMA con t(8;21) presentan un desenlace más favorable que los niños con una LMA caracterizada por cariotipos normales o complejos,[47 ,57 ,58 ,59 ] y tasas de supervivencia general (SG) a 5 años del 74 % al 90 %.[48 ,49 ,60 ] La translocación t(8;21) se presenta en cerca del 12 % de los niños con LMA.[48 ,49 ,60 ] - LMA con fusiones génicas CBFB::MYH11 (inv(16)(p13.1;q22) o t(16;16)(p13.1;q22)). En las leucemias con inv(16), el gen CBFB de la banda cromosómica 16q22 se fusiona con el gen MYH11 de la banda cromosómica 16p13. La translocación inv(16) se relaciona con el subtipo FAB M4Eo. La inv(16) confiere un pronóstico favorable para adultos y niños con LMA,[
47 ,57 ,58 ,59 ] y una tasa de SG a 5 años de casi el 85 %.[48 ,49 ] La inv(16) se presenta en el 7 % al 9 % de los niños con LMA.[48 ,49 ,60 ] Como se indicó antes, los casos con fusiones CBFB::MYH11 y los casos con fusiones RUNX1::RUNX1T1 tienen mutaciones secundarias particulares; las mutaciones secundarias tipo CBFB::MYH11 se restringen sobre todo a los genes que activan la señalización del receptores tirosina–cinasa (NRAS, FLT3 y KIT).[61 ,62 ] - LMA con fusiones génicas RUNX1::CBFA2T3 (t(16;21)(q24;q22)). En las leucemias con t(16;21)(q24;q22), el gen RUNX1 se fusiona con el gen CBFA2T3 y el perfil de expresión génica se relaciona de forma estrecha con los casos de LMA con t(8;21) y fusiones RUNX1::RUNX1T1.[
63 ] Este tipo de leucemia se presenta a una mediana de 7 años de edad y es poco frecuente; representa entre el 0,1 % y el 0,3 % de los casos pediátricos de LMA. De los 23 pacientes con fusiones RUNX1::CBFA2T3, 5 tuvieron LMA secundaria, que incluyó a 2 pacientes con diagnóstico primario de sarcoma de Ewing. La cohorte de 23 pacientes tuvo un desenlace favorable, con una tasa de SSC a 4 años del 77 % y una tasa de incidencia acumulada de recaída del 0 %.[63 ]
Los subtipos con fusiones génicas RUNX1::RUNX1T1 y CBFB::MYH11 por lo común exhiben mutaciones en los genes que activan la señalización de receptores tirosina–cinasa (por ejemplo, NRAS, FLT3 y KIT); los genes NRAS y KIT son los que más a menudo presentan mutaciones en ambos subtipos. Se ha estudiado la importancia pronóstica de las mutaciones activadoras en KIT en adultos con LMA CBF y los resultados han sido contradictorios. En un metanálisis se encontró que las mutaciones en KIT aumentan el riesgo de recaída sin un efecto en la SG de los adultos con LMA y fusiones RUNX1::RUNX1T1.[
64 ] En niños y adultos con LMA CBF, las mutaciones en KIT son a menudo subclonales;[65 ,66 ] en adultos con LMA y fusiones RUNX1::RUNX1T1, una proporción más alta de alelos mutados de KIT se relaciona con un riesgo más alto de fracaso del tratamiento.[61 ,65 ] Aún está por aclararse la importancia pronóstica de las mutaciones en KIT en los casos de LMA CBF infantil; en algunos estudios, no se encontró un efecto de las mutaciones en KIT sobre el desenlace,[67 ,68 ,69 ] mientras que en otros estudios se notificó un riesgo más alto de fracaso del tratamiento cuando se presentan mutaciones en KIT.[66 ,70 ,71 ,72 ,73 ]Aunque tanto los genes de fusión RUNX1::RUNX1T1 como CBFB::MYH11 alteran la actividad del CBF, los casos que presentan estas alteraciones genómicas exhiben mutaciones secundarias características.[
61 ,62 ]- Los pacientes con fusiones RUNX1::RUNX1T1 también presentan mutaciones frecuentes en los genes que regulan la conformación de la cromatina (por ejemplo, ASXL1 y ASXL2) (40 % de los casos) y los genes que codifican componentes del complejo de la cohesina (20 % de los casos). Las mutaciones en ASXL1 y las mutaciones en ASXL2 de los componentes del complejo de la cohesina son infrecuentes en los casos de leucemias con fusiones génicas CBFB::MYH11.[
61 ,62 ]En un estudio de 204 adultos con LMA y fusiones RUNX1::RUNX1T1, se encontró que las mutaciones en ASXL2 (presentes en el 17 % de los casos) y las mutaciones en ASXL1 o ASXL2 (presentes en el 25 % de los casos) carecen de importancia pronóstica.[
74 ] Se informaron resultados similares, aunque con números más pequeños, en niños con LMA y fusiones RUNX1::RUNX1T1 que además presentan mutaciones en ASXL1 y ASXL2.[75 ]
- LMA con fusiones génicas RUNX1::RUNX1T1 (t(8;21)(q22;q22.1)). En las leucemias con t(8;21), el gen RUNX1 (AML1) del cromosoma 21 se fusiona con el gen RUNX1T1 (ETO) del cromosoma 8. La translocación t(8;21) se relaciona con el subtipo FAB M2 y con los sarcomas granulocíticos. Los adultos que tienen LMA con t(8;21) tienen un pronóstico más favorable que los adultos que tienen otros tipos de LMA.[
- Leucemia promielocítica aguda (LPA) con fusiones génicas PML::RARA. La LPA da cuenta de cerca del 7 % de los niños con LMA.[
49 ,76 ] La LMA con t(15;17) se relaciona de manera invariable con la LPA, un subtipo específico de LMA que se trata de forma diferente a otros tipos de LMA debido a su marcada sensibilidad al trióxido de arsénico y los efectos diferenciadores de la tretinoína. Es posible que la translocación t(15;17) u otros reordenamientos cromosómicos más complejos conduzcan a la producción de una proteína de fusión que afectan el receptor α del ácido retinoico y PML. En la revisión de 2016 de la clasificación de la OMS no se incluye la designación citogenética t(15;17) para enfatizar la importancia de la fusión PML::RARA, que tal vez sea críptica o surja de cambios cariotípicos complejos.[12 ]Se ha vuelto una práctica estándar el empleo de la reacción en cadena de la polimerasa con retrotranscripción (RCP-RT) cuantitativa para identificar los transcritos de PML::RARA.[
77 ] La RCP-RT cuantitativa permite identificar tres variantes frecuentes de los transcritos; se usa para vigilar la reacción al tratamiento y con el fin de detectar temprano una recidiva molecular. Otras translocaciones mucho menos comunes que afectan el receptor α del ácido retinoico también pueden conducir a la LPA (por ejemplo, t(11;17)(q23;q21) que compromete el gen PLZF).[78 ,79 ] Es importante identificar los casos que tienen t(11;17)(q23;q21) debido a que presentan una disminución de la sensibilidad a la tretinoína.[78 ] - LMA con mutación en NPM1. La NPM1 es una proteína que se ha relacionado con el ensamblaje y trasporte proteico en los ribosomas; además es una chaperona molecular que previene la agregación proteica en el nucléolo. Se pueden utilizar métodos inmunohistoquímicos para identificar con precisión a los pacientes que tienen mutaciones en NPM1 cuando se demuestra la ubicación citoplasmática de NPM. Las mutaciones en la proteína NPM1 que reducen su ubicación nuclear se relacionan de manera primaria con un subconjunto de LMA con un cariotipo normal, que no expresa CD34, y presenta mejor pronóstico cuando no hay mutaciones tipo FLT3-ITD en adultos y adultos jóvenes.[
80 ,81 ,82 ,83 ,84 ,85 ]Los estudios de niños con LMA indican una menor tasa de mutaciones en NPM1 en los niños en comparación con los adultos que tienen características citogenéticas normales. Las mutaciones en NPM1 afectan a casi el 8 % de los pacientes de LMA infantil y son infrecuentes en niños menores de 2 años.[
51 ,52 ,86 ,87 ] Las mutaciones en NPM1 se relacionan con un pronóstico favorable en pacientes de LMA caracterizada por un cariotipo normal.[51 ,52 ,87 ] Se publicaron informes contradictorios sobre la importancia pronóstica en la población pediátrica de una mutación en NPM1 cuando también hay mutación FLT3-ITD. En un estudio se informó que una mutación en NPM1 no anula por completo el pronóstico precario que acarrea la mutación FLT3-ITD;[51 ,88 ] sin embargo, en otros estudios se observó que no hay efecto de la mutación FLT3-ITD sobre el pronóstico favorable relacionado con la mutación en NPM1.[45 ,52 ,87 ] - LMA con mutaciones bialélicas en CEBPA. Las mutaciones en el gen CEBPA se producen en un subgrupo de niños y adultos que tienen LMA con características citogenéticas normales.[
89 ,90 ] En los adultos menores de 60 años, cerca del 15 % de los casos de LMA con características citogenéticas normales tienen mutaciones en CEBPA.[84 ] El desenlace para los adultos de LMA con mutaciones en CEBPA es relativamente favorable y similar al de los pacientes que tienen leucemias con CBF.[84 ,91 ] En los estudios de adultos con LMA se demostró que la mutación doble en CEBPA, pero no la mutación en un solo alelo, se relacionó de modo independiente con un pronóstico favorable de la LMA;[92 ,93 ,94 ,95 ] ello llevó a que, en la revisión de 2016 de la OMS, se incluyeran las mutaciones bialélicas como una característica distinta para definir la enfermedad.[12 ] Sin embargo, en un estudio de más de 4700 adultos con LMA se observó que los pacientes con mutación en un solo alelo de CEBPA en el dominio C-terminal bZip tienen características clínicas y desenlaces favorables similares a los de los pacientes de LMA con mutación en ambos alelos.[96 ]Hay mutaciones en CEBPA en alrededor del 5 % de los niños con LMA y se encuentran casi siempre en el subtipo de LMA con características citogenéticas normales, tipo FAB M1 o M2.
- Los pacientes con mutaciones en ambos alelos de CEBPA o mutaciones bZip en uno de los alelos de CEBPA tienen una mediana de edad de presentación de 12 a 13 años y perfiles de expresión muy parecidos entre sí.[
90 ] - Cerca del 80 % de los pacientes pediátricos tienen ambos alelos mutados (es decir, casos que tienen al mismo tiempo una mutación en el dominio TAD de CEBPA y una mutación en el dominio bZip de CEBPA), lo que predice una supervivencia significativamente mejor, similar al efecto observado en los estudios de adultos.[
90 ,97 ] - En un estudio de cerca de 3000 niños con LMA, se observó que los pacientes con mutaciones en ambos alelos de CEBPA y aquellos con solo una mutación en el dominio bZip tenían un pronóstico favorable, en comparación con los pacientes con el tipo natural de CEBPA.[
90 ] - Las mutaciones en CSF3R se presentan en el 10 % al 15 % de los pacientes de LMA con mutación en CEBPA. La presencia de las mutaciones en CSF3R se asocia a un mayor riesgo de recaída, pero sin consecuencias en la supervivencia general.[
90 ,98 ] - En pacientes de LMA con mutación en ambos alelos de CEBPA recién diagnosticada, se debe considerar el análisis de la línea germinal además de las preguntas habituales sobre los antecedentes familiares, porque se ha notificado que el 5 % al 10 % de estos pacientes tiene una mutación germinal en CEBPA.[
89 ]
- Los pacientes con mutaciones en ambos alelos de CEBPA o mutaciones bZip en uno de los alelos de CEBPA tienen una mediana de edad de presentación de 12 a 13 años y perfiles de expresión muy parecidos entre sí.[
- Leucemia mieloide relacionada con el síndrome de Down (mutaciones en GATA1). Hay mutaciones en GATA1 en la mayoría, si no en todos, los niños con síndrome de Down que tienen mielopoyesis anormal transitoria (MAT) o leucemia megacarioblástica aguda (LMCA).[
99 ,100 ,101 ,102 ] También se encuentran mutaciones en GATA1 en el 9 % de los niños con LMCA que no tienen síndrome de Down y el 4 % de los adultos con LMCA (se presentó de manera simultánea con una amplificación del gen RCAN1 [DSCR1] en el cromosoma 21 en 9 de 10 casos).[103 ] El gen GATA1 forma un factor de transcripción necesario para el desarrollo normal de los eritrocitos, megacariocitos, eosinófilos y mastocitos.Las mutaciones en GATA1 confieren un aumento de la sensibilidad a la citarabina por el descenso regulado en la expresión de la citidina desaminasa, lo que quizá proporcione una explicación para el desenlace superior de los niños con síndrome de Down y LMA M7 que reciben tratamiento con regímenes que contienen citarabina.[
104 ]
Anomalías genéticas relacionadas con un pronóstico desfavorable
Las anomalías genéticas relacionadas con un pronóstico desfavorable son las siguientes:
- Cromosomas 5 y 7. Las anomalías cromosómicas relacionadas con un pronóstico precario en adultos con LMA incluyen las que afectan el cromosoma 5 (del(5q)) y el cromosoma 7 (monosomía 7).[
47 ,105 ,106 ] Estos subgrupos citogenéticos representan entre el 2 % y el 4 % de los casos de LMA infantil, respectivamente, y también se relacionan con un pronóstico precario en los niños.[48 ,105 ,106 ,107 ,108 ,109 ]En el pasado, se consideró que los pacientes con del(7q) también tenían un riesgo alto de fracaso del tratamiento; además, los datos de los adultos con LMA apoyan un pronóstico precario tanto para la del(7q) como para la monosomía 7.[
50 ] Sin embargo, los desenlaces en niños con del(7q), pero sin monosomía 7, son comparables a los de otros niños con LMA.[49 ,108 ] La presencia de la del(7q) no anula la importancia pronóstica de las características citogenéticas favorables (por ejemplo, inv(16) y t(8;21)).[47 ,108 ]Las anomalías en los cromosomas 5 y 7 carecen de importancia pronóstica para los pacientes de LMA con síndrome de Down de 4 años de edad y menos.[
110 ] - Hipodiploidía. La hipodiploidía se define como un número modal de cromosomas igual o inferior a 45. Esta situación es infrecuente en los pacientes pediátricos con LMA. En un análisis de una cohorte retrospectiva, el Berlin-Frankfurt-Münster AML Study Group se enfocó en caracterizar la hipodiploidía en pacientes pediátricos con LMA. Se excluyeron varios grupos de pacientes, como aquellos con LPA, síndrome de Down y pérdida de cromosoma 7.[
111 ] Se notificaron las siguientes observaciones:- Se observó hipodiploidía en el 1,3 % de los niños con LMA. Alrededor del 80 % de los pacientes tenía un número modal de cromosomas de 45, y el 20 % restante presentaba un número modal de cromosomas de 43 o 44.
- La mayoría de los pacientes (>80 %) con un número modal de cromosomas de 43 o 44 también cumplió con los criterios de cariotipo complejo. En este estudio, el cariotipo complejo se definió como la presencia de por lo menos 3 anormalidades cromosómicas diferentes, ya fueran anormalidades estructurales o defectos en el número de cromosomas, además de la ausencia de anomalías recurrentes definidas por la OMS.
- Los pacientes con un número modal de cromosomas de 43 o 44 presentaron tasas de SSC y SG inferiores, en comparación con los pacientes con 45 cromosomas (tasa de SSC, 21 vs. 37 %; P = 0,07; tasa de SG, 33 vs. 56 %; P = 0,1).
- LMA con alteración en GATA2 o MECOM (inv(3)(q21.3;q26.2) o t(3;3)(q21.3;q26.2)). El gen MECOM del cromosoma 3q26 codifica dos proteínas que regulan la transcripción: la EVI1 y la MDS1EVI1. Las anomalías inv(3) y t(3;3) producen sobreexpresión de EVI1 y disminuyen la expresión de GATA2.[
112 ,113 ] Estas anomalías se vinculan con un pronóstico precario en adultos con LMA,[47 ,105 ,114 ] pero son muy infrecuentes en niños (<1 % de casos de LMA infantil).[48 ,58 ,115 ]Las anomalías que afectan MECOM se detectan en algunos casos de LMA que tienen otras anomalías 3q y también se relacionaron con un pronóstico precario.
- Mutaciones en FLT3. La presencia de una mutación FLT3-ITD parece relacionarse con un pronóstico precario en los adultos con LMA;[
116 ] en particular, cuando ambos alelos están mutados o la proporción entre el alelo mutado y el alelo normal es alta.[117 ] Las mutaciones FLT3-ITD también conllevan un pronóstico precario en niños con LMA.[54 ,88 ,118 ,119 ,120 ] La frecuencia de las mutaciones FLT3-ITD en los niños es inferior a la de los adultos; en especial, para los niños menores de 10 años que en el 5 % al 10 % de los casos tienen la mutación (en comparación con casi el 30 % de los adultos).[119 ,120 ]La importancia pronóstica de FLT3-ITD se modifica por la presencia de otras alteraciones genómicas recurrentes. La prevalencia de FLT3-ITD aumenta en ciertos subtipos genómicos de LMA infantil, incluso en los casos que tienen la fusión génica NUP98::NSD1; de ellos, el 80 % al 90 % presentan FLT3-ITD.[
121 ,122 ] Cerca del 15 % de los pacientes con FLT3-ITD también tienen fusiones NUP98::NSD1; los pacientes con ambas alteraciones, FLT3-ITD y NUP98::NSD1, tienen un pronóstico más precario que los pacientes que presentan FLT3-ITD sin NUP98::NSD1.[122 ] Para los pacientes con FLT3-ITD, la presencia de mutaciones en WT1 o fusiones NUP98::NSD1 se relaciona con un desenlace más precario (tasas de SSC inferiores al 25 %) que el de los pacientes con FLT3-ITD, pero sin estas alteraciones.[45 ] Por el contrario, cuando FLT3-ITD se acompaña de mutaciones en NPM1, el desenlace es relativamente favorable y similar al de los casos de LMA infantil sin FLT3-ITD.[45 ]En la LPA, las mutaciones FLT3-ITD y las mutaciones puntuales se producen en el 30 % al 40 % de los niños y adultos.[
117 ,119 ,123 ,124 ] La presencia de las mutaciones FLT3-ITD tiene una relación sólida con la variante microgranular (M3v) de la LPA y con la hiperleucocitosis.[123 ,125 ,126 ,127 ] Todavía no está claro si las mutaciones en FLT3 entrañan un pronóstico más precario para los pacientes de LPA que reciben el tratamiento contemporáneo con tretinoína y trióxido de arsénico.[124 ,126 ,128 ,129 ,130 ,131 ]En niños y adultos con LMA también se identificaron mutaciones activadoras puntuales en FLT3, aunque la importancia clínica de estas mutaciones no está bien definida. Algunas de estas mutaciones puntuales parecen ser específicas de pacientes pediátricos.[
45 ] - LMA con fusiones génicas FUS::ERG (t(16;21)(p11;q22)). En las leucemias con t(16;21)(p11;q22), el gen FUS se fusiona con el gen ERG y produce un subtipo distinto de LMA con un perfil de expresión génica que se agrupa por separado de otros subgrupos citogéneticos.[
63 ] Este tipo de leucemia se presenta con una mediana de edad entre los 8 y 9 años, y es poco frecuente; representa entre el 0,3 % y el 0,5 % de los casos pediátricos de LMA. En una cohorte de 31 pacientes de LMA con fusiones FUS::ERG, el desenlace fue precario; la tasa de SSC a 4 años fue del 7 % y la tasa de incidencia acumulada de recaída fue del 74 %.[63 ]
Otras anomalías genéticas de la leucemia mieloide infantil
Otras anomalías genéticas de la LMA infantil son las siguientes:
- Reordenamientos del gen KMT2A (MLL). En casi el 20 % de los niños con LMA hay un reordenamiento del gen KMT2A.[
48 ,49 ] Estos casos, incluso la mayoría de las LMA secundarias a la exposición a la epipodofilotoxina,[132 ] por lo general se relacionan con una diferenciación monocítica (FAB M4 y M5). También se notificaron reordenamientos de KMT2A en casi el 10 % de los pacientes de LMCA con FAB M7 (consultar a continuación).[103 ,133 ]En la población de LMA infantil, la translocación más frecuente, que representa casi el 50 % de los casos con reordenamiento del gen KMT2A, es la t(9;11)(p22;q23); en esta, el gen KMT2A se fusiona con el gen MLLT3.[
134 ] En la revisión de 2016 de la clasificación de la OMS, se definió la LMA con fusiones génicas MLLT3::KMT2A (t(9;11)(p21.3;q23.3) como una entidad diferenciada. Sin embargo, se han identificado más de 50 parejas de fusión diferentes para el gen KMT2A en pacientes de LMA.En el entorno de la LMA infantil, la mediana de edad de los casos con reordenamiento 11q23/KMT2A es de cerca de 2 años; la mayoría de los subgrupos de translocaciones tienen una mediana de edad de menos de 5 años en el momento del cuadro clínico inicial.[
134 ] No obstante, se notificaron medianas de edad mucho más altas en el momento del cuadro clínico inicial de casos pediátricos que tienen t(6;11)(q27;q23) (12 años) y t(11;17)(q23;q21) (9 años).[134 ]Por lo general, se notifica que el desenlace para los pacientes de LMA de novo y reordenamientos del gen KMT2A es similar o un poco más precario que el observado en otros pacientes de LMA.[
47 ,48 ,134 ,135 ,136 ] Como el gen KMT2A puede participar en translocaciones con muchas parejas de genes de fusión, la pareja específica de gen de fusión quizá influya en el pronóstico, como se demostró en un gran estudio retrospectivo internacional de evaluación del desenlace de la LMA en 756 niños con 11q23- o reordenamiento de KMT2A.[134 ,136 ] Esto también se observó en los pacientes del ensayo del COG AAML0531 (NCT00372593) (n = 215), lo que derivó en una amplia variedad de desenlaces.[136 ] Este desenlace general menos favorable se anuló en uno de los grupos del ensayo AAML0531, en pacientes cuyo tratamiento incluyó gemtuzumab ozogamicina. La tasa de SSC en los pacientes de LMA con reordenamientos en KMT2A fue superior para el tratamiento con gemtuzumab ozogamicina (tasa de SSC, 48 % con gemtuzumab ozogamicina vs. 29 % sin este; P = 0,003). Los desenlaces de los pacientes con reordenamientos en KMT2A que recibieron gemtuzumab ozogamicina fueron similares a los desenlaces observados en los pacientes sin reordenamientos en KMT2A.[136 ]En los pacientes con el subtipo más prevalente de LMA con reordenamiento en KMT2A, t(9;11)(p21.3;q23.3)/fusiones génicas MLLT3::KMT2A, los grupos de ensayos clínicos individuales han descrito, de manera desigual, un pronóstico más favorable; sin embargo, ni en el estudio retrospectivo internacional ni en el estudio del COG se confirmó el pronóstico favorable para este subgrupo.[
47 ,48 ,134 ,136 ] Además, en un estudio de colaboración internacional para evaluar la LMCA infantil, se observó que la presencia de t(9;11), que se identificó en casi el 5 % de los casos de LMCA, se relacionó con un desenlace inferior al de otros casos de LMCA.[133 ]Los subgrupos de LMA con reordenamiento de KMT2A que se vinculan con desenlaces más precarios son los siguientes:
- Los casos con la translocación t(10;11) conforman un grupo de riesgo alto de recaída en la médula ósea y el SNC.[
47 ,49 ] Algunos casos con la translocación t(10;11) tienen una fusión del gen KMT2A con MLLT10 en 10p12, mientras que otros tienen una fusión del gen KMT2A con ABI1 en 10p11.2. En un estudio retrospectivo internacional se encontró que estos casos, que se manifiestan a una mediana de edad de alrededor de 1 a 3 años, tienen una tasa de SSC a 5 años del 17 % al 30 %.[134 ,136 ] - Los pacientes con t(6;11)(q27;q23) tienen un pronóstico precario, con una tasa de SSC a 5 años del 11 % al 15 %.[
136 ] - Los pacientes con t(4;11)(q21;q23) a menudo presentan hiperleucocitosis y también tienen un pronóstico precario, con una tasa de SSC a 5 años del 0 % al 29 %.[
134 ,136 ] - Los pacientes con t(11;19)(q23;p13.3) tienen un pronóstico precario, con una tasa de SSC a 5 años del 14 %.[
136 ] - En un estudio de seguimiento llevado a cabo por un grupo de colaboración internacional, se demostró que otras anomalías citogenéticas también afectan los desenlaces de los niños con translocaciones de KMT2A; los cariotipos complejos y la trisomía 19 predicen un desenlace precario y la trisomía 8 predice un desenlace más favorable.[
137 ] - La adición de la terapia con gemtuzumab ozogamicina mejoró el desenlace precario de estos pacientes que presentan reordenamientos en KMT2A con parejas de translocaciones de riesgo alto (27 % [intervalo de confianza (IC) 95%, 14–41%] vs. 6 % [ IC 95%, 1–18%]; P = 0,013).[
136 ]
- Los casos con la translocación t(10;11) conforman un grupo de riesgo alto de recaída en la médula ósea y el SNC.[
- LMA con fusiones génicas DEK::NUP214 (t(6;9)(p23;q34.1)). La t(6;9) conduce a la formación de la proteína de fusión DEK-NUP214 que se relaciona con la leucemia.[
138 ,139 ] Este subgrupo de LMA se vinculó con un pronóstico precario en adultos con LMA [138 ,140 ,141 ] y se presenta con poca frecuencia en los niños (menos del 1 % de los casos de LMA). La mediana de edad de los niños con LMA que tienen fusiones génicas DEK::NUP214 es de 10 a 11 años; cerca del 40 % de los pacientes pediátricos tienen FLT3-ITD.[142 ,143 ]La LMA con t(6;9) se relaciona con un riesgo alto de fracaso del tratamiento en los niños, sobre todo para aquellos que no pasan a recibir un trasplante alogénico de células madre.[
48 ,139 ,142 ,143 ] - Subgrupos moleculares de leucemia megacarioblástica aguda (LMCA) sin síndrome de Down. La LMCA representa cerca del 10 % de las LMA infantiles y tiene gran heterogeneidad molecular. A continuación, se enumeran los subtipos moleculares de LMCA.
- Fusiones génicas CBFA2T3::GLIS2. La fusión CBFA2T3::GLIS2 surge de una inversión críptica del cromosoma 16 (inv(16)(p13;3q24.3)).[
144 ,145 ,146 ,147 ,148 ] Por lo común, se presenta en la LMCA sin síndrome de Down; representa entre el 16 % y el 27 % de las LMCA infantiles y se manifiesta a una mediana de edad de 1 año.[103 ,146 ,149 ,150 ] Parece relacionarse con un desenlace desfavorable.[103 ,144 ,148 ,149 ,150 ]En un estudio de casi 2000 niños con LMA, la fusión CBFA2T3::GLIS2 se identificó en 39 casos (1,9 %), con una mediana de edad en el momento de la presentación de 1,5 años; todos los casos se observaron en niños menores de 3 años.[
151 ] Cerca de la mitad de los casos exhibían morfología megacarioblástica M7 y el 29 % de los pacientes eran negros o afroamericanos (excediendo la frecuencia del 12,8 % en pacientes sin la fusión). La fusión CBFA2T3::GLIS2 fue un factor pronóstico independiente tanto de la SG como de la SSC, La tasa de SG a 5 años fue del 22 % en los pacientes con fusiones CBFA2T3::GLIS2 versus el 63 % en los pacientes sin esa fusión. Las células leucémicas con fusiones CBFA2T3::GLIS2 tienen un inmunofenotipo característico (que en el inicio se notificó como fenotipo RAM),[152 ,153 ] con CD56 alto, expresión débil o negativa de CD45 y CD38, y ausencia de expresión de HLA-DR. - Reordenamientos de KMT2A. Los casos con translocaciones de KMT2A representan el 10 % al 17 % de las LMCA infantiles; el gen MLLT3 es la pareja de fusión más frecuente del gen KMT2A.[
103 ,133 ,149 ] Los pacientes con reordenamientos de KMT2A presentan un desenlace más precario, con una tasa de SG a los 4 o 5 años de casi el 30 %.[103 ,133 ,149 ] En una colaboración internacional sobre la LMCA infantil, se observó que la presencia de t(9;11)/fusiones MLLT3::KMT2A, que ocurre en cerca del 5 % de los casos de LMCA (n = 21), se vincula con un desenlace más precario (tasa de SG a 5 años, casi el 20 %) en comparación con otros casos de LMCA y otros reordenamientos de KMT2A (n = 17), cada uno con una tasa de SG a 5 años del 50 % al 55 %.[133 ] No se observó un desenlace más precario para los pacientes con otros reordenamientos de KMT2A (n = 17). - Fusiones génicas NUP98::KDM5A. Se observan fusiones NUP98::KDM5A en cerca del 10 % de los casos de LMCA infantil,[
103 ,149 ] y en tasas más bajas para los casos con otros tipos diferentes a LMCA.[150 ] Sin embargo, cerca de dos tercios de los niños con fusiones NUP98::KDM5A tienen un subtipo FAB diferente a LMCA (ver más adelante).[154 ] Los pacientes con fusiones NUP98::KDM5A presentaron una tendencia a un pronóstico más precario, aunque el número pequeño de casos estudiados restringió la confianza de esta determinación.[103 ,149 ] - Fusiones génicas RBM15::MKL1. La translocación t(1;22)(p13;q13) que produce las fusiones RBM15::MKL1 es infrecuente (<1 % de las LMA infantiles) y se limita a la leucemia megacariocítica aguda (LMCA).[
48 ,150 ,155 ,156 ,157 ,158 ] En estudios se observó que la t(1;22)(p13;q13) se encuentra en el 10 % al 18 % de los niños con LMCA en quienes se pueden evaluar las características citogenéticas o de genética molecular.[103 ,133 ,149 ] La mayoría de los casos de LMCA con t(1;22) se presentan en lactantes con una mediana de edad en el momento del cuadro clínico inicial (4 a 7 meses) menor que la de otros niños con LMCA.[133 ,146 ,159 ] También se han notificado casos en los que se detectan los transcritos de la fusión RBM15::MKL1 en ausencia de t(1;22) porque estos pacientes jóvenes por lo general tienen una médula ósea hipoplásica.[156 ]En un estudio retrospectivo de colaboración internacional de 51 casos con t(1;22), se informó que los pacientes con esta anomalía tuvieron una tasa de SSC a 5 años del 54,5 % y una tasa de SG del 58,2 %, similar a las tasas de otros niños con LMCA.[
133 ] En otro análisis retrospectivo internacional de 153 casos de LMCA sin síndrome de Down para los que se contaba con muestras para análisis molecular, la tasa de SSC a 4 años para los pacientes con t(1;22) fue del 59 % y la tasa de SG fue del 70 %; estas fueron significativamente mejores que las de los pacientes con LMCA que tenían otras anomalías genéticas específicas (fusiones CBFA2T3::GUS2, fusiones NUP98::KDM5A, reordenamientos de KMT2A y monosomía 7).[149 ] - Reordenamientos de HOX. En un informe, los casos con una fusión génica que afecta el complejo génico HOX representaron el 15 % de las LMCA infantiles.[
103 ] En este informe se observó que estos pacientes parecen tener un pronóstico relativamente favorable, aunque el número pequeño de casos estudiados restringió la confianza de esta determinación. - Mutaciones en GATA1. En los niños pequeños (mediana de edad, 1–2 años) con LMCA sin síndrome de Down surgen mutaciones interruptoras en GATA1 que se relacionan con amplificación del gen RCAN1 (DSCR1) en el cromosoma 21.[
103 ] Estos pacientes representan cerca del 10 % de las LMCA sin síndrome de Down y tienen un pronóstico favorable si no hay, de manera simultánea, genes de fusión con pronóstico desfavorable; aunque el número de pacientes estudiados fue bajo (n = 8).[103 ]
- Fusiones génicas CBFA2T3::GLIS2. La fusión CBFA2T3::GLIS2 surge de una inversión críptica del cromosoma 16 (inv(16)(p13;3q24.3)).[
- Fusión MYST3::CREBBP (t(8;16)). La translocación t(8;16) fusiona el gen MYST3 del cromosoma 8p11 con el gen CREBBP del cromosoma 16p13. La LMA con t(8;16) es infrecuente en niños. En un estudio del grupo Internacional del Berlin-Frankfurt-Münster (IBFM) con 62 niños que tenían LMA, la presencia de esta translocación se relacionó con una edad menor en el momento del diagnóstico (mediana, 1,2 años), fenotipo FAB M4/M5, eritrofagocitosis, leucemia cutánea y coagulación intravascular diseminada.[
160 ] El desenlace para los niños que tienen LMA con t(8;16) es similar al de otros tipos de LMA.Una proporción importante de los lactantes que reciben un diagnóstico de LMA con t(8;16) durante el primer mes de vida remiten de manera espontánea, aunque es posible que la enfermedad recidive meses o años después.[
160 ,161 ,162 ,163 ] Estas observaciones indican que se podría considerar una estrategia de observar y esperar para los casos de LMA con t(8;16) diagnosticada en el período neonatal si se puede garantizar una vigilancia estrecha a largo plazo.[160 ] - Alteración t(7;12)(q36;p13). La translocación t(7;12)(q36;p13) afecta el gen ETV6 en el cromosoma 12p13 y puntos de ruptura variables de la región MNX1 en el cromosoma 7q36 (HLXB9). Es posible que la translocación sea críptica en un cariotipado convencional y, en ocasiones, solo se confirma mediante FISH.[
164 ,165 ] Esta alteración se produce de manera casi exclusiva en niños menores de 2 años, es mutuamente excluyente del reordenamiento de KMT2A y se relaciona con un riesgo alto de fracaso del tratamiento.[48 ,49 ,87 ,164 ,166 ,167 ] - Fusiones génicas NUP98. Se notificó que NUP98 forma fusiones génicas leucemógenas con más de 20 parejas de genes diferentes.[
168 ] En el entorno de la LMA infantil, las dos fusiones génicas más comunes son NUP98-::NSD1 y NUP98::KDM5A; el primero se observó en un informe en cerca del 15 % de casos de LMA infantil con características citogenéticas normales y el segundo se observó en cerca del 10 % de las LMCA infantiles (consultar más arriba).[103 ,121 ,146 ] Los casos de LMA con cualquier fusión génica de NUP98 exhiben una expresión alta de los genes HOXA y HOXB; ello indica un fenotipo de células madre.[139 ,146 ]La fusión génica NUP98::NSD1, que a menudo es críptico en el análisis citogenético, resulta de la fusión de NUP98 (cromosoma 11p15) con NSD1 (cromosoma 5q35).[
121 ,122 ,139 ,169 ] Esta alteración se produce en cerca del 4 % al 7 % de los casos de LMA infantil.[12 ,55 ,121 ,139 ,170 ]- La frecuencia más alta de fusiones NUP98::NSD1 en la población pediátrica se observa en niños de 5 a 9 años (casi el 8 %); la frecuencia más baja se observa en niños más pequeños (casi el 2 % en niños menores de 2 años).
- Los pacientes con fusiones NUP98::NSD1 presentaron al inicio un recuento alto de glóbulos blancos (GB) (mediana, 147 × 109 /l en un estudio).[
121 ,122 ] La mayoría de los pacientes de LMA con fusiones NUP98::NSD1 no exhiben anomalías citogenéticas.[121 ,139 ] - Un porcentaje alto de los pacientes con fusiones NUP98::NSD1 (74 a 90 %) exhiben FLT3-ITD.[
55 ,121 ,122 ] - En un estudio de 12 niños con LMA y fusiones NUP98::NSD1 se notificó que, aunque todos los pacientes alcanzaron un respuesta completa (RC), la presencia de fusiones NUP98::NSD1 predijo de modo independiente un pronóstico precario:. Los niños con LMA y fusiones NUP98::NSD1 tenían un riesgo alto de recaída que resultó en una tasa de SSC a 4 años de cerca del 10 %.[
121 ] En otros estudio de niños (n = 38) y adultos (n = 7) con LMA y fusión NUP98::NSD1, la presencia de las fusiones NUP98NSD1 y FLT3-ITD predijo de manera independiente un pronóstico precario; los pacientes con ambas lesiones exhibieron una tasa de RC baja (alrededor de un 30 %) y una tasa baja de SSC a 3 años (alrededor de un 15 %).[122 ] - En un estudio de niños con LMA resistente al tratamiento, NUP98 estuvo sobrerrepresentada en comparación con una cohorte que sí alcanzó la remisión (21 % [6 de 28 pacientes vs. <4 %).[
171 ]
La fusión génica NUP98::KDM5A resulta de la fusión del gen NUP98 con el gen KDM5A, que a su vez surge a raíz de un translocación críptica en las pruebas citogenéticas, t(11;12)(p15;p13).[
172 ] Cerca del 2 % de los pacientes de LMA infantil tienen NUP98::KDM5A; estos casos tienden a presentarse a una edad temprana (mediana, 3 años).[154 ]- Los casos con NUP98::KDM5A tienden a ser de LMCA (34 %), seguidos de FAB M5 (21 %) y FAB M6 (17 %).[
154 ] Las fusiones NUP98::KDM5A se observa en cerca del 10 % de los casos de LMCA infantil.[103 ,149 ] - Otras anomalías genéticas relacionadas con la LMA infantil, incluso las mutaciones en FLT3, son poco frecuentes en los pacientes con NUP98::KDM5A.[
154 ] - El pronóstico de los niños con NUP98::KDM5A es más precario que el de otros niños con LMA (tasa de SSC a 5 años del 29,6 ± 14,6 % y una tasa de SG del 34,1 ± 16,1 %).[
154 ]
- Mutaciones en RUNX1. La LMA con mutación en RUNX1, que es una entidad provisional en la clasificación de la OMS de 2016 de la LMA y neoplasias relacionadas, es más común en adultos que en niños. En adultos, la mutación en RUNX1 se relaciona con riesgo alto de fracaso terapéutico. En un estudio de niños con LMA, las mutaciones en RUNX1 se observaron en 11 de 503 pacientes (alrededor del 2 %). De los 11 pacientes de LMA con la mutación en RUNX1, 6 no obtuvieron remisión y su tasa de SSC a 5 años fue del 9 %, lo que sugiere que la mutación RUNX1 confiere un pronóstico precario tanto en niños como en adultos.[
173 ] - Mutaciones en RAS. Aunque se identificaron mutaciones en RAS en el 20 % al 25 % de los pacientes de LMA, la importancia pronóstica de estas mutaciones no se conoce bien.[
87 ,174 ] En casos de LMA infantil se observaron más mutaciones en NRAS que en KRAS.[87 ,175 ] Las mutaciones en RAS se producen con una frecuencia similar a todos los subtipos de alteraciones de tipo II, excepto para la LPA: en esta, casi nunca se encuentran mutaciones en RAS.[87 ] - Mutaciones en KIT. Las mutaciones en KIT se producen en casi el 5 % de los casos de LMA, pero en el 10 % al 40 % de los casos de LMA con anomalías en el CBF.[
73 ,87 ,175 ,176 ]Se ha estudiado la importancia pronóstica de las mutaciones activadoras en KIT en adultos con LMA CBF y los resultados han sido contradictorios. En un metanálisis se encontró que las mutaciones en KIT aumentan el riesgo de recaída sin un efecto en la SG de los adultos con LMA y fusiones RUNX1::RUNX1T1.[
64 ] En niños y adultos con LMC CBF, las mutaciones en KIT son subclonales;[65 ,66 ] en adultos con LMA con mutaciones RUNX1::RUNX1T1, una proporción más alta de alelos mutados de KIT se relaciona con un riesgo más alto de fracaso del tratamiento.[61 ,65 ] Aún está por aclararse la importancia pronóstica de las mutaciones en KIT en los casos de LMA CBF infantil CBF; en algunos estudios, no se encontró un efecto de las mutaciones en KIT en el desenlace,[67 ,68 ,69 ] mientras que en otros estudios se notificó un riesgo más alto de fracaso del tratamiento cuando se presentan mutaciones en KIT.[66 ,70 ,71 ,72 ,73 ] - Mutaciones en WT1. En los adultos, WT1, una proteína con dedos de zinc que regula la transcripción génica, está mutada en cerca del 10 % de los casos de LMA con características citogenéticas normales.[
177 ,178 ,179 ,180 ] En algunos estudios, pero no en todos, se observó que la mutación en WT1[177 ,178 ,180 ] es [179 ] un predictor independiente de una supervivencia sin enfermedad, SSC, y SG más precarias en los adultos.En los niños con LMA, se observan mutaciones en WT1 en cerca del 10 % de los casos.[
181 ,182 ] Los casos con mutaciones en WT1 ocurren con mucha frecuencia en los niños con características citogenéticas normales y FLT3-ITD, pero son menos comunes en los niños menores de 3 años.[181 ,182 ] Los casos de LMA con fusiones NUP98::NSD1 tienen abundantes mutaciones FLT3-ITD y mutaciones en WT1.[121 ] En análisis univariantes, las mutaciones en WT1 predicen un desenlace más precario en los pacientes pediátricos; sin embargo, no está clara la importancia como factor de pronóstico independiente del estado de la mutación en WT1 porque este estado tiene una relación sólida con FLT3-ITD y su relación con NUP98::NSD1.[121 ,181 ,182 ] En el estudio más grande sobre mutaciones en WT1 de niños con LMA, se observó que los niños que tienen mutaciones en WT1 pero no exhiben FLT3-ITD presentan desenlaces similares a los niños que tienen mutaciones en WT1; por otra parte, otros niños que tienen al mismo tiempo una mutación en WT1 y una mutación FLT3-ITD presentaron tasas de supervivencia de menos del 20 %.[181 ]En un estudio de niños con LMA resistente al tratamiento, WT1 estaba sobrerrepresentado en comparación con la cohorte que obtuvo la remisión (54 % [15 de 28 pacientes] vs. 15 %).[
171 ] - Mutaciones en DNMT3A. Se identificaron mutaciones en el gen DNMT3A en cerca del 20 % de los adultos de LMA; estas mutaciones son infrecuentes en los pacientes con características citogenéticas favorables, pero se presentan en un tercio de los adultos con características citogenéticas de riesgo intermedio.[
183 ] Las mutaciones en este gen tienen una relación independiente con un desenlace precario.[183 ,184 ,185 ] Las mutaciones en DNMT3A prácticamente no se presentan en los niños.[186 ] - Mutaciones en IDH1 y IDH2. Las mutaciones en IDH1 y IDH2, que codifican la isocitrato deshidrogenasa, se presentan en casi el 20 % de los adultos con LMA [
187 ,188 ,189 ,190 ,191 ] y son muy frecuentes en los pacientes que también tienen mutaciones en NPM1.[188 ,189 ,192 ] Las mutaciones específicas que se producen en IDH1 e IDH2 crean una actividad enzimática nueva que promueve la conversión del α-cetoglutarato en 2-hidroxiglutarato.[193 ,194 ] Esta actividad nueva induce un fenotipo de hipermetilación del DNA similar al que se observa en los casos de LMA con mutaciones de pérdida de la función en TET2.[192 ]Las mutaciones en IDH1 y IDH2 son infrecuentes en la LMA infantil: se presentan en el 0 % al 4 % de los casos.[
186 ,195 ,196 ,197 ,198 ,199 ] No hay indicación de un efecto pronóstico negativo de las mutaciones en IDH1 e IDH2 en los niños con LMA.[195 ] - Mutaciones en CSF3R. El gen CSF3R codifica el receptor del factor estimulante de colonias de granulocitos (G-CSF); se observan mutaciones activadoras en CSF3R en el 2 % a 3 % de los casos de LMA infantil.[
200 ] Estas mutaciones aumentan la señalización mediada por el receptor G-CSF; se presentan sobre todo en la LMA con mutaciones en CEBPA o anomalías de CBF (fusiones RUNX1::RUNX1T1 y CBFB::MYH11).[200 ] En un estudio de 2150 pacientes con LMA infantil, se encontró que 35 pacientes (1,6 %) tenían mutaciones en CSF3R; 30 de estos casos (89 %) tenían fusiones RUNX1::RUNX1T1 (n = 18) o CEBPA (n = 12).[98 ] El riesgo de recaída fue significativamente más alto en los pacientes con mutaciones simultáneas en CSF3R y en CEBPA en comparación con los pacientes de LMA con fusiones RUNX1::RUNX1T1 y mutaciones en CSF3R.[98 ] Si bien las tasas de recaídas en los pacientes de LMA con mutaciones simultáneas en CSF3R y CEBPA son más altas, la supervivencia general no se afecta de manera desfavorable, lo que indica una tasa de rescate alta con la terapia de reinducción y el trasplante de células madre.[90 ]También se observan mutaciones activadoras en CSF3R en los pacientes con neutropenia congénita grave. Estas mutaciones no causan la neutropenia congénita grave; más bien, surgen como mutaciones somáticas y pueden reflejar un paso inicial en la vía que lleva a la LMA.[
201 ] En un estudio de pacientes con neutropenia congénita grave, el 34 % de los pacientes que no tenían una neoplasia maligna mieloide exhibieron mutaciones en CSF3R en neutrófilos y células mononucleares de sangre periférica, mientras que el 78 % de los pacientes con una neoplasia maligna mieloide exhibieron mutaciones en CSF3R.[201 ] En un estudio de 31 pacientes con neutropenia congénita grave que padecían de LMA o SMD, se observaron mutaciones en CSF3R en cerca del 80 % de los pacientes; también se observó una frecuencia alta de mutaciones en RUNX1 (cerca del 60 %); ello indica cooperación entre las mutaciones en CSF3R y RUNX1 para que sobrevenga una leucemia en el contexto de una neutropenia congénita grave.[202 ]
Referencias:
- Bennett JM, Catovsky D, Daniel MT, et al.: Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 33 (4): 451-8, 1976.
- Bennett JM, Catovsky D, Daniel MT, et al.: Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med 103 (4): 620-5, 1985.
- Bennett JM, Catovsky D, Daniel MT, et al.: Criteria for the diagnosis of acute leukemia of megakaryocyte lineage (M7). A report of the French-American-British Cooperative Group. Ann Intern Med 103 (3): 460-2, 1985.
- Bennett JM, Catovsky D, Daniel MT, et al.: A variant form of hypergranular promyelocytic leukaemia (M3) Br J Haematol 44 (1): 169-70, 1980.
- Cheson BD, Bennett JM, Kopecky KJ, et al.: Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 21 (24): 4642-9, 2003.
- Bennett JM, Catovsky D, Daniel MT, et al.: Proposal for the recognition of minimally differentiated acute myeloid leukaemia (AML-MO) Br J Haematol 78 (3): 325-9, 1991.
- Kaleem Z, White G: Diagnostic criteria for minimally differentiated acute myeloid leukemia (AML-M0). Evaluation and a proposal. Am J Clin Pathol 115 (6): 876-84, 2001.
- Vardiman JW, Harris NL, Brunning RD: The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 100 (7): 2292-302, 2002.
- Jaffe ES, Harris NL, Stein H, et al., eds.: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press, 2001. World Health Organization Classification of Tumours, 3.
- Hasle H, Niemeyer CM, Chessells JM, et al.: A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia 17 (2): 277-82, 2003.
- Arber DA, Vardiman JW, Brunning RD: Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 110-23.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.
- Borowitz MJ, Béné MC, Harris NL: Acute leukaemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 150-5.
- Alexander TB, Gu Z, Iacobucci I, et al.: The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 562 (7727): 373-379, 2018.
- Gerr H, Zimmermann M, Schrappe M, et al.: Acute leukaemias of ambiguous lineage in children: characterization, prognosis and therapy recommendations. Br J Haematol 149 (1): 84-92, 2010.
- Rubnitz JE, Onciu M, Pounds S, et al.: Acute mixed lineage leukemia in children: the experience of St Jude Children's Research Hospital. Blood 113 (21): 5083-9, 2009.
- Al-Seraihy AS, Owaidah TM, Ayas M, et al.: Clinical characteristics and outcome of children with biphenotypic acute leukemia. Haematologica 94 (12): 1682-90, 2009.
- Matutes E, Pickl WF, Van't Veer M, et al.: Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood 117 (11): 3163-71, 2011.
- Hrusak O, de Haas V, Stancikova J, et al.: International cooperative study identifies treatment strategy in childhood ambiguous lineage leukemia. Blood 132 (3): 264-276, 2018.
- Orgel E, Alexander TB, Wood BL, et al.: Mixed-phenotype acute leukemia: A cohort and consensus research strategy from the Children's Oncology Group Acute Leukemia of Ambiguous Lineage Task Force. Cancer 126 (3): 593-601, 2020.
- Bennett JM, Catovsky D, Daniel MT, et al.: Proposals for the classification of the myelodysplastic syndromes. Br J Haematol 51 (2): 189-99, 1982.
- Mandel K, Dror Y, Poon A, et al.: A practical, comprehensive classification for pediatric myelodysplastic syndromes: the CCC system. J Pediatr Hematol Oncol 24 (7): 596-605, 2002.
- Bennett JM: World Health Organization classification of the acute leukemias and myelodysplastic syndrome. Int J Hematol 72 (2): 131-3, 2000.
- Head DR: Proposed changes in the definitions of acute myeloid leukemia and myelodysplastic syndrome: are they helpful? Curr Opin Oncol 14 (1): 19-23, 2002.
- Nösslinger T, Reisner R, Koller E, et al.: Myelodysplastic syndromes, from French-American-British to World Health Organization: comparison of classifications on 431 unselected patients from a single institution. Blood 98 (10): 2935-41, 2001.
- Brunning RD, Porwit A, Orazi A, et al.: Myelodysplastic syndromes/neoplasms overview. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 88-93.
- Wlodarski MW, Sahoo SS, Niemeyer CM: Monosomy 7 in Pediatric Myelodysplastic Syndromes. Hematol Oncol Clin North Am 32 (4): 729-743, 2018.
- Hasle H, Baumann I, Bergsträsser E, et al.: The International Prognostic Scoring System (IPSS) for childhood myelodysplastic syndrome (MDS) and juvenile myelomonocytic leukemia (JMML). Leukemia 18 (12): 2008-14, 2004.
- Wlodarski MW, Hirabayashi S, Pastor V, et al.: Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 127 (11): 1387-97; quiz 1518, 2016.
- Narumi S, Amano N, Ishii T, et al.: SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet 48 (7): 792-7, 2016.
- Schwartz JR, Ma J, Lamprecht T, et al.: The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun 8 (1): 1557, 2017.
- Davidsson J, Puschmann A, Tedgård U, et al.: SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 32 (5): 1106-1115, 2018.
- Pastor V, Hirabayashi S, Karow A, et al.: Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia 31 (3): 759-762, 2017.
- Baumann I, Niemeyer CM, Bennett JM, et al.: Childhood myelodysplastic syndrome. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 104-7.
- Kuerbitz SJ, Civin CI, Krischer JP, et al.: Expression of myeloid-associated and lymphoid-associated cell-surface antigens in acute myeloid leukemia of childhood: a Pediatric Oncology Group study. J Clin Oncol 10 (9): 1419-29, 1992.
- Smith FO, Lampkin BC, Versteeg C, et al.: Expression of lymphoid-associated cell surface antigens by childhood acute myeloid leukemia cells lacks prognostic significance. Blood 79 (9): 2415-22, 1992.
- Dinndorf PA, Andrews RG, Benjamin D, et al.: Expression of normal myeloid-associated antigens by acute leukemia cells. Blood 67 (4): 1048-53, 1986.
- Zhou Y, Jorgensen JL, Wang SA, et al.: Usefulness of CD11a and CD18 in flow cytometric immunophenotypic analysis for diagnosis of acute promyelocytic leukemia. Am J Clin Pathol 138 (5): 744-50, 2012.
- Paietta E, Goloubeva O, Neuberg D, et al.: A surrogate marker profile for PML/RAR alpha expressing acute promyelocytic leukemia and the association of immunophenotypic markers with morphologic and molecular subtypes. Cytometry B Clin Cytom 59B (1): 1-9, 2004.
- Lin P, Hao S, Medeiros LJ, et al.: Expression of CD2 in acute promyelocytic leukemia correlates with short form of PML-RARalpha transcripts and poorer prognosis. Am J Clin Pathol 121 (3): 402-7, 2004.
- Bene MC, Castoldi G, Knapp W, et al.: Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia 9 (10): 1783-6, 1995.
- Vardiman JW, Thiele J, Arber DA, et al.: The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114 (5): 937-51, 2009.
- Tarlock K, Meshinchi S: Pediatric acute myeloid leukemia: biology and therapeutic implications of genomic variants. Pediatr Clin North Am 62 (1): 75-93, 2015.
- Bolouri H, Farrar JE, Triche T, et al.: The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med 24 (1): 103-112, 2018.
- Creutzig U, van den Heuvel-Eibrink MM, Gibson B, et al.: Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 120 (16): 3187-205, 2012.
- Grimwade D, Walker H, Oliver F, et al.: The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood 92 (7): 2322-33, 1998.
- Harrison CJ, Hills RK, Moorman AV, et al.: Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J Clin Oncol 28 (16): 2674-81, 2010.
- von Neuhoff C, Reinhardt D, Sander A, et al.: Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J Clin Oncol 28 (16): 2682-9, 2010.
- Grimwade D, Hills RK, Moorman AV, et al.: Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 116 (3): 354-65, 2010.
- Brown P, McIntyre E, Rau R, et al.: The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood 110 (3): 979-85, 2007.
- Hollink IH, Zwaan CM, Zimmermann M, et al.: Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia 23 (2): 262-70, 2009.
- Ho PA, Alonzo TA, Gerbing RB, et al.: Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): a report from the Children's Oncology Group. Blood 113 (26): 6558-66, 2009.
- Meshinchi S, Alonzo TA, Stirewalt DL, et al.: Clinical implications of FLT3 mutations in pediatric AML. Blood 108 (12): 3654-61, 2006.
- Struski S, Lagarde S, Bories P, et al.: NUP98 is rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group with a poor prognosis. Leukemia 31 (3): 565-572, 2017.
- Farrar JE, Schuback HL, Ries RE, et al.: Genomic Profiling of Pediatric Acute Myeloid Leukemia Reveals a Changing Mutational Landscape from Disease Diagnosis to Relapse. Cancer Res 76 (8): 2197-205, 2016.
- Creutzig U, Zimmermann M, Ritter J, et al.: Definition of a standard-risk group in children with AML. Br J Haematol 104 (3): 630-9, 1999.
- Raimondi SC, Chang MN, Ravindranath Y, et al.: Chromosomal abnormalities in 478 children with acute myeloid leukemia: clinical characteristics and treatment outcome in a cooperative pediatric oncology group study-POG 8821. Blood 94 (11): 3707-16, 1999.
- Lie SO, Abrahamsson J, Clausen N, et al.: Treatment stratification based on initial in vivo response in acute myeloid leukaemia in children without Down's syndrome: results of NOPHO-AML trials. Br J Haematol 122 (2): 217-25, 2003.
- Klein K, Kaspers G, Harrison CJ, et al.: Clinical Impact of Additional Cytogenetic Aberrations, cKIT and RAS Mutations, and Treatment Elements in Pediatric t(8;21)-AML: Results From an International Retrospective Study by the International Berlin-Frankfurt-Münster Study Group. J Clin Oncol 33 (36): 4247-58, 2015.
- Duployez N, Marceau-Renaut A, Boissel N, et al.: Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood 127 (20): 2451-9, 2016.
- Faber ZJ, Chen X, Gedman AL, et al.: The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet 48 (12): 1551-1556, 2016.
- Noort S, Zimmermann M, Reinhardt D, et al.: Prognostic impact of t(16;21)(p11;q22) and t(16;21)(q24;q22) in pediatric AML: a retrospective study by the I-BFM Study Group. Blood 132 (15): 1584-1592, 2018.
- Chen W, Xie H, Wang H, et al.: Prognostic Significance of KIT Mutations in Core-Binding Factor Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis. PLoS One 11 (1): e0146614, 2016.
- Christen F, Hoyer K, Yoshida K, et al.: Genomic landscape and clonal evolution of acute myeloid leukemia with t(8;21): an international study on 331 patients. Blood 133 (10): 1140-1151, 2019.
- Tarlock K, Alonzo TA, Wang YC, et al.: Functional Properties of KIT Mutations Are Associated with Differential Clinical Outcomes and Response to Targeted Therapeutics in CBF Acute Myeloid Leukemia. Clin Cancer Res 25 (16): 5038-5048, 2019.
- Shih LY, Liang DC, Huang CF, et al.: Cooperating mutations of receptor tyrosine kinases and Ras genes in childhood core-binding factor acute myeloid leukemia and a comparative analysis on paired diagnosis and relapse samples. Leukemia 22 (2): 303-7, 2008.
- Goemans BF, Zwaan CM, Miller M, et al.: Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia 19 (9): 1536-42, 2005.
- Pollard JA, Alonzo TA, Gerbing RB, et al.: Prevalence and prognostic significance of KIT mutations in pediatric patients with core binding factor AML enrolled on serial pediatric cooperative trials for de novo AML. Blood 115 (12): 2372-9, 2010.
- Shimada A, Taki T, Tabuchi K, et al.: KIT mutations, and not FLT3 internal tandem duplication, are strongly associated with a poor prognosis in pediatric acute myeloid leukemia with t(8;21): a study of the Japanese Childhood AML Cooperative Study Group. Blood 107 (5): 1806-9, 2006.
- Manara E, Bisio V, Masetti R, et al.: Core-binding factor acute myeloid leukemia in pediatric patients enrolled in the AIEOP AML 2002/01 trial: screening and prognostic impact of c-KIT mutations. Leukemia 28 (5): 1132-4, 2014.
- Chen X, Dou H, Wang X, et al.: KIT mutations correlate with adverse survival in children with core-binding factor acute myeloid leukemia. Leuk Lymphoma 59 (4): 829-836, 2018.
- Tokumasu M, Murata C, Shimada A, et al.: Adverse prognostic impact of KIT mutations in childhood CBF-AML: the results of the Japanese Pediatric Leukemia/Lymphoma Study Group AML-05 trial. Leukemia 29 (12): 2438-41, 2015.
- Jahn N, Agrawal M, Bullinger L, et al.: Incidence and prognostic impact of ASXL2 mutations in adult acute myeloid leukemia patients with t(8;21)(q22;q22): a study of the German-Austrian AML Study Group. Leukemia 31 (4): 1012-1015, 2017.
- Yamato G, Shiba N, Yoshida K, et al.: ASXL2 mutations are frequently found in pediatric AML patients with t(8;21)/ RUNX1-RUNX1T1 and associated with a better prognosis. Genes Chromosomes Cancer 56 (5): 382-393, 2017.
- Smith MA, Ries LA, Gurney JG, et al.: Leukemia. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 17-34. Also available online. Last accessed August 11, 2022.
- Sanz MA, Grimwade D, Tallman MS, et al.: Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 113 (9): 1875-91, 2009.
- Licht JD, Chomienne C, Goy A, et al.: Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood 85 (4): 1083-94, 1995.
- Yan W, Zhang G: Molecular Characteristics and Clinical Significance of 12 Fusion Genes in Acute Promyelocytic Leukemia: A Systematic Review. Acta Haematol 136 (1): 1-15, 2016.
- Döhner K, Schlenk RF, Habdank M, et al.: Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood 106 (12): 3740-6, 2005.
- Verhaak RG, Goudswaard CS, van Putten W, et al.: Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood 106 (12): 3747-54, 2005.
- Schnittger S, Schoch C, Kern W, et al.: Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 106 (12): 3733-9, 2005.
- Falini B, Mecucci C, Tiacci E, et al.: Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 352 (3): 254-66, 2005.
- Schlenk RF, Döhner K, Krauter J, et al.: Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 358 (18): 1909-18, 2008.
- Gale RE, Green C, Allen C, et al.: The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 111 (5): 2776-84, 2008.
- Cazzaniga G, Dell'Oro MG, Mecucci C, et al.: Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood 106 (4): 1419-22, 2005.
- Balgobind BV, Hollink IH, Arentsen-Peters ST, et al.: Integrative analysis of type-I and type-II aberrations underscores the genetic heterogeneity of pediatric acute myeloid leukemia. Haematologica 96 (10): 1478-87, 2011.
- Staffas A, Kanduri M, Hovland R, et al.: Presence of FLT3-ITD and high BAALC expression are independent prognostic markers in childhood acute myeloid leukemia. Blood 118 (22): 5905-13, 2011.
- Tawana K, Wang J, Renneville A, et al.: Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood 126 (10): 1214-23, 2015.
- Tarlock K, Lamble AJ, Wang YC, et al.: CEBPA-bZip mutations are associated with favorable prognosis in de novo AML: a report from the Children's Oncology Group. Blood 138 (13): 1137-1147, 2021.
- Marcucci G, Maharry K, Radmacher MD, et al.: Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B Study. J Clin Oncol 26 (31): 5078-87, 2008.
- Wouters BJ, Löwenberg B, Erpelinck-Verschueren CA, et al.: Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 113 (13): 3088-91, 2009.
- Dufour A, Schneider F, Metzeler KH, et al.: Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol 28 (4): 570-7, 2010.
- Taskesen E, Bullinger L, Corbacioglu A, et al.: Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood 117 (8): 2469-75, 2011.
- Fasan A, Haferlach C, Alpermann T, et al.: The role of different genetic subtypes of CEBPA mutated AML. Leukemia 28 (4): 794-803, 2014.
- Taube F, Georgi JA, Kramer M, et al.: CEBPA mutations in 4708 patients with acute myeloid leukemia: differential impact of bZIP and TAD mutations on outcome. Blood 139 (1): 87-103, 2022.
- Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, et al.: Characterization of CEBPA mutations and promoter hypermethylation in pediatric acute myeloid leukemia. Haematologica 96 (3): 384-92, 2011.
- Tarlock K, Alonzo T, Wang YC, et al.: Prognostic impact of CSF3R mutations in favorable risk childhood acute myeloid leukemia. Blood 135 (18): 1603-1606, 2020.
- Groet J, McElwaine S, Spinelli M, et al.: Acquired mutations in GATA1 in neonates with Down's syndrome with transient myeloid disorder. Lancet 361 (9369): 1617-20, 2003.
- Hitzler JK, Cheung J, Li Y, et al.: GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood 101 (11): 4301-4, 2003.
- Rainis L, Bercovich D, Strehl S, et al.: Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood 102 (3): 981-6, 2003.
- Wechsler J, Greene M, McDevitt MA, et al.: Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet 32 (1): 148-52, 2002.
- de Rooij JD, Branstetter C, Ma J, et al.: Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet 49 (3): 451-456, 2017.
- Ge Y, Stout ML, Tatman DA, et al.: GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst 97 (3): 226-31, 2005.
- Mrózek K, Heerema NA, Bloomfield CD: Cytogenetics in acute leukemia. Blood Rev 18 (2): 115-36, 2004.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Outcome of pediatric patients with acute myeloid leukemia (AML) and -5/5q- abnormalities from five pediatric AML treatment protocols: a report from the Children's Oncology Group. Pediatr Blood Cancer 60 (12): 2073-8, 2013.
- Stevens RF, Hann IM, Wheatley K, et al.: Marked improvements in outcome with chemotherapy alone in paediatric acute myeloid leukemia: results of the United Kingdom Medical Research Council's 10th AML trial. MRC Childhood Leukaemia Working Party. Br J Haematol 101 (1): 130-40, 1998.
- Hasle H, Alonzo TA, Auvrignon A, et al.: Monosomy 7 and deletion 7q in children and adolescents with acute myeloid leukemia: an international retrospective study. Blood 109 (11): 4641-7, 2007.
- Rasche M, von Neuhoff C, Dworzak M, et al.: Genotype-outcome correlations in pediatric AML: the impact of a monosomal karyotype in trial AML-BFM 2004. Leukemia 31 (12): 2807-2814, 2017.
- Blink M, Zimmermann M, von Neuhoff C, et al.: Normal karyotype is a poor prognostic factor in myeloid leukemia of Down syndrome: a retrospective, international study. Haematologica 99 (2): 299-307, 2014.
- Hammer ASB, Juul-Dam KL, Sandahl JD, et al.: Hypodiploidy has unfavorable impact on survival in pediatric acute myeloid leukemia: an I-BFM Study Group collaboration. Blood Adv 7 (6): 1045-1055, 2023.
- Gröschel S, Sanders MA, Hoogenboezem R, et al.: A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157 (2): 369-81, 2014.
- Yamazaki H, Suzuki M, Otsuki A, et al.: A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 25 (4): 415-27, 2014.
- Lugthart S, Gröschel S, Beverloo HB, et al.: Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J Clin Oncol 28 (24): 3890-8, 2010.
- Balgobind BV, Lugthart S, Hollink IH, et al.: EVI1 overexpression in distinct subtypes of pediatric acute myeloid leukemia. Leukemia 24 (5): 942-9, 2010.
- Schnittger S, Schoch C, Dugas M, et al.: Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 100 (1): 59-66, 2002.
- Thiede C, Steudel C, Mohr B, et al.: Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 99 (12): 4326-35, 2002.
- Iwai T, Yokota S, Nakao M, et al.: Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. The Children's Cancer and Leukemia Study Group, Japan. Leukemia 13 (1): 38-43, 1999.
- Meshinchi S, Stirewalt DL, Alonzo TA, et al.: Activating mutations of RTK/ras signal transduction pathway in pediatric acute myeloid leukemia. Blood 102 (4): 1474-9, 2003.
- Zwaan CM, Meshinchi S, Radich JP, et al.: FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood 102 (7): 2387-94, 2003.
- Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, et al.: NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 118 (13): 3645-56, 2011.
- Ostronoff F, Othus M, Gerbing RB, et al.: NUP98/NSD1 and FLT3/ITD coexpression is more prevalent in younger AML patients and leads to induction failure: a COG and SWOG report. Blood 124 (15): 2400-7, 2014.
- Arrigoni P, Beretta C, Silvestri D, et al.: FLT3 internal tandem duplication in childhood acute myeloid leukaemia: association with hyperleucocytosis in acute promyelocytic leukaemia. Br J Haematol 120 (1): 89-92, 2003.
- Kutny MA, Moser BK, Laumann K, et al.: FLT3 mutation status is a predictor of early death in pediatric acute promyelocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 59 (4): 662-7, 2012.
- Gale RE, Hills R, Pizzey AR, et al.: Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood 106 (12): 3768-76, 2005.
- Tallman MS, Kim HT, Montesinos P, et al.: Does microgranular variant morphology of acute promyelocytic leukemia independently predict a less favorable outcome compared with classical M3 APL? A joint study of the North American Intergroup and the PETHEMA Group. Blood 116 (25): 5650-9, 2010.
- Sung L, Aplenc R, Alonzo TA, et al.: Predictors and short-term outcomes of hyperleukocytosis in children with acute myeloid leukemia: a report from the Children's Oncology Group. Haematologica 97 (11): 1770-3, 2012.
- Callens C, Chevret S, Cayuela JM, et al.: Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia 19 (7): 1153-60, 2005.
- Schnittger S, Bacher U, Haferlach C, et al.: Clinical impact of FLT3 mutation load in acute promyelocytic leukemia with t(15;17)/PML-RARA. Haematologica 96 (12): 1799-807, 2011.
- Breccia M, Loglisci G, Loglisci MG, et al.: FLT3-ITD confers poor prognosis in patients with acute promyelocytic leukemia treated with AIDA protocols: long-term follow-up analysis. Haematologica 98 (12): e161-3, 2013.
- Poiré X, Moser BK, Gallagher RE, et al.: Arsenic trioxide in front-line therapy of acute promyelocytic leukemia (C9710): prognostic significance of FLT3 mutations and complex karyotype. Leuk Lymphoma 55 (7): 1523-32, 2014.
- Pui CH, Relling MV, Rivera GK, et al.: Epipodophyllotoxin-related acute myeloid leukemia: a study of 35 cases. Leukemia 9 (12): 1990-6, 1995.
- Inaba H, Zhou Y, Abla O, et al.: Heterogeneous cytogenetic subgroups and outcomes in childhood acute megakaryoblastic leukemia: a retrospective international study. Blood 126 (13): 1575-84, 2015.
- Balgobind BV, Raimondi SC, Harbott J, et al.: Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood 114 (12): 2489-96, 2009.
- Swansbury GJ, Slater R, Bain BJ, et al.: Hematological malignancies with t(9;11)(p21-22;q23)--a laboratory and clinical study of 125 cases. European 11q23 Workshop participants. Leukemia 12 (5): 792-800, 1998.
- Pollard JA, Guest E, Alonzo TA, et al.: Gemtuzumab Ozogamicin Improves Event-Free Survival and Reduces Relapse in Pediatric KMT2A-Rearranged AML: Results From the Phase III Children's Oncology Group Trial AAML0531. J Clin Oncol 39 (28): 3149-3160, 2021.
- Coenen EA, Raimondi SC, Harbott J, et al.: Prognostic significance of additional cytogenetic aberrations in 733 de novo pediatric 11q23/MLL-rearranged AML patients: results of an international study. Blood 117 (26): 7102-11, 2011.
- Ageberg M, Drott K, Olofsson T, et al.: Identification of a novel and myeloid specific role of the leukemia-associated fusion protein DEK-NUP214 leading to increased protein synthesis. Genes Chromosomes Cancer 47 (4): 276-87, 2008.
- Shiba N, Ichikawa H, Taki T, et al.: NUP98-NSD1 gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia. Genes Chromosomes Cancer 52 (7): 683-93, 2013.
- Slovak ML, Gundacker H, Bloomfield CD, et al.: A retrospective study of 69 patients with t(6;9)(p23;q34) AML emphasizes the need for a prospective, multicenter initiative for rare 'poor prognosis' myeloid malignancies. Leukemia 20 (7): 1295-7, 2006.
- Alsabeh R, Brynes RK, Slovak ML, et al.: Acute myeloid leukemia with t(6;9) (p23;q34): association with myelodysplasia, basophilia, and initial CD34 negative immunophenotype. Am J Clin Pathol 107 (4): 430-7, 1997.
- Sandahl JD, Coenen EA, Forestier E, et al.: t(6;9)(p22;q34)/DEK-NUP214-rearranged pediatric myeloid leukemia: an international study of 62 patients. Haematologica 99 (5): 865-72, 2014.
- Tarlock K, Alonzo TA, Moraleda PP, et al.: Acute myeloid leukaemia (AML) with t(6;9)(p23;q34) is associated with poor outcome in childhood AML regardless of FLT3-ITD status: a report from the Children's Oncology Group. Br J Haematol 166 (2): 254-9, 2014.
- Gruber TA, Larson Gedman A, Zhang J, et al.: An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell 22 (5): 683-97, 2012.
- Thiollier C, Lopez CK, Gerby B, et al.: Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J Exp Med 209 (11): 2017-31, 2012.
- de Rooij JD, Hollink IH, Arentsen-Peters ST, et al.: NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 27 (12): 2280-8, 2013.
- Masetti R, Pigazzi M, Togni M, et al.: CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 121 (17): 3469-72, 2013.
- Masetti R, Rondelli R, Fagioli F, et al.: Infants with acute myeloid leukemia treated according to the Associazione Italiana di Ematologia e Oncologia Pediatrica 2002/01 protocol have an outcome comparable to that of older children. Haematologica 99 (8): e127-9, 2014.
- de Rooij JD, Masetti R, van den Heuvel-Eibrink MM, et al.: Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective intergroup study. Blood 127 (26): 3424-30, 2016.
- Hara Y, Shiba N, Ohki K, et al.: Prognostic impact of specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down syndrome. Genes Chromosomes Cancer 56 (5): 394-404, 2017.
- Smith JL, Ries RE, Hylkema T, et al.: Comprehensive Transcriptome Profiling of Cryptic CBFA2T3-GLIS2 Fusion-Positive AML Defines Novel Therapeutic Options: A COG and TARGET Pediatric AML Study. Clin Cancer Res 26 (3): 726-737, 2020.
- Eidenschink Brodersen L, Alonzo TA, Menssen AJ, et al.: A recurrent immunophenotype at diagnosis independently identifies high-risk pediatric acute myeloid leukemia: a report from Children's Oncology Group. Leukemia 30 (10): 2077-2080, 2016.
- Pardo LM, Voigt AP, Alonzo TA, et al.: Deciphering the Significance of CD56 Expression in Pediatric Acute Myeloid Leukemia: A Report from the Children's Oncology Group. Cytometry B Clin Cytom 98 (1): 52-56, 2020.
- Noort S, Wander P, Alonzo TA, et al.: The clinical and biological characteristics of NUP98-KDM5A in pediatric acute myeloid leukemia. Haematologica 106 (2): 630-634, 2021.
- Lion T, Haas OA: Acute megakaryocytic leukemia with the t(1;22)(p13;q13). Leuk Lymphoma 11 (1-2): 15-20, 1993.
- Duchayne E, Fenneteau O, Pages MP, et al.: Acute megakaryoblastic leukaemia: a national clinical and biological study of 53 adult and childhood cases by the Groupe Français d'Hématologie Cellulaire (GFHC). Leuk Lymphoma 44 (1): 49-58, 2003.
- Ma Z, Morris SW, Valentine V, et al.: Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat Genet 28 (3): 220-1, 2001.
- Mercher T, Coniat MB, Monni R, et al.: Involvement of a human gene related to the Drosophila spen gene in the recurrent t(1;22) translocation of acute megakaryocytic leukemia. Proc Natl Acad Sci U S A 98 (10): 5776-9, 2001.
- Bernstein J, Dastugue N, Haas OA, et al.: Nineteen cases of the t(1;22)(p13;q13) acute megakaryblastic leukaemia of infants/children and a review of 39 cases: report from a t(1;22) study group. Leukemia 14 (1): 216-8, 2000.
- Coenen EA, Zwaan CM, Reinhardt D, et al.: Pediatric acute myeloid leukemia with t(8;16)(p11;p13), a distinct clinical and biological entity: a collaborative study by the International-Berlin-Frankfurt-Munster AML-study group. Blood 122 (15): 2704-13, 2013.
- Wong KF, Yuen HL, Siu LL, et al.: t(8;16)(p11;p13) predisposes to a transient but potentially recurring neonatal leukemia. Hum Pathol 39 (11): 1702-7, 2008.
- Wu X, Sulavik D, Roulston D, et al.: Spontaneous remission of congenital acute myeloid leukemia with t(8;16)(p11;13). Pediatr Blood Cancer 56 (2): 331-2, 2011.
- Terui K, Sato T, Sasaki S, et al.: Two novel variants of MOZ-CBP fusion transcripts in spontaneously remitted infant leukemia with t(1;16;8)(p13;p13;p11), a new variant of t(8;16)(p11;p13). Haematologica 93 (10): 1591-3, 2008.
- von Bergh AR, van Drunen E, van Wering ER, et al.: High incidence of t(7;12)(q36;p13) in infant AML but not in infant ALL, with a dismal outcome and ectopic expression of HLXB9. Genes Chromosomes Cancer 45 (8): 731-9, 2006.
- Tosi S, Harbott J, Teigler-Schlegel A, et al.: t(7;12)(q36;p13), a new recurrent translocation involving ETV6 in infant leukemia. Genes Chromosomes Cancer 29 (4): 325-32, 2000.
- Slater RM, von Drunen E, Kroes WG, et al.: t(7;12)(q36;p13) and t(7;12)(q32;p13)--translocations involving ETV6 in children 18 months of age or younger with myeloid disorders. Leukemia 15 (6): 915-20, 2001.
- Park J, Kim M, Lim J, et al.: Three-way complex translocations in infant acute myeloid leukemia with t(7;12)(q36;p13): the incidence and correlation of a HLXB9 overexpression. Cancer Genet Cytogenet 191 (2): 102-5, 2009.
- Takeda A, Yaseen NR: Nucleoporins and nucleocytoplasmic transport in hematologic malignancies. Semin Cancer Biol 27: 3-10, 2014.
- Panarello C, Rosanda C, Morerio C: Cryptic translocation t(5;11)(q35;p15.5) with involvement of the NSD1 and NUP98 genes without 5q deletion in childhood acute myeloid leukemia. Genes Chromosomes Cancer 35 (3): 277-81, 2002.
- Cerveira N, Correia C, Dória S, et al.: Frequency of NUP98-NSD1 fusion transcript in childhood acute myeloid leukaemia. Leukemia 17 (11): 2244-7, 2003.
- McNeer NA, Philip J, Geiger H, et al.: Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia 33 (8): 1934-1943, 2019.
- van Zutven LJ, Onen E, Velthuizen SC, et al.: Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Genes Chromosomes Cancer 45 (5): 437-46, 2006.
- Yamato G, Shiba N, Yoshida K, et al.: RUNX1 mutations in pediatric acute myeloid leukemia are associated with distinct genetic features and an inferior prognosis. Blood 131 (20): 2266-2270, 2018.
- Berman JN, Gerbing RB, Alonzo TA, et al.: Prevalence and clinical implications of NRAS mutations in childhood AML: a report from the Children's Oncology Group. Leukemia 25 (6): 1039-42, 2011.
- Kühn MW, Radtke I, Bullinger L, et al.: High-resolution genomic profiling of adult and pediatric core-binding factor acute myeloid leukemia reveals new recurrent genomic alterations. Blood 119 (10): e67-75, 2012.
- Schnittger S, Kohl TM, Haferlach T, et al.: KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood 107 (5): 1791-9, 2006.
- Paschka P, Marcucci G, Ruppert AS, et al.: Wilms' tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol 26 (28): 4595-602, 2008.
- Virappane P, Gale R, Hills R, et al.: Mutation of the Wilms' tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: the United Kingdom Medical Research Council Adult Leukaemia Working Party. J Clin Oncol 26 (33): 5429-35, 2008.
- Gaidzik VI, Schlenk RF, Moschny S, et al.: Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: a study of the German-Austrian AML Study Group. Blood 113 (19): 4505-11, 2009.
- Renneville A, Boissel N, Zurawski V, et al.: Wilms tumor 1 gene mutations are associated with a higher risk of recurrence in young adults with acute myeloid leukemia: a study from the Acute Leukemia French Association. Cancer 115 (16): 3719-27, 2009.
- Ho PA, Zeng R, Alonzo TA, et al.: Prevalence and prognostic implications of WT1 mutations in pediatric acute myeloid leukemia (AML): a report from the Children's Oncology Group. Blood 116 (5): 702-10, 2010.
- Hollink IH, van den Heuvel-Eibrink MM, Zimmermann M, et al.: Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood 113 (23): 5951-60, 2009.
- Ley TJ, Ding L, Walter MJ, et al.: DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363 (25): 2424-33, 2010.
- Yan XJ, Xu J, Gu ZH, et al.: Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet 43 (4): 309-15, 2011.
- Thol F, Damm F, Lüdeking A, et al.: Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol 29 (21): 2889-96, 2011.
- Ho PA, Kutny MA, Alonzo TA, et al.: Leukemic mutations in the methylation-associated genes DNMT3A and IDH2 are rare events in pediatric AML: a report from the Children's Oncology Group. Pediatr Blood Cancer 57 (2): 204-9, 2011.
- Green CL, Evans CM, Hills RK, et al.: The prognostic significance of IDH1 mutations in younger adult patients with acute myeloid leukemia is dependent on FLT3/ITD status. Blood 116 (15): 2779-82, 2010.
- Paschka P, Schlenk RF, Gaidzik VI, et al.: IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol 28 (22): 3636-43, 2010.
- Abbas S, Lugthart S, Kavelaars FG, et al.: Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 116 (12): 2122-6, 2010.
- Marcucci G, Maharry K, Wu YZ, et al.: IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 28 (14): 2348-55, 2010.
- Wagner K, Damm F, Göhring G, et al.: Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol 28 (14): 2356-64, 2010.
- Figueroa ME, Abdel-Wahab O, Lu C, et al.: Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18 (6): 553-67, 2010.
- Ward PS, Patel J, Wise DR, et al.: The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17 (3): 225-34, 2010.
- Dang L, White DW, Gross S, et al.: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462 (7274): 739-44, 2009.
- Damm F, Thol F, Hollink I, et al.: Prevalence and prognostic value of IDH1 and IDH2 mutations in childhood AML: a study of the AML-BFM and DCOG study groups. Leukemia 25 (11): 1704-10, 2011.
- Oki K, Takita J, Hiwatari M, et al.: IDH1 and IDH2 mutations are rare in pediatric myeloid malignancies. Leukemia 25 (2): 382-4, 2011.
- Pigazzi M, Ferrari G, Masetti R, et al.: Low prevalence of IDH1 gene mutation in childhood AML in Italy. Leukemia 25 (1): 173-4, 2011.
- Ho PA, Alonzo TA, Kopecky KJ, et al.: Molecular alterations of the IDH1 gene in AML: a Children's Oncology Group and Southwest Oncology Group study. Leukemia 24 (5): 909-13, 2010.
- Andersson AK, Miller DW, Lynch JA, et al.: IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia 25 (10): 1570-7, 2011.
- Maxson JE, Ries RE, Wang YC, et al.: CSF3R mutations have a high degree of overlap with CEBPA mutations in pediatric AML. Blood 127 (24): 3094-8, 2016.
- Germeshausen M, Kratz CP, Ballmaier M, et al.: RAS and CSF3R mutations in severe congenital neutropenia. Blood 114 (16): 3504-5, 2009.
- Skokowa J, Steinemann D, Katsman-Kuipers JE, et al.: Cooperativity of RUNX1 and CSF3R mutations in severe congenital neutropenia: a unique pathway in myeloid leukemogenesis. Blood 123 (14): 2229-37, 2014.
Aspectos generales de las opciones de tratamiento de la leucemia mieloide aguda infantil
Se considera que la leucemia ya está diseminada por el sistema hematopoyético en el momento del diagnóstico, incluso en los niños que presentan al inicio leucemia mieloide aguda (LMA) con cloromas aislados (también llamados sarcomas granulocíticos o mieloides). Si estos niños no reciben quimioterapia sistémica, siempre surge una LMA después de meses o años. Es posible que la LMA invada tejidos no hematopoyéticos (extramedulares) como las meninges, el parénquima encefálico, los testículos, los ovarios o la piel (leucemia cutánea). La leucemia extramedular es más frecuente en los lactantes que en los niños más grandes con LMA.[
La LMA infantil se diagnostica cuando hay más de un 20 % de blastocitos en la médula ósea. Los blastocitos tienen las características morfológicas e histoquímicas compatibles con uno de los subtipos de LMA de la clasificación French-American-British (FAB). También se puede diagnosticar con una biopsia de un cloroma. Para efectos del tratamiento, se considera que los pacientes con anomalías citogenéticas clonales que se suelen relacionar con la LMA, como t(8:21)(fusiones génicas RUNX1::RUNX1T1), inv(16)(fusiones génicas CBFB::MYH11), t(9;11)(fusiones génicas MLLT3::KMT2A) o t(15;17)(fusiones génicas PML::RARA), y con menos de un 20 % de blastocitos en la médula ósea, tienen LMA en lugar de un síndrome mielodisplásico.[
En los Estados Unidos, la remisión completa (RC) se ha definido de forma tradicional mediante criterios morfológicos como los siguientes:
- Recuentos de sangre periférica (recuento de glóbulos blancos [GB], recuento diferencial [recuento absoluto de neutrófilos >1000/μl] y recuento de plaquetas >100 000/μl) que aumentan y se acercan a los valores normales.
- Médula moderadamente hipocelular o con celularidad normal, y menos de un 5 % de blastocitos.
- Ausencia de signos o síntomas clínicos de enfermedad, incluso en el sistema nervioso central (SNC) u otros sitios extramedulares.[
3 ]
Para la LMA se emplean otras definiciones de remisión de acuerdo con las características morfológicas debido a la mielodepresión persistente que causa la quimioterapia intensiva e incluye la RC con recuperación incompleta de plaquetas (RCp) y la RC con recuperación incompleta de la médula (por lo general, del recuento absoluto de neutrófilos) (RCi). Aunque el uso de la RCp proporciona una respuesta importante desde el punto de vista clínico, la definición tradicional de RC sigue siendo el criterio de referencia porque los pacientes en RC tienen una mayor probabilidad de sobrevivir por más tiempo que aquellos en RCp.[
El logro de una médula ósea hipoplásica (usando criterios morfológicos) suele ser el primer paso para la remisión de la LMA, excepto en el subtipo M3 (leucemia promielocítica aguda [LPA]); a menudo no se necesita una fase de médula hipoplásica antes de la remisión de la LPA. Además, la recuperación temprana de la médula en cualquier subtipo de LMA quizá sea difícil de diferenciar de una leucemia persistente, aunque la aplicación de la inmunofenotipificación por citometría de flujo o pruebas citogenéticas o moleculares facilita esta diferenciación. Es imprescindible la correlación de los recuentos de células sanguíneas y el estado clínico antes de emitir el dictamen definitivo de los resultados de las observaciones iniciales en la médula ósea en la LMA.[
Además de las características morfológicas, se usan métodos más precisos (por ejemplo, citometría de flujo multiparamétrica o reacción en cadena de la polimerasa con retrotranscripción [PCR-RT]) con el fin de evaluar la respuesta y se ha observado que tienen mayor importancia pronóstica que las características morfológicas. Para obtener más información sobre estos métodos, consultar la sección Factores pronósticos en la leucemia mieloide aguda infantil.
Abordaje de tratamiento
El componente principal del abordaje terapéutico es la administración sistémica de quimioterapia combinada. Se están probando abordajes que incluyen estratificación por grupos de riesgo y terapias dirigidas de tipo biológico con el fin de mejorar el tratamiento antileucémico sin afectar el tejido sano. El tratamiento óptimo de la LMA exige el control de la médula ósea y la enfermedad sistémica. El tratamiento del SNC, por lo general con administración intratecal de los medicamentos, es un componente de casi todos los protocolos de tratamiento de la LMA infantil; no obstante, hasta el momento no se ha observado que contribuya de manera directa a mejorar la supervivencia. Los pacientes no necesitan irradiación del SNC, ni como profilaxis ni para quienes exhiben al inicio leucemia en el líquido cefalorraquídeo que desaparece después de la quimioterapia intratecal o sistémica.
Por lo general, el tratamiento se divide en las dos fases siguientes:
- Inducción (para inducir la remisión).
- Consolidación o intensificación posterior a la remisión (para reducir el riesgo de recaída).
Es posible que el tratamiento posterior a la remisión incluya diferentes número de cursos de quimioterapia intensiva o trasplante alogénico de células madre hematopoyéticas (TCMH) alogénico. Por ejemplo, en los ensayos en curso del Children's Oncology Group (COG) y el United Kingdom Medical Research Council (MRC) se utilizan regímenes de quimioterapia similares que constan de 2 cursos de quimioterapia de inducción seguidos de otros 2 cursos de quimioterapia de intensificación.[
La terapia de mantenimiento no se incluye en la mayoría de los protocolos de LMA infantil porque en dos ensayos clínicos aleatorizados no se logró demostrar un beneficio de esta terapia cuando se administra después de una quimioterapia intensiva contemporánea.[
Es fundamental vigilar la presencia de complicaciones agudas y a largo plazo en los niños con LMA. Los abordajes contemporáneos de tratamiento de la LMA por lo general se vinculan con mielodepresión prolongada y grave con complicaciones relacionadas. Los niños con LMA deben recibir una atención dirigida por oncólogos pediatras en centros oncológicos u hospitales con instalaciones apropiadas para brindar cuidados de apoyo (por ejemplo, productos sanguíneos especializados; cuidados intensivos pediátricos; servicios de apoyo emocional y del desarrollo). A medida que se mejoran los cuidados médicos de apoyo, las muertes por causas tóxicas constituyen una proporción menor de los fracasos terapéuticos iniciales en comparación con el pasado.[
En la actualidad los niños que reciben tratamiento por una LMA viven más y necesitan una vigilancia minuciosa de los efectos secundarios del tratamiento del cáncer, que a veces persisten o se presentan meses o años después del tratamiento. Las dosis acumuladas altas de antraciclinas requieren vigilancia a largo plazo del funcionamiento cardíaco. El uso de algunas modalidades se ha reducido; entre ellas, la irradiación corporal total con TCMH porque aumenta el riesgo de retraso del crecimiento, disfunción gonadal y tiroidea, formación de cataratas y neoplasias malignas secundarias.[
Factores pronósticos de la leucemia mieloide aguda infantil
Los factores pronósticos de la LMA infantil se clasifican de la siguiente manera:
- Factores de riesgo del anfitrión.
- Factores de riesgo de la leucemia.
- Factores de riesgo de la respuesta terapéutica.
Factores de riesgo del anfitrión
- Edad: en varios informes se identificó la edad avanzada como un factor de pronóstico adverso.[
7 ,16 ,17 ,18 ,19 ,20 ] El efecto de la edad no es muy grande con respecto a la SG, pero en general se observa que los desenlaces adversos de los adolescentes en comparación con los niños pequeños son ante todo producto del aumento en la mortalidad por causas tóxicas.[21 ] En el estudio del COG AAML1031 (NCT01371981) , la edad mayor de 11 años fue un factor de predicción independiente de SSC en el análisis multivariante.[22 ]Aunque el desenlace para los lactantes con LLA sigue siendo inferior al de los niños mayores, el desenlace para los lactantes con LMA es similar al de los niños mayores cuando se tratan con regímenes de LMA estándar.[
16 ,23 ,24 ,25 ] Se ha notificado que los lactantes tienen una tasa de supervivencia a 5 años del 60 % al 70 %, aunque con aumento de la toxicidad relacionada con el tratamiento; en particular, durante la inducción.[16 ,23 ,24 ,25 ,26 ] - Raza o etnia: en los estudios del Children's Cancer Group (CCG) CCG-2891 y COG-2961 (NCT00002798), los niños blancos tuvieron tasas de SG más altas que los niños negros o hispanos.[
18 ,27 ,28 ] Los niños negros también presentaron tasas de supervivencia más bajas que los niños blancos en los ensayos clínicos de LMA del St. Jude Children's Research Hospital.[29 ] - Síndrome de Down: por lo general, los niños con síndrome de Down que presentan LMA tienen una supervivencia favorable cuando se diagnostica a una edad temprana.[
30 ,31 ,32 ] El pronóstico es particularmente bueno (tasa de SSC de más del 80 %) para los niños menores de 4 años en el momento del diagnóstico, que es el grupo de edad que incluye a la gran mayoría de los pacientes con síndrome de Down y LMA, mientras que los mayores de 4 años tienen desenlaces similares a los de los niños sin síndrome de Down.[32 ,33 ,34 ,35 ,36 ] - Índice de masa corporal: la obesidad (índice de masa corporal por encima del percentil 95 según la edad) predice una supervivencia más baja.[
18 ,37 ] La mortalidad prematura relacionada con el tratamiento, en su mayoría por complicaciones infecciosas, explicó esta supervivencia más baja.[37 ,38 ]
Factores de riesgo de leucemia
- Recuento de glóbulos blancos (GB): se ha observado de manera uniforme que el recuento de GB en el momento del diagnóstico se relaciona de manera inversa con la supervivencia.[
7 ,22 ,39 ,40 ] Los pacientes que tienen recuentos altos de leucocitos durante la presentación inicial tienen un riesgo más alto de padecer complicaciones pulmonares y del SNC; además, tienen un riesgo más alto de morir durante la inducción.[41 ]En la LPA, se usa solo el recuento de GB en el momento del diagnóstico inicial para diferenciar la LPA de riesgo estándar y riesgo alto. Un recuento de GB de 10 000 células/μl o superior implica riesgo alto, y estos pacientes tienen un riesgo elevado de muerte prematura y recaída.[
42 ] Sin embargo, con los regímenes más nuevos que incluyen trióxido de arsénico se observan tasas bajas de recaída que no difieren significativamente entre los grupos de riesgo alto y bajo.[43 ] - Subtipo FAB: las relaciones entre el subtipo FAB y el pronóstico han sido más variables.
- Subtipo M3. El subtipo M3 (LPA) tiene un desenlace favorable en los estudios en los que se usa tretinoína en combinación con quimioterapia y consolidación con trióxido de arsénico.[
42 ,43 ,44 ,45 ] - Subtipo M6. En el sistema de clasificación de la OMS de 2016, el subtipo M6 se limitó a la leucemia eritroide pura. En la combinación de los estudios del COG AAML0531 y AAML1031, se demostró que es un subtipo raro (5 de 1934 casos; 0,2 %), que ocurre en pacientes más jóvenes (mediana de edad, 2,3 años), y se asoció con un desenlace precario (tasas de SSC y SG a 5, 20 % ± 36 %).[
46 ] - Subtipo M7. En algunos estudios se indicó que el desenlace es relativamente precario para el subtipo M7 (leucemia megacariocítica) en los pacientes que no tienen síndrome de Down;[
30 ] sin embargo, en otros informes se indica un pronóstico intermedio para este grupo de pacientes cuando se usan abordajes de tratamiento contemporáneos.[6 ,47 ,48 ]En un estudio retrospectivo de pacientes con subtipo M7 sin síndrome de Down que tenían muestras disponibles para análisis molecular, la presencia de anomalías genéticas específicas (fusiones génicas CBFA2T3::GLIS2 [inv(16)(p13q24) críptica], fusiones génicas NUP98::KDM5A, t(11;12)(p15;p13), reordenamientos de KMT2A [MLL] y monosomía 7) se relacionó con un desenlace significativamente más precario que el de otros pacientes del subtipo M7.[
49 ,50 ] Por el contrario, el 10 % de los pacientes con LMCA sin síndrome de Down con mutaciones en GATA1 presentaron un desenlace favorable cuando no se encontraban fusiones génicas con pronóstico desfavorable. Esto también ocurría en los pacientes que exhibían reordenamientos de HOX.[50 ] - Subtipo M0. El M0, o subtipo con diferenciación mínima, se ha relacionado con un desenlace precario.[
51 ]
- Subtipo M3. El subtipo M3 (LPA) tiene un desenlace favorable en los estudios en los que se usa tretinoína en combinación con quimioterapia y consolidación con trióxido de arsénico.[
- Enfermedad en el SNC: el compromiso del SNC en el momento del diagnóstico se clasifica según la presencia o ausencia de blastocitos en el líquido cefalorraquídeo (LCR). Los grupos cooperativos europeos han utilizado definiciones similares a las de la leucemia linfoblástica aguda para la LMA, de la siguiente manera:
- SNC1: LCR con resultado negativo para blastocitos en la prueba con citocentrífuga, con independencia del recuento de GB en el LCR.
- SNC2 se divide en los 3 grupos que se definen a continuación:
- SNC2a: LCR con menos de 5 GB/μl y resultado positivo para blastocitos en la prueba con citocentrífuga de una muestra de punción atraumática (<10 glóbulos rojos [GR]/μl).
- SNC2b: LCR con menos de 5 GB/µl y resultado positivo para blastocitos en la prueba con citocentrífuga de una muestra de punción traumática (≥10 GR/μl).
- SNC2c: LCR con 5 o más GB/μl y resultado positivo para blastocitos en la prueba con citocentrífuga de una muestra de punción traumática (≥10 GR/μl), en la que la proporción de GB/GR en el LCR es menos del doble de la proporción en sangre periférica.
- SNC3 incluye los 3 subgrupos que se definen a continuación:
- SNC3a: LCR con 5 o más GB/µl y resultado positivo para blastocitos en la prueba con citocentrífuga de una muestra de punción atraumática (<10 GR/μl).
- SNC3b: LCR con 5 o más GB/μl y resultado positivo para blastocitos en la prueba con citocentrífuga de una muestra de punción traumática (≥10 GR/μl), en la que la proporción de GB/GR en el LCR es mayor o igual al doble de la proporción en sangre periférica.
- SNC3c: signos clínicos de leucemia en el SNC (por ejemplo, parálisis de un nervio craneal, compromiso encefálico u ocular, o indicios radiográficos de cloroma intracraneal o intradural).
En los ensayos del COG (como el AAML03P1 [NCT00070174], el AAML0531 [NCT00372593]y el AAML1031 [NCT01371981]) se usó una versión modificada de las definiciones de enfermedad en el SNC para efectos del tratamiento, con una clasificación dicotómica de los pacientes según el resultado positivo o negativo para enfermedad en el SNC. El grupo que obtuvo un resultado positivo para la enfermedad en el SNC abarcó a todos los pacientes con blastocitos en la prueba de citocentrífuga (con independencia de los GB en el LCR) salvo que presentaran más de 100 GR/μl en el LCR. Los pacientes con 100 GR/μl en el LCR se clasificaban en el grupo de resultado positivo para la enfermedad en el SNC si la proporción de GB/GR en el LCR era mayor o igual al doble de la proporción en sangre periférica. En los estudios del COG, los desenlaces en el SNC se analizaron utilizando las definiciones más tradicionales de SNC1, SNC2 y SNC3.[
52 ]La enfermedad SNC2 se observó en cerca del 13 % al 16 % de los niños con LMA y la enfermedad SNC3 se observó en alrededor del 11 % al 17 % de los niños con LMA.[
52 ,53 ] En los estudios, se observó de manera variable que los pacientes con SNC2 o SNC3 eran más jóvenes, a menudo tenían hiperleucocitosis, y presentaban una incidencia más alta de t(9;11), t(8;21) o inv(16).[52 ,53 ]Si bien el compromiso del SNC (SNC2 o SNC3) en el momento del diagnóstico no se correlaciona con la SG en la mayoría de los estudios, en un análisis del COG de niños con LMA inscritos entre 2003 y 2010 en dos ensayos consecutivos e idénticos sobre el tratamiento de base, se encontró que el compromiso del SNC, en especial, el estado SNC3, se relacionó con desenlaces inferiores; incluso en la tasa de remisión completa, la SSC, la supervivencia sin enfermedad, y un aumento del riesgo de recaída con compromiso del SNC.[
52 ] En otro ensayo se observó que se relacionaba con un aumento de riesgo de recaída aislada en el SNC.[54 ] Por último, en el estudio del COG no se encontró un efecto adverso sobre el desenlace de las punciones lumbares traumáticas que se hacen en el momento del diagnóstico.[52 ]
- Características citogenéticas y moleculares: las características citogenéticas y moleculares también se relacionan con el pronóstico. Para obtener información detallada, consultar la sección Evaluación molecular. Las características citogenéticas y moleculares que ahora se usan en los ensayos clínicos para la asignación de tratamiento son las siguientes:
- Favorable:: inv(16)/t(16;16), t(8;21), t(15;17), mutaciones bialélicas en CEBPA y mutaciones en NPM1.
- Desfavorable:: monosomía 7, monosomía 5/del(5q), anomalías de 3q, duplicaciones internas en tándem (ITD) en FLT3 con proporción alélica alta, fusiones génicas de KMT2A con las siguientes translocaciones recíprocas t(4;11)(q21;q23), t(6;11)(q27;q23), t(10;11)(p11.2;q23), t(10;11)(p12;q23), o t(11;19)(q23;p13.3), NUP98 (11p15.5), 12p (reordenamiento o pérdida de ETV6), t(16;21)(p11;q22)(fusiones génicas FUS::ERG), fusiones génicas CBFA2T3::GLIS2, KAT6A (8p11.21) (si tiene más de 90 días de edad), y fusiones génicas diferentes a la fusión KMT2A::MLLT10.[
55 ,56 ]
- Inmunofenotipo:
- Un inmunofenotipo distintivo (inicialmente reportado como fenotipo RAM), con concentraciones altas de CD56, expresión debil o negativa de CD45 y CD38, y una falta de expresión de HLA-DR se relacionó con un pronóstico precario (tasa de SSC a 5 años de alrededor del 20 %).[
57 ,58 ] La mayoría de los pacientes con el fenotipo RAM tienen la fusión CBFA2T3::GLIS2.[58 ,59 ] - La expresión elevada de CD123 (cuartil 4 vs. cuartiles 1–3), en la regresión multivariable de Cox, demostró ser un factor de riesgo de pronóstico adverso independiente para la SG, la SSC y el riesgo de recaída (RR), aunque no afectó el éxito de la remisión. La expresión elevada de CD123 fue más frecuente en pacientes con muchas características citogenéticas y moleculares de riesgo alto. La expresión elevada de CD123 también afectó de manera negativa la SG y la SSC, pero no el RR. En los pacientes con características citogenéticas y moleculares de riesgo bajo, aquellos con una expresión elevada de CD123 (cuartil 4) tuvieron una SG, una SSC y una RR significativamente más precarios.[
60 ]
- Un inmunofenotipo distintivo (inicialmente reportado como fenotipo RAM), con concentraciones altas de CD56, expresión debil o negativa de CD45 y CD38, y una falta de expresión de HLA-DR se relacionó con un pronóstico precario (tasa de SSC a 5 años de alrededor del 20 %).[
Factores de riesgo de la respuesta terapéutica
- Respuesta al tratamiento o enfermedad residual mínima (ERM): la respuesta temprana a la terapia, que por lo general se evalúa después del primer curso de terapia de inducción, es un factor que predice el desenlace y se evalúa mediante un examen morfológico estándar de la médula ósea,[
39 ,61 ] análisis citogenético, hibridación fluorescente in situ, o técnicas más sofisticadas para identificar la ERM (por ejemplo, citometría de flujo multiparamétrica, RT-PCR cuantitativa).[62 ,63 ,64 ] En múltiples grupos se observó que el grado de la ERM después de un curso de terapia de inducción es un factor independiente que permite predecir el pronóstico.[62 ,63 ,64 ,65 ,66 ,67 ]Los abordajes moleculares para evaluar la ERM en la LMA (por ejemplo, el uso de la RT-PCR cuantitativa) han sido difíciles de aplicar debido a la heterogeneidad genómica de la LMA infantil y la inestabilidad de algunas alteraciones genómicas. La detección mediante RT-PCR cuantitativa de los transcritos de fusión RUNX1::RUNX1T1 puede predecir con eficacia un riesgo más alto de recaída para los pacientes en remisión clínica.[
68 ,69 ,70 ] Otras alteraciones moleculares, como las mutaciones en NPM1[71 ] y los transcritos de la fusión CBFB::MYH11, [72 ] también se han empleado con éxito como marcadores moleculares específicos de la leucemia en los ensayos de ERM; para estas alteraciones, el grado de ERM tiene importancia pronóstica. Se demostró que la presencia de mutaciones FLT3 ITD es discordante en el momento del diagnóstico y la recaída, aunque cuando persiste (con frecuencia, en relación con un cociente alélico alto en el momento del diagnóstico) puede ser útil para detectar una leucemia residual.[73 ]En la LPA, la detección de la ERM al final de la terapia de inducción carece de importancia pronóstica; es probable que se relacione con la demora en la eliminación de las células leucémicas en diferenciación destinadas a desparecer después de un tiempo.[
74 ,75 ] No obstante, el comportamiento cinético de la remisión molecular después de completar la terapia de inducción tiene carácter pronóstico: la persistencia de enfermedad mínima después de tres cursos de terapia anticipa un mayor riesgo de recaída.[75 ,76 ,77 ]Los métodos de citometría de flujo han resultado útiles para detectar la ERM y sirven para detectar los blastocitos leucémicos a partir de la expresión de antígenos de superficie anómalos que difieren del patrón que se observa en los progenitores normales. En un análisis del COG (AAML0531 [NCT00372593]) de 784 pacientes, el 69 % de los pacientes (n = 544) obtuvieron resultados negativos para la ERM (definida como <0,02 %) en la médula ósea al final de la inducción 1. Esos pacientes tuvieron mejores tasas de supervivencia sin enfermedad (57 %; IC 95 %, 53–61 %; P < 0,001) y mejores tasas de supervivencia general (73 %; IC 95 %, 69–76 %; P < 0,001) que los pacientes con resultados positivos para la ERM (SSE: 30 %; IC 95 %, 25–36 % y SG: 48 %; IC 95 %, 42–54 %).[
67 ] Además, del 76 % de los pacientes que estaban en remisión morfológica al final de la inducción 1, el 20 % presentaban resultados positivos para la ERM y tuvieron desenlaces significativamente peores que los pacientes con resultados negativos para la ERM y la remisión morfológica. Del 24 % de pacientes que no estaban en remisión morfológica, un 36 % de ellos, en realidad, tenían resultados negativos para la ERM y presentaron desenlaces significativamente mejores que los pacientes con resultados positivos para la ERM y la remisión morfológica. Esto también se observó en pacientes con porcentajes de blastocitos en médula ósea superiores al 15 %, el 27 % de ellos tuvieron resultados negativos para la ERM en la médula ósea y desenlaces significativamente mejores.[67 ] En un estudio del CCG de 252 pacientes pediátricos con LMA en remisión morfológica, se observó en un análisis multivariante que la ERM según la evaluación de citometría de flujo fue el factor pronóstico más sólido para predecir el desenlace.[78 ] En otros informes se confirmó tanto la utilidad de los métodos citométricos de flujo para la detección de la ERM en el entorno de la LMA infantil como la importancia pronóstica de la ERM en diferentes momentos después de empezar el tratamiento.[62 ,63 ,65 ]
Sistemas de clasificación de riesgo
Varios grupos cooperativos han usado la clasificación de riesgo para la asignación de tratamiento en los ensayos clínicos de niños con LMA. En el COG, un abordaje relativamente reciente es clasificar las opciones terapéuticas a partir de los factores de riesgo de los pacientes que no tienen LPA ni síndrome de Down. La clasificación se obtuvo de manera directa de las observaciones de SSC y SG del ensayo MRC AML 10 [
A continuación se presentan los ensayos del COG en los que se usa un sistema de clasificación de riesgo para estratificar las opciones de tratamiento:
- En el ensayo AAML0531 (NCT00372593), el primero del COG en el que se estratificó el tratamiento según el grupo de riesgo, se estratificó a los pacientes en 3 grupos de riesgo según las características citogenéticas en el momento del diagnóstico y la respuesta después de la inducción 1.[
8 ]- Los pacientes de riesgo bajo eran aquellos con diagnóstico de LMA con factor de unión nuclear (t(8;21) o inv(16)).
- Los pacientes de riesgo alto eran los que tenían monosomía 7, monosomía 5 o del5q, anomalías del cromosoma 3, o respuesta inadecuada a la terapia de inducción 1 con características morfológicas de blastocitos leucémicos en la médula (>15 %).
- El resto de los pacientes se asignó a la categoría de riesgo intermedio.
- Esto produjo una distribución del 24 % para el riesgo bajo, del 59 % para el riesgo intermedio y del 17 % para el riesgo alto.
- En el ensayo posterior del COG, COG-AAML1031 (NCT01371981), los grupos de riesgo se restringieron a 2 a partir de la observación de que los pacientes de la categoría de riesgo intermedio se podrían definir de manera más específica y con mayor valor pronóstico si se añadía la identificación de la ERM por citometría de flujo multiparamétrica.[
22 ,80 ]- Los pacientes con características citogenéticas o de genética molecular que no otorgaban información importante (es decir, riesgo intermedio tradicional) y que obtuvieron un resultado negativo para ERM (<0,1 %) se incluyeron en la categoría de riesgo bajo.
- Los pacientes que obtuvieron un resultado positivo para ERM (≥0,1 %) se incluyeron en la categoría de riesgo alto.
- En el ensayo del COG-AAML1031, la estratificación del estudio se basó además en las características citogenéticas, los marcadores moleculares y la ERM durante la recuperación de la médula ósea después de la posinducción 1; se dividió a los pacientes en grupos de riesgo bajo o riesgo alto como se explica a continuación:[
22 ]- El grupo de riesgo bajo incluyó al 78 % de los pacientes, tuvo una tasa de SG a 3 años desde el final de la inducción 1 del 74,1 % (±3,4 %) y se definió de la siguiente manera:
- inv(16), t(8;21), mutaciones en NPM1 o mutaciones en CEBPA con independencia de la ERM u otras características citogenéticas.
- Características citogenéticas de riesgo intermedio (definidas como ausencia de características citogenéticas de riesgo bajo o riesgo alto), resultado negativo para ERM (<0,1 % por citometría de flujo) al final de la inducción 1.
- El grupo de riesgo alto incluyó al 22 % restante de los pacientes, tuvo una tasa de SG a 3 años desde el final de la inducción 1 del 36,9 % (±7,6 %) y se definió de la siguiente manera:
- Proporción alélica alta de mutaciones FLT3 ITD con cualquier estado de ERM.
- Monosomía 7 con cualquier estado de ERM.
- Monosomía 5/del(5q) con cualquier estado de ERM.
- Características citogenéticas de riesgo intermedio con resultado positivo para ERM al final de la inducción 1.
Cuando los factores de riesgo se contradecían entre sí, se usó el siguiente cuadro basado en la evidencia (consultar el Cuadro 7).
Cuadro 7. Evaluación de riesgo en el estudio AAML1031a,b Evaluación de riesgo: Riesgo bajo Riesgo alto Grupo de riesgo bajo 1 Grupo de riesgo bajo 2 Grupo de riesgo alto 1 Grupo de riesgo alto 2 Grupo de riesgo alto 3 ITD = duplicaciones internas en tándem. a Los grupos se basan en la combinación de factores de riesgo que se encuentran en cualquier paciente individual. b La letra ennegrita indica el factor de riesgo dominante en la evaluación del grupo de riesgo. c NPM1,CEBPA, t(8;21), inv(16). d Monosomía 7, monosomía 5, del(5q). Proporción alélica de mutacionesFLT3ITD Baja o negativa Baja o negativa Alta Baja o negativa Baja o negativa Marcadores moleculares de riesgo bajoc Presentes Ausentes Cualquiera Ausentes Ausentes Marcadores citogenéticos de riesgo desfavorabled Cualquiera Ausente Cualquiera Presente Ausente Enfermedad residual mínima Cualquiera Ausente Cualquiera Cualquiera Presente
- El grupo de riesgo bajo incluyó al 78 % de los pacientes, tuvo una tasa de SG a 3 años desde el final de la inducción 1 del 74,1 % (±3,4 %) y se definió de la siguiente manera:
Al grupo de pacientes de riesgo alto se los dirigió a recibir un trasplante del donante más apropiado una vez que alcanzaron la primera remisión. A los pacientes del grupo de riesgo bajo se les indicó proseguir con un trasplante en caso de recaída.[
Los factores de riesgo que se usan para la estratificación varían según los grupos de ensayos cooperativos de niños y adultos; la repercusión pronóstica de determinado factor de riesgo quizás varíe de acuerdo con la terapia de base que se use. Otros grupos cooperativos pediátricos usan solo algunos o todos estos factores; por lo general, eligen los factores de riesgo que se han podido reproducir en múltiples ensayos clínicos y, a veces, incluyen otros factores de riesgo que ya usaron antes en su propio abordaje de estratificación de grupos de riesgo.
Referencias:
- Ebb DH, Weinstein HJ: Diagnosis and treatment of childhood acute myelogenous leukemia. Pediatr Clin North Am 44 (4): 847-62, 1997.
- Chan GC, Wang WC, Raimondi SC, et al.: Myelodysplastic syndrome in children: differentiation from acute myeloid leukemia with a low blast count. Leukemia 11 (2): 206-11, 1997.
- Cheson BD, Bennett JM, Kopecky KJ, et al.: Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 21 (24): 4642-9, 2003.
- Walter RB, Kantarjian HM, Huang X, et al.: Effect of complete remission and responses less than complete remission on survival in acute myeloid leukemia: a combined Eastern Cooperative Oncology Group, Southwest Oncology Group, and M. D. Anderson Cancer Center Study. J Clin Oncol 28 (10): 1766-71, 2010.
- Konopleva M, Cheng SC, Cortes JE, et al.: Independent prognostic significance of day 21 cytogenetic findings in newly-diagnosed acute myeloid leukemia or refractory anemia with excess blasts. Haematologica 88 (7): 733-6, 2003.
- Hann IM, Webb DK, Gibson BE, et al.: MRC trials in childhood acute myeloid leukaemia. Ann Hematol 83 (Suppl 1): S108-12, 2004.
- Gibson BE, Webb DK, Howman AJ, et al.: Results of a randomized trial in children with Acute Myeloid Leukaemia: medical research council AML12 trial. Br J Haematol 155 (3): 366-76, 2011.
- Gamis AS, Alonzo TA, Meshinchi S, et al.: Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children's Oncology Group trial AAML0531. J Clin Oncol 32 (27): 3021-32, 2014.
- Wells RJ, Woods WG, Buckley JD, et al.: Treatment of newly diagnosed children and adolescents with acute myeloid leukemia: a Childrens Cancer Group study. J Clin Oncol 12 (11): 2367-77, 1994.
- Perel Y, Auvrignon A, Leblanc T, et al.: Impact of addition of maintenance therapy to intensive induction and consolidation chemotherapy for childhood acute myeloblastic leukemia: results of a prospective randomized trial, LAME 89/91. Leucámie Aiqüe Myéloïde Enfant. J Clin Oncol 20 (12): 2774-82, 2002.
- Fenaux P, Chastang C, Chevret S, et al.: A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood 94 (4): 1192-200, 1999.
- Lo-Coco F, Avvisati G, Vignetti M, et al.: Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 369 (2): 111-21, 2013.
- Avvisati G, Lo-Coco F, Paoloni FP, et al.: AIDA 0493 protocol for newly diagnosed acute promyelocytic leukemia: very long-term results and role of maintenance. Blood 117 (18): 4716-25, 2011.
- Cooper TM, Franklin J, Gerbing RB, et al.: AAML03P1, a pilot study of the safety of gemtuzumab ozogamicin in combination with chemotherapy for newly diagnosed childhood acute myeloid leukemia: a report from the Children's Oncology Group. Cancer 118 (3): 761-9, 2012.
- Leung W, Hudson MM, Strickland DK, et al.: Late effects of treatment in survivors of childhood acute myeloid leukemia. J Clin Oncol 18 (18): 3273-9, 2000.
- Webb DK, Harrison G, Stevens RF, et al.: Relationships between age at diagnosis, clinical features, and outcome of therapy in children treated in the Medical Research Council AML 10 and 12 trials for acute myeloid leukemia. Blood 98 (6): 1714-20, 2001.
- Razzouk BI, Estey E, Pounds S, et al.: Impact of age on outcome of pediatric acute myeloid leukemia: a report from 2 institutions. Cancer 106 (11): 2495-502, 2006.
- Lange BJ, Smith FO, Feusner J, et al.: Outcomes in CCG-2961, a children's oncology group phase 3 trial for untreated pediatric acute myeloid leukemia: a report from the children's oncology group. Blood 111 (3): 1044-53, 2008.
- Creutzig U, Büchner T, Sauerland MC, et al.: Significance of age in acute myeloid leukemia patients younger than 30 years: a common analysis of the pediatric trials AML-BFM 93/98 and the adult trials AMLCG 92/99 and AMLSG HD93/98A. Cancer 112 (3): 562-71, 2008.
- Woods WG, Franklin AR, Alonzo TA, et al.: Outcome of adolescents and young adults with acute myeloid leukemia treated on COG trials compared to CALGB and SWOG trials. Cancer 119 (23): 4170-9, 2013.
- Canner J, Alonzo TA, Franklin J, et al.: Differences in outcomes of newly diagnosed acute myeloid leukemia for adolescent/young adult and younger patients: a report from the Children's Oncology Group. Cancer 119 (23): 4162-9, 2013.
- Aplenc R, Meshinchi S, Sung L, et al.: Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: a report from the Children's Oncology Group. Haematologica 105 (7): 1879-1886, 2020.
- Creutzig U, Zimmermann M, Bourquin JP, et al.: Favorable outcome in infants with AML after intensive first- and second-line treatment: an AML-BFM study group report. Leukemia 26 (4): 654-61, 2012.
- Kawasaki H, Isoyama K, Eguchi M, et al.: Superior outcome of infant acute myeloid leukemia with intensive chemotherapy: results of the Japan Infant Leukemia Study Group. Blood 98 (13): 3589-94, 2001.
- Masetti R, Rondelli R, Fagioli F, et al.: Infants with acute myeloid leukemia treated according to the Associazione Italiana di Ematologia e Oncologia Pediatrica 2002/01 protocol have an outcome comparable to that of older children. Haematologica 99 (8): e127-9, 2014.
- Guest EM, Aplenc R, Sung L, et al.: Gemtuzumab ozogamicin in infants with AML: results from the Children's Oncology Group trials AAML03P1 and AAML0531. Blood 130 (7): 943-945, 2017.
- Aplenc R, Alonzo TA, Gerbing RB, et al.: Ethnicity and survival in childhood acute myeloid leukemia: a report from the Children's Oncology Group. Blood 108 (1): 74-80, 2006.
- Conneely SE, McAtee CL, Gupta R, et al.: Association of race and ethnicity with clinical phenotype, genetics, and survival in pediatric acute myeloid leukemia. Blood Adv 5 (23): 4992-5001, 2021.
- Rubnitz JE, Lensing S, Razzouk BI, et al.: Effect of race on outcome of white and black children with acute myeloid leukemia: the St. Jude experience. Pediatr Blood Cancer 48 (1): 10-5, 2007.
- Lange BJ, Kobrinsky N, Barnard DR, et al.: Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: Children's Cancer Group Studies 2861 and 2891. Blood 91 (2): 608-15, 1998.
- Sorrell AD, Alonzo TA, Hilden JM, et al.: Favorable survival maintained in children who have myeloid leukemia associated with Down syndrome using reduced-dose chemotherapy on Children's Oncology Group trial A2971: a report from the Children's Oncology Group. Cancer 118 (19): 4806-14, 2012.
- Taub JW, Berman JN, Hitzler JK, et al.: Improved outcomes for myeloid leukemia of Down syndrome: a report from the Children's Oncology Group AAML0431 trial. Blood 129 (25): 3304-3313, 2017.
- Creutzig U, Reinhardt D, Diekamp S, et al.: AML patients with Down syndrome have a high cure rate with AML-BFM therapy with reduced dose intensity. Leukemia 19 (8): 1355-60, 2005.
- Massey GV, Zipursky A, Chang MN, et al.: A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481. Blood 107 (12): 4606-13, 2006.
- Gamis AS, Woods WG, Alonzo TA, et al.: Increased age at diagnosis has a significantly negative effect on outcome in children with Down syndrome and acute myeloid leukemia: a report from the Children's Cancer Group Study 2891. J Clin Oncol 21 (18): 3415-22, 2003.
- Uffmann M, Rasche M, Zimmermann M, et al.: Therapy reduction in patients with Down syndrome and myeloid leukemia: the international ML-DS 2006 trial. Blood 129 (25): 3314-3321, 2017.
- Lange BJ, Gerbing RB, Feusner J, et al.: Mortality in overweight and underweight children with acute myeloid leukemia. JAMA 293 (2): 203-11, 2005.
- Inaba H, Surprise HC, Pounds S, et al.: Effect of body mass index on the outcome of children with acute myeloid leukemia. Cancer 118 (23): 5989-96, 2012.
- Creutzig U, Zimmermann M, Ritter J, et al.: Definition of a standard-risk group in children with AML. Br J Haematol 104 (3): 630-9, 1999.
- Pession A, Masetti R, Rizzari C, et al.: Results of the AIEOP AML 2002/01 multicenter prospective trial for the treatment of children with acute myeloid leukemia. Blood 122 (2): 170-8, 2013.
- Sung L, Aplenc R, Alonzo TA, et al.: Predictors and short-term outcomes of hyperleukocytosis in children with acute myeloid leukemia: a report from the Children's Oncology Group. Haematologica 97 (11): 1770-3, 2012.
- Testi AM, Biondi A, Lo Coco F, et al.: GIMEMA-AIEOPAIDA protocol for the treatment of newly diagnosed acute promyelocytic leukemia (APL) in children. Blood 106 (2): 447-53, 2005.
- Kutny MA, Alonzo TA, Gerbing RB, et al.: Arsenic Trioxide Consolidation Allows Anthracycline Dose Reduction for Pediatric Patients With Acute Promyelocytic Leukemia: Report From the Children's Oncology Group Phase III Historically Controlled Trial AAML0631. J Clin Oncol 35 (26): 3021-3029, 2017.
- de Botton S, Coiteux V, Chevret S, et al.: Outcome of childhood acute promyelocytic leukemia with all-trans-retinoic acid and chemotherapy. J Clin Oncol 22 (8): 1404-12, 2004.
- Ortega JJ, Madero L, Martín G, et al.: Treatment with all-trans retinoic acid and anthracycline monochemotherapy for children with acute promyelocytic leukemia: a multicenter study by the PETHEMA Group. J Clin Oncol 23 (30): 7632-40, 2005.
- Chisholm KM, Heerema-McKenney AE, Choi JK, et al.: Acute erythroid leukemia is enriched in NUP98 fusions: a report from the Children's Oncology Group. Blood Adv 4 (23): 6000-6008, 2020.
- Reinhardt D, Diekamp S, Langebrake C, et al.: Acute megakaryoblastic leukemia in children and adolescents, excluding Down's syndrome: improved outcome with intensified induction treatment. Leukemia 19 (8): 1495-6, 2005.
- Schweitzer J, Zimmermann M, Rasche M, et al.: Improved outcome of pediatric patients with acute megakaryoblastic leukemia in the AML-BFM 04 trial. Ann Hematol 94 (8): 1327-36, 2015.
- de Rooij JD, Masetti R, van den Heuvel-Eibrink MM, et al.: Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective intergroup study. Blood 127 (26): 3424-30, 2016.
- de Rooij JD, Branstetter C, Ma J, et al.: Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet 49 (3): 451-456, 2017.
- Barbaric D, Alonzo TA, Gerbing RB, et al.: Minimally differentiated acute myeloid leukemia (FAB AML-M0) is associated with an adverse outcome in children: a report from the Children's Oncology Group, studies CCG-2891 and CCG-2961. Blood 109 (6): 2314-21, 2007.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Central nervous system disease in pediatric acute myeloid leukemia: A report from the Children's Oncology Group. Pediatr Blood Cancer 64 (12): , 2017.
- Abbott BL, Rubnitz JE, Tong X, et al.: Clinical significance of central nervous system involvement at diagnosis of pediatric acute myeloid leukemia: a single institution's experience. Leukemia 17 (11): 2090-6, 2003.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: The presence of central nervous system disease at diagnosis in pediatric acute myeloid leukemia does not affect survival: a Children's Oncology Group study. Pediatr Blood Cancer 55 (3): 414-20, 2010.
- Lugthart S, Gröschel S, Beverloo HB, et al.: Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J Clin Oncol 28 (24): 3890-8, 2010.
- Creutzig U, van den Heuvel-Eibrink MM, Gibson B, et al.: Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 120 (16): 3187-205, 2012.
- Eidenschink Brodersen L, Alonzo TA, Menssen AJ, et al.: A recurrent immunophenotype at diagnosis independently identifies high-risk pediatric acute myeloid leukemia: a report from Children's Oncology Group. Leukemia 30 (10): 2077-2080, 2016.
- Pardo LM, Voigt AP, Alonzo TA, et al.: Deciphering the Significance of CD56 Expression in Pediatric Acute Myeloid Leukemia: A Report from the Children's Oncology Group. Cytometry B Clin Cytom 98 (1): 52-56, 2020.
- Smith JL, Ries RE, Hylkema T, et al.: Comprehensive Transcriptome Profiling of Cryptic CBFA2T3-GLIS2 Fusion-Positive AML Defines Novel Therapeutic Options: A COG and TARGET Pediatric AML Study. Clin Cancer Res 26 (3): 726-737, 2020.
- Lamble AJ, Eidenschink Brodersen L, Alonzo TA, et al.: CD123 Expression Is Associated With High-Risk Disease Characteristics in Childhood Acute Myeloid Leukemia: A Report From the Children's Oncology Group. J Clin Oncol 40 (3): 252-261, 2022.
- Wheatley K, Burnett AK, Goldstone AH, et al.: A simple, robust, validated and highly predictive index for the determination of risk-directed therapy in acute myeloid leukaemia derived from the MRC AML 10 trial. United Kingdom Medical Research Council's Adult and Childhood Leukaemia Working Parties. Br J Haematol 107 (1): 69-79, 1999.
- van der Velden VH, van der Sluijs-Geling A, Gibson BE, et al.: Clinical significance of flowcytometric minimal residual disease detection in pediatric acute myeloid leukemia patients treated according to the DCOG ANLL97/MRC AML12 protocol. Leukemia 24 (9): 1599-606, 2010.
- Loken MR, Alonzo TA, Pardo L, et al.: Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukemia: a report from Children's Oncology Group. Blood 120 (8): 1581-8, 2012.
- Buldini B, Rizzati F, Masetti R, et al.: Prognostic significance of flow-cytometry evaluation of minimal residual disease in children with acute myeloid leukaemia treated according to the AIEOP-AML 2002/01 study protocol. Br J Haematol 177 (1): 116-126, 2017.
- Rubnitz JE, Inaba H, Dahl G, et al.: Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol 11 (6): 543-52, 2010.
- Tierens A, Bjørklund E, Siitonen S, et al.: Residual disease detected by flow cytometry is an independent predictor of survival in childhood acute myeloid leukaemia; results of the NOPHO-AML 2004 study. Br J Haematol 174 (4): 600-9, 2016.
- Brodersen LE, Gerbing RB, Pardo ML, et al.: Morphologic remission status is limited compared to ΔN flow cytometry: a Children's Oncology Group AAML0531 report. Blood Adv 4 (20): 5050-5061, 2020.
- Buonamici S, Ottaviani E, Testoni N, et al.: Real-time quantitation of minimal residual disease in inv(16)-positive acute myeloid leukemia may indicate risk for clinical relapse and may identify patients in a curable state. Blood 99 (2): 443-9, 2002.
- Viehmann S, Teigler-Schlegel A, Bruch J, et al.: Monitoring of minimal residual disease (MRD) by real-time quantitative reverse transcription PCR (RQ-RT-PCR) in childhood acute myeloid leukemia with AML1/ETO rearrangement. Leukemia 17 (6): 1130-6, 2003.
- Weisser M, Haferlach C, Hiddemann W, et al.: The quality of molecular response to chemotherapy is predictive for the outcome of AML1-ETO-positive AML and is independent of pretreatment risk factors. Leukemia 21 (6): 1177-82, 2007.
- Krönke J, Schlenk RF, Jensen KO, et al.: Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: a study from the German-Austrian acute myeloid leukemia study group. J Clin Oncol 29 (19): 2709-16, 2011.
- Corbacioglu A, Scholl C, Schlenk RF, et al.: Prognostic impact of minimal residual disease in CBFB-MYH11-positive acute myeloid leukemia. J Clin Oncol 28 (23): 3724-9, 2010.
- Cloos J, Goemans BF, Hess CJ, et al.: Stability and prognostic influence of FLT3 mutations in paired initial and relapsed AML samples. Leukemia 20 (7): 1217-20, 2006.
- Mandelli F, Diverio D, Avvisati G, et al.: Molecular remission in PML/RAR alpha-positive acute promyelocytic leukemia by combined all-trans retinoic acid and idarubicin (AIDA) therapy. Gruppo Italiano-Malattie Ematologiche Maligne dell'Adulto and Associazione Italiana di Ematologia ed Oncologia Pediatrica Cooperative Groups. Blood 90 (3): 1014-21, 1997.
- Burnett AK, Grimwade D, Solomon E, et al.: Presenting white blood cell count and kinetics of molecular remission predict prognosis in acute promyelocytic leukemia treated with all-trans retinoic acid: result of the Randomized MRC Trial. Blood 93 (12): 4131-43, 1999.
- Diverio D, Rossi V, Avvisati G, et al.: Early detection of relapse by prospective reverse transcriptase-polymerase chain reaction analysis of the PML/RARalpha fusion gene in patients with acute promyelocytic leukemia enrolled in the GIMEMA-AIEOP multicenter "AIDA" trial. GIMEMA-AIEOP Multicenter "AIDA" Trial. Blood 92 (3): 784-9, 1998.
- Martinelli G, Ottaviani E, Testoni N, et al.: Disappearance of PML/RAR alpha acute promyelocytic leukemia-associated transcript during consolidation chemotherapy. Haematologica 83 (11): 985-8, 1998.
- Sievers EL, Lange BJ, Alonzo TA, et al.: Immunophenotypic evidence of leukemia after induction therapy predicts relapse: results from a prospective Children's Cancer Group study of 252 patients with acute myeloid leukemia. Blood 101 (9): 3398-406, 2003.
- Webb DK, Wheatley K, Harrison G, et al.: Outcome for children with relapsed acute myeloid leukaemia following initial therapy in the Medical Research Council (MRC) AML 10 trial. MRC Childhood Leukaemia Working Party. Leukemia 13 (1): 25-31, 1999.
- Tarlock K, Meshinchi S: Pediatric acute myeloid leukemia: biology and therapeutic implications of genomic variants. Pediatr Clin North Am 62 (1): 75-93, 2015.
- Pui CH, Carroll WL, Meshinchi S, et al.: Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol 29 (5): 551-65, 2011.
Consideraciones especiales para el tratamiento de niños con cáncer
El cáncer en niños y adolescentes es infrecuente, aunque desde 1975 se ha observado un aumento gradual de la incidencia general.[
- Médicos de atención primaria.
- Subespecialistas en cirugía pediátrica.
- Radioncólogos.
- Oncólogos o hematólogos pediatras.
- Especialistas en rehabilitación.
- Enfermeros especializados en pediatría.
- Trabajadores sociales.
Para obtener información específica sobre los cuidados médicos de apoyo para niños y adolescentes con cáncer, consultar los sumarios de Cuidados médicos de apoyo y cuidados paliativos.
La American Academy of Pediatrics estableció pautas para los centros de oncología pediátrica y su función en el tratamiento de los pacientes de cáncer infantil.[
Referencias:
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Wolfson J, Sun CL, Wyatt L, et al.: Adolescents and Young Adults with Acute Lymphoblastic Leukemia and Acute Myeloid Leukemia: Impact of Care at Specialized Cancer Centers on Survival Outcome. Cancer Epidemiol Biomarkers Prev 26 (3): 312-320, 2017.
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed May 19, 2023.
Tratamiento de la leucemia mieloide aguda infantil
Los principios generales del tratamiento de los niños y adolescentes con leucemia mieloide aguda (LMA) se examinan a continuación, y se siguen de un análisis más específico del tratamiento de los niños con síndrome de Down y leucemia promielocítica aguda (LPA).
Las tasas de supervivencia general (SG) han mejorado en los últimos 30 años para los niños con LMA, con tasas de supervivencia a 5 años que ahora oscilan entre el 55 % y el 65 %.[
Terapia de inducción
Los protocolos contemporáneos para la LMA infantil producen tasas de remisión completa (RC) del 85 % al 90 %.[
Las opciones de tratamiento para los niños con LMA durante la fase de inducción son las siguientes:
- Quimioterapia.
- Abordajes inmunoterapéuticos (por ejemplo, gemtuzumab ozogamicina).
- Terapia dirigida (por ejemplo, inhibidores de FLT3).
- Cuidados médicos de apoyo.
Quimioterapia
Los dos fármacos más eficaces e imprescindibles que se usan para inducir la remisión en niños con LMA son la citarabina y una antraciclina. Los regímenes de terapia de inducción que se usan con más frecuencia en pediatría incluyen la citarabina y una antraciclina en combinación con otros fármacos como etopósido o tioguanina.[
Evidencia (régimen de quimioterapia de inducción):
- En el ensayo del United Kingdom Medical Research Council (MRC) AML10, se comparó la inducción con citarabina, daunorrubicina y etopósido (ADE) versus citarabina, daunorrubicina y tioguanina (DAT).[
12 ]- En los desenlaces, no se observaron diferencias entre los grupos de tioguanina y etopósido en la tasa de remisión o supervivencia sin enfermedad (SSE), aunque el régimen con tioguanina se relacionó con aumento de la toxicidad.
- En el ensayo MRC AML15 se demostró que la inducción con daunorrubicina y citarabina (DA) produjo tasas de supervivencia equivalentes cuando se la comparó con la inducción con ADE.[
13 ]
La antraciclina que se ha usado con mayor frecuencia en los regímenes de inducción para los niños con LMA es la daunorrubicina,[
Evidencia (antraciclina):
- En el estudio AML-BFM 93 del grupo alemán Berlin-Frankfurt-Münster (BFM) se evaluó la citarabina y el etopósido combinadas con la daunorrubicina o la idarrubicina (ADE o AIE).[
11 ,14 ]- Se observaron tasas de SSC y SG similares con ambos tratamientos de inducción.
- En el ensayo clínico MRC-LEUK-AML12 (NCT00002658) , se estudió la inducción con citarabina, mitoxantrona y etopósido (MAE) en niños y adultos de LMA comparados con un régimen similar en el que se usó daunorrubicina (ADE).[
6 ,16 ]- Para todos los pacientes, el régimen MAE redujo el riesgo de recaída, pero el aumento de la tasa de mortalidad relacionada con el tratamiento que se observó en los pacientes que recibieron el régimen MAE no derivó en diferencias significativas en la SSE o SG cuando se lo comparó con el régimen ADE.[
16 ] - Se observaron resultados similares cuando el análisis se restringió a los pacientes pediátricos.[
6 ]
- Para todos los pacientes, el régimen MAE redujo el riesgo de recaída, pero el aumento de la tasa de mortalidad relacionada con el tratamiento que se observó en los pacientes que recibieron el régimen MAE no derivó en diferencias significativas en la SSE o SG cuando se lo comparó con el régimen ADE.[
- En el ensayo clínico AML-BFM 2004 (NCT00111345), se comparó la daunorrubicina liposomal (L-DNR) con la idarrubicina en una dosis mayor que la dosis equivalente (80 mg/m2 vs. 12 mg/m2 diarios por 3 días) durante la inducción.[
17 ]- Las tasas de SG y SSC a 5 años fueron similares en ambos grupos de tratamiento.
- La mortalidad relacionada con el tratamiento fue significativamente más baja con L-DNR que con idarrubicina (2 de 257 pacientes vs. 10 de 264 pacientes).
- En el ensayo del COG AAML1031 (NCT01371981) se usó mitoxantrona con dosis altas de citarabina en una fase de inducción de 2 ciclos para pacientes con LMA de riesgo alto.[
18 ]- En una comparación planificada con el ensayo AAML0531 (NCT00372593), en el que se usó un régimen ADE estándar en el segundo ciclo de inducción en pacientes parecidos, no se encontró una mejora de la supervivencia, pero sí un aumento de la toxicidad en los pacientes que recibieron la mitoxantrona.
Evidencia (régimen de inducción con reducción de antraciclina):
- Aunque la combinación de una antraciclina y citarabina es la base de la terapia inicial de inducción estándar para adultos y niños, hay evidencia de que se pueden usar otros fármacos para reducir el uso de antraciclinas cuando sea necesario. En el protocolo del St. Jude Children's Research Hospital (SJCRH) AML08 (NCT00703820) , los pacientes se asignaron al azar para recibir clofarabina y citarabina (CA) o citarabina en dosis alta en combinación con daunorrubicina y etopósido (HD-ADE) para la inducción I; luego, todos los pacientes recibieron el régimen ADE en dosis estándar que contiene antraciclina para la inducción II.[
19 ]- Pese a la tasa más alta de enfermedad residual mínima (ERM) en el grupo de CA en el día 22 de la inducción I (47 vs. 35 %, P = 0,04), las tasas de SSC y SG a 3 años fueron similares en los dos grupos.
La intensidad de la terapia de inducción influye en el resultado general del tratamiento. En el estudio CCG-2891, se demostró que la terapia de inducción de programación intensa (cursos de tratamiento de 4 días separados solo por 6 días) produjo una SSC superior que la terapia de inducción de programación estándar (cursos de tratamiento de 4 días separados por 2 semanas o más).[
En adultos, otro método para intensificar la terapia de inducción es administrar dosis altas de citarabina. Aunque en los estudios de adultos de edad mediana se indica una ventaja de la terapia de inducción intensificada con dosis altas de citarabina (2–3 g/m2 /dosis) en comparación con la dosis estándar de citarabina,[
Abordajes inmunoterapéuticos
Se han examinado otros abordajes, como el uso de gemtuzumab ozogamicina, debido a que la intensificación adicional de los regímenes de inducción aumenta la toxicidad con poca mejora de la SSC o la SG.
Terapia con un conjugado anticuerpo-fármaco (gemtuzumab ozogamicina)
El gemtuzumab ozogamicina es un anticuerpo monoclonal dirigido a CD33 unido a la caliqueamicina, un fármaco citotóxico.
Evidencia (gemtuzumab ozogamicina durante la inducción):
- El Children's Oncology Group (COG) completó una serie de ensayos —un estudio piloto, AAML03P1 (NCT00070174) y un ensayo aleatorizado, AAML0531 (NCT00372593)— en los que se evaluó la incorporación de gemtuzumab ozogamicina a la terapia de inducción.[
8 ,9 ]- Con el uso de gemtuzumab ozogamicina durante el ciclo de inducción 1, en dosis de 3 mg/m2 en el día 6; en el ensayo aleatorizado se observó una mejora de la SSC pero no en la SG. Esto obedeció a una reducción en la recaída general después de la remisión y, en particular, en ciertos subgrupos de pacientes. En estos subconjuntos, se incluyeron pacientes con características citogenéticas de riesgo bajo, pacientes de LMA con reordenamientos en KMT2A (translocaciones de riesgo alto y translocaciones que no generan riesgo alto) de riesgo intermedio que pasaron a recibir un trasplante de células madre (TCM) de un donante emparentado compatible [
24 ] y pacientes con LMA de riesgo alto (con proporción alélica alta (>0,4) de duplicaciones internas en tándem (ITD) en FLT3 que luego se sometieron a un TCM de cualquier tipo de donante.[25 ] - En estos ensayos se determinó la eficacia e inocuidad del uso de gemtuzumab ozogamicina en niños, incluso en lactantes mayores de 1 mes.[
26 ]
- Con el uso de gemtuzumab ozogamicina durante el ciclo de inducción 1, en dosis de 3 mg/m2 en el día 6; en el ensayo aleatorizado se observó una mejora de la SSC pero no en la SG. Esto obedeció a una reducción en la recaída general después de la remisión y, en particular, en ciertos subgrupos de pacientes. En estos subconjuntos, se incluyeron pacientes con características citogenéticas de riesgo bajo, pacientes de LMA con reordenamientos en KMT2A (translocaciones de riesgo alto y translocaciones que no generan riesgo alto) de riesgo intermedio que pasaron a recibir un trasplante de células madre (TCM) de un donante emparentado compatible [
- La dosificación fraccionada de gemtuzumab ozogamicina (3 mg/m2 por dosis los días 1, 4, y 7; dosis máxima, 5 mg), que se demostró que es inocua y eficaz en pacientes adultos con LMA de novo, es una alternativa a la administración de dosis única durante la inducción.[
27 ] Debido a que este es el método de dosificación recomendado para adultos, este abordaje se está evaluando en pacientes pediátricos con LMA de novo en el estudio de fase III MyeChild 01 (NCT02724163) en el Reino Unido. - Se examinaron las características de CD33, que es la diana del gemtuzumab ozogamicina, para identificar a los pacientes que se beneficiarán más con este fármaco.
- La intensidad de la expresión de CD33 en las células leucémicas predijo qué pacientes se beneficiaron con el gemtuzumab ozogamicina en el ensayo clínico COG AAML0531.[
28 ][Nivel de evidencia B1] Los pacientes cuya intensidad de CD33 se ubicaba en los 3 cuartiles más altos de la población se beneficiaron del tratamiento con gemtuzumab ozogamicina (es decir, mejora del riesgo de recaída, SSE y SSC), mientras que aquellos con intensidad en el cuartil más bajo no presentaron una disminución del riesgo de recaída, SSC o SG. Este efecto se observó en los pacientes de los grupos de riesgo bajo, intermedio y alto. - En un análisis retrospectivo del ensayo ALFA-0701 (NCT00927498) con adultos mayores, la expresión alta de CD33 se correspondió con un mayor beneficio del tratamiento con gemtuzumab ozogamicina.[
29 ] - El receptor de CD33 en las células de la LMA exhibió variación estructural (polimorfismo) que se tradujo en que el 51 % de los pacientes expresaba un polimorfismo de un solo nucleótido (SNP) rs12459419 (designado CC); en estos pacientes se produjo una reducción significativa de las recaídas con el uso de gemtuzumab ozogamicina en comparación con los pacientes que no se trataron con este fármaco (26 vs. 49 %; P < 0,001). La alteración de este SNP produjo una isoforma de CD33 que carece del dominio para la IgV en CD33, sitio al que se une el gemtuzumab ozogamicina y que se usa para el diagnóstico por inmunofenotipificación.[
30 ] - Los pacientes con mutación C>T monoalélica o bialélica (fenotipos CT y TT, respectivamente) en este SNP, no presentaron reducción de las recaídas cuando se agregó la terapia con gemtuzumab ozogamicina (incidencia acumulada de recaída a 5 años, 39 vs. 40 %; P = 0,85).
- La intensidad de la expresión de CD33 en las células leucémicas predijo qué pacientes se beneficiaron con el gemtuzumab ozogamicina en el ensayo clínico COG AAML0531.[
- En un metanálisis de cinco ensayos clínicos aleatorizados en los que se evaluó el uso de gemtuzumab ozogamicina para adultos con LMA, se observó lo siguiente:[
31 ]- El mayor beneficio de SG se observó en los pacientes con características citogenéticas de riesgo bajo (t(8;21)(q22;q22) e inv(16)(p13;q22)/t(16;16)(p13;q22)).
- Los pacientes adultos de LMA con características citogenéticas de riesgo intermedio que recibieron gemtuzumab ozogamicina presentaron una mejora significativa, pero más baja, de la SG.
- No se encontró evidencia de beneficio para los pacientes con características citogenéticas adversas.
- Hubo discrepancia en la evidencia de beneficio para los pacientes con mutaciones FLT3 ITD; en el ensayo francés ALFA-0701 (NCT00927498) se observó tendencia hacia el beneficio, mientras que en el metanálisis de cinco ensayos no se encontró beneficio.[
27 ,31 ] En estos ensayos no se evaluaron los desenlaces específicos de la combinación de gemtuzumab ozogamicina seguido de trasplante de células madre que notificó el COG.[25 ]
Terapia dirigida
Al igual que con los abordajes inmunoterapéuticos, el uso de terapia dirigida intenta evitar la toxicidad grave de la quimioterapia tradicional al emplear fármacos que se dirigen a mutaciones específicas de la leucemia y a sus derivados anormales presentes o ausentes. Al diferencia de la LMA en adultos (excepto en la LAP como se describe más adelante en otra sección), en los ensayos clínicos aleatorizados no se ha demostrado todavía que las terapias dirigidas mejoren los desenlaces en los niños con LMA recién diagnosticada; por lo tanto, las terapias dirigidas no se han incorporado a los regímenes terapéuticos de inducción estándar fuera de los ensayos clínicos. Dado que gran parte de los datos sobre el uso de fármacos dirigidos provienen de ensayos clínicos con adultos, primero se describe la experiencia con adultos, seguida de una descripción de la experiencia más limitada con niños.
Inhibidores de FLT3 en la LMAde novo
Debido a la alta prevalencia de mutaciones en FLT3 de la LMA en adultos y el efecto negativo en los pacientes de LMA de todas las edades, el FLT3 ha recibido la mayor atención en el desarrollo de fármacos específicos para una molécula diana en la LMA. Entre los diferentes inhibidores de FLT3 que se desarrollaron y se estudiaron de forma clínica, la midostaurina, un inhibidor multicinasa, es el único aprobado por la Administración de Alimentos y Medicamentos (FDA) para la LMA de novo en adultos; se aprobó en 2017 para el uso con quimioterapia de base convencional, pero no como monoterapia.[
Midostaurina
Evidencia (midostaurina para adultos con LMA de novo):
- En un estudio aleatorizado, controlado con placebo, de fase III (CALGB10603/RATIFY [NCT00651261]) de 717 adultos entre 18 a 59 años con LMA que presentaba mutaciones FLT3 ITD o TKD, se administró quimioterapia estándar con midostaurina o sin esta (50 mg/dosis 2 veces al día) seguida de midostaurina de mantenimiento o placebo para los pacientes que no recibieron a continuación un TCM.[
33 ]- La SG (criterio principal de valoración) fue significativamente mejor en los pacientes que recibieron midostaurina (mediana de SG, 74,7 meses [intervalo de confianza (IC) 95 %, 31,5–no alcanzado] vs. 25,6 meses [IC 95 %, 18,6–42,9]; cociente de riesgos instantáneos [CRI], 0,78 [IC 95 %, 0,63–0,96]; P = 0,009), al igual que la SSC (mediana de SSC, 8,2 meses [IC 95 %, 5,4–10,7] vs. 3,0 meses [IC 95 %, 1,9–5,9]; CRI, 0,78 [IC 95 %, 0,66–0,93]; P = 0,002).
- Este beneficio se observó en todos los subgrupos de mutaciones en FLT3 independientemente de que se hubiera utilizado un TCM alogénico durante la consolidación.
- En un segundo ensayo de grupo único con 284 pacientes adultos (edad,18–70 años) de LMA con mutaciones FLT3 ITD, se añadió midostaurina (50 mg/dosis 2 veces al día) a la quimioterapia intensiva seguida de TCM alogénico o consolidación, y todos los pacientes tuvieron una fase de mantenimiento con midostaurina.[
34 ]- La tasa de SSC a 2 años fue del 37,7 % (IC 95 %, 32–44,3 %) y la tasa de SG fue del 50,9 % (IC 95 %, 44,9–57,6 %).
- En una comparación donde se utilizó un grupo de control histórico se notificó una mejora significativa de la SSC (CRI, 0,58; IC 95 %, 0,48–0,70; P < 0,001).
La midostaurina se ha estudiado en niños con LMA en recaída o resistente al tratamiento,[
Sorafenib
El sorafenib, otro inhibidor multicinasa, se aprobó para el tratamiento de otras neoplasias malignas, pero no para la LMA. Se ha evaluado el uso de este medicamento en pacientes con LMA de novo con mutación en FLT3.
Evidencia (sorafenib):
- En el estudio AAML1031 (NCT01371981) del COG de pacientes pediátricos con LMA de novo y con proporción alélica alta (HAR) (es decir, >0,4) de mutaciones FLT3 ITD, se demostró que el sorafenib mejoró la SSC. Se pudieron evaluar por respuesta a 72 de los pacientes que recibieron sorafenib. Los pacientes de este estudio se compararon con pacientes con LMA y HAR FLT3 ITD (N = 76) de los ensayos AAML1031 y COG AAML0531 que no recibieron sorafenib.[
36 ]- La tasa morfológica de RC después del ciclo de inducción I mejoró significativamente en los pacientes que recibieron sorafenib (75 vs. 57 %; P = 0,028).
- Sin embargo, hubo una prevalencia similar de ERM residual en ambos grupos de pacientes (48 vs. 45 %; P = 0,724).
- Los pacientes que recibieron sorafenib presentaron tasas de SSC a 3 años (55,9 vs. 31,9 %; P = 0,001), tasas de SSE (70,9 vs. 49,4 %; P = 0,032) y recaídas luego de RC (17,6 vs. 44,1 %; P = 0,012) significativamente mejores.
- La tasa de SG no mejoró después del tratamiento con sorafenib (65,8 vs. 55,3 %; P = 0,244).
- A pesar de que se observaron tendencias similares en los pacientes con LMA que presentaban HAR de mutaciones FLT3 ITD y mutaciones en NPM1, estos no obtuvieron un grado de beneficio significativo.
- Los datos estadísticos demostraron que se mantuvo un beneficio del tratamiento con sorafenib después de los análisis multivariantes en los que se controló el estado de NPM1 y del trasplante de células hematopoyéticas (TCMH) como una covariable en el tiempo.
Cuidados médicos de apoyo
En los niños con LMA que reciben el tratamiento intensivo contemporáneo, el cálculo de incidencia de las infecciones bacterianas graves es del 50 % al 60 %, y el cálculo de incidencia de infecciones fúngicas es del 7,0 % al 12,5 %.[
Factores de crecimiento hematopoyéticos
En múltiples estudios controlados con placebo se evaluó el uso de los factores de crecimiento hematopoyéticos como el factor estimulante de colonias de granulocitos y macrófagos (GM-CSF) o el factor estimulante de colonias de granulocitos (G-CSF) durante la terapia de inducción en adultos con LMA para reducir la toxicidad relacionada con la mielodepresión prolongada.[
No se recomienda el uso profiláctico rutinario de factores de crecimiento hematopoyético para los niños con LMA.
Evidencia (en contra del uso de factores de crecimiento hematopoyéticos):
- En un estudio aleatorizado de niños con LMA en el que se evaluó el G-CSF administrado después de la quimioterapia de inducción, se observó una reducción en la duración de la neutropenia, pero no se observaron diferencias en las complicaciones infecciosas o la mortalidad.[
41 ] - Se notificó una tasa de recaída más alta en los niños con LMA que expresan diferenciación defectuosa de la isoforma IV del receptor del G-CSF.[
42 ]
Profilaxis antimicrobiana
En varios estudios se ha respaldado la administración profiláctica de antibióticos para los niños que se someten a tratamiento de la LMA. En los estudios, incluso en un ensayo aleatorizado prospectivo, se indica un beneficio del uso de profilaxis con antibiótico.
Evidencia (profilaxis antimicrobiana):
- En un estudio retrospectivo del SJCRH con pacientes de LMA, se informó que el uso de cefepima intravenosa (IV) o vancomicina junto con ciprofloxacino oral o una cefalosporina disminuye de manera significativa la incidencia de infecciones bacterianas y sepsis en comparación con los pacientes que solo reciben profilaxis con antibiótico oral o no reciben profilaxis.[
43 ] - Los resultados del SJCRH se confirmaron en un estudio posterior.[
44 ] - En un informe retrospectivo del ensayo del COG AAML0531 (NCT00372593), se demostraron reducciones significativas de las infecciones bacterianas en sitios estériles y, en particular, de infecciones en sitios estériles causadas por microorganismos grampositivos cuando se usó la profilaxis con antibióticos.[
45 ] En este estudio también se notificó que el uso profiláctico de G-CSF redujo las infecciones bacterianas y por Clostridium difficile.[45 ] - En un estudio en el que se comparó el porcentaje de infecciones sanguíneas o infecciones fúngicas invasivas en niños con leucemia linfoblástica aguda (LLA) o LMA sometidos a quimioterapia y que recibieron profilaxis con antibacterianos o antifúngicos, se observó una reducción significativa de ambas variables cuando se compararon con un grupo de control histórico que no recibió ningún tipo de profilaxis.[
46 ] - En el estudio prospectivo del COG ACCL0934 para niños que estaban recibiendo quimioterapia intensiva, los pacientes se inscribieron en dos grupos diferentes: pacientes de leucemia aguda (LMA o LLA recidivante) y pacientes sometidos a un TCM. Los pacientes con leucemia aguda se asignaron al azar para recibir levofloxacino (n = 96) o no recibir antibiótico profiláctico (n = 99) durante el período de neutropenia en 1 o 2 ciclos de quimioterapia.[
47 ]- En el análisis de los 195 niños con leucemia aguda se observó una reducción significativa de las bacteriemias (43,4 % a 21,9 %, P = 0,001) y de la fiebre neutropénica (82,1 % a 71,2 %, P = 0,002) en el grupo de profilaxis con levofloxacino en comparación con el grupo de control, sin aumento de las infecciones por hongos, diarrea relacionada con C. difficile o efectos tóxicos osteomusculares.
- No hubo una disminución significativa de las infecciones graves (3,6 % vs. 5,9 %, P = 0,20) y no hubo muertes a causa de infecciones bacterianas en ninguno de los dos grupos.
- La profilaxis con levofloxacino concuerda con las pautas publicadas en 2018 por la American Society of Clinical Oncology y la Infectious Diseases Society of America para adultos con cáncer que se consideran en riesgo alto de infección como resultado de una neutropenia (<100 neutrófilos/µl) de más de 7 días.[
48 ]
Profilaxis antifúngica
La profilaxis antifúngica es importante en el abordaje de los pacientes con LMA.
Evidencia (profilaxis antifúngica):
- En dos informes de metanálisis, se indicó que, en los pacientes pediátricos con LMA, la administración de profilaxis antifúngica durante la neutropenia inducida por el tratamiento o durante el trasplante de médula ósea reduce la frecuencia de las infecciones fúngicas invasivas y, en algunos casos, la mortalidad no relacionada con las recaídas.[
49 ,50 ] - En otro estudio se llevó a cabo una encuesta en instituciones que inscribieron pacientes en el ensayo del COG AAML0531 (NCT00372593) y se evaluó si estas instituciones prescribían profilaxis antiúngica de forma habitual.[
45 ]- En el estudio se observó que la profilaxis antifúngica no redujo las infecciones fúngicas ni la mortalidad no relacionada con una recaída.
- Sin embargo, el estudio tuvo limitaciones porque los investigadores no analizaron si los pacientes, a nivel individual, recibieron profilaxis antifúngica, independientemente de las directrices institucionales.
- En múltiples ensayos clínicos aleatorizados de adultos con LMA se notificaron beneficios significativos del uso de profilaxis antifúngica para reducir las infecciones fúngicas invasivas. En dichos estudios, también se compararon los efectos adversos en función del costo; cuando la eficacia de reducir las infecciones fúngicas invasivas se equilibra con estos factores adicionales, el posaconazol, el voriconazol, la caspofungina y la micafungina se consideran opciones aceptables.[
46 ,51 ,52 ,53 ,54 ,55 ] - Hay un solo estudio aleatorizado en el que se comparan 2 antifúngicos para la profilaxis de pacientes pediátricos con LMA. En el ensayo del COG ACCL0933 (NCT01307579) , los pacientes se asignaron al azar para recibir tratamiento profiláctico con fluconazol o caspofungina (equinocandina con mayor actividad frente a levaduras y mohos que el fluconazol).[
56 ]- La caspofungina fue superior al fluconazol para reducir la incidencia acumulada a 5 meses de enfermedad fúngica invasiva comprobada o probable (3,1 % vs. 7,2 %; P = 0,03) y la aspergilosis invasiva comprobada o probable (0,5 % vs. 3,1 %; P = 0,046).
Vigilancia cardíaca
La bacteriemia o septicemia y el uso de antraciclinas se han identificado como importantes factores de riesgo de cardiotoxicidad, que se manifiesta como una reducción de la función ventricular izquierda.[
Evidencia (vigilancia cardíaca y repercusión del dexrazoxano):
- En el ensayo del COG AAML0531 (NCT00372593), 8,6 % de los pacientes presentaron disfunción sistólica ventricular izquierda (DSVI) durante la terapia del protocolo; la incidencia acumulada de DSVI dentro de los 5 años posteriores a la finalización de la terapia fue de 12 %.[
57 ]- Los factores de riesgo de DSVI durante la terapia incluyeron raza negra, edad avanzada, peso corporal insuficiente y bacteriemia.
- La presencia de DSVI afectó de manera adversa la SSC a 5 años (CRI, 1,57; IC 95 %, 1,16–2,14; P = 0,004) y la SG (CRI, 1,59; IC 95 %, 1,15–2,19; P = 0,005), como resultado sobre todo de mortalidad que no se debió a una recaída.
- Los pacientes que experimentaron DSVI durante la terapia, tuvieron un riesgo 12 veces mayor de presentar DSVI en los 5 años siguientes a la finalización de la terapia.
- Se evaluó el uso de dexrazoxano en pacientes inscritos en el ensayo del COG AAML1031 (NCT01371981).[
59 ]- Este ensayo requirió vigilancia cardíaca prospectiva en cada ciclo y durante el seguimiento. Se encontró una incidencia de DSVI más alta (39 %) al cabo de una mediana de 3,8 meses desde la inscripción (intervalo intercuartilo, 2–6,2 meses) que la observada en el ensayo anterior.
- En cerca del 10 % de los niños (96 de 1014) que recibieron dexrazoxano optativo con cada dosis de antraciclina, la incidencia de DSVI (definida como una fracción de eyección <55 % o una fracción de acortamiento <28 %) fue significativamente menor (26,5 % vs. 42,2 %; CRI, 0,55; IC 95 %, 0,36–0,86; P = 0,009) que en aquellos que optaron por no recibir dexrazoxano. Esto también ocurrió con el riesgo de DSVI de grado 2 o superior (60 % más bajo). Los pacientes que recibieron dexrazoxano obtuvieron una mejoría constante del funcionamiento cardíaco después de la terapia (mediana de seguimiento, 3,5 años).
- Los pacientes que recibieron dexrazoxano tuvieron una menor mortalidad relacionada con el tratamiento (5,7 % vs. 12,7 %; P = 0,068), sin embargo la mejora en los desenlaces de SG, SSC y RR no alcanzó significación estadística.
Hospitalización
Se ha usado la hospitalización hasta que se logra una recuperación adecuada de los granulocitos (recuento absoluto de neutrófilos o de fagocitos) para reducir la mortalidad relacionada con el tratamiento. El ensayo clínico COG-2961 (NCT00002798), fue el primero en el que se observó una disminución significativa de la mortalidad relacionada con el tratamiento (19 % antes de que se estableciera la hospitalización obligatoria en el ensayo junto con otros cambios de los cuidados de apoyo vs. 12 % después de estos cambios); la SG también mejoró en este ensayo (P <0,001).[
Fracaso de la inducción (LMA resistente al tratamiento)
El fracaso de la inducción (presencia morfológica de un 5 % o más de blastocitos en la médula ósea al final de todos los cursos de inducción) se observa en el 10 % al 15 % de los niños con LMA. Los desenlaces posteriores para los pacientes con fracaso de la inducción son similares a los de los pacientes con LMA que presentan una recaída temprana (<12 meses después de la remisión).[
Sarcoma granulocítico o cloroma
El sarcoma granulocítico (cloroma) describe la acumulación extramedular de células leucémicas. Estas acumulaciones pueden ser la única manifestación de una leucemia, aunque esto es infrecuente. En una revisión de tres estudios de LMA dirigidos por el antiguo Children's Cancer Group, menos del 1 % de los pacientes tenían un sarcoma granulocítico aislado, y el 11 % tenían sarcoma granulocítico con enfermedad en la médula ósea en el momento del diagnóstico.[
Se destaca que el paciente que presenta al inicio un tumor aislado, sin indicios de compromiso de la médula ósea, se debe tratar como si presentara una enfermedad sistémica. Los pacientes con sarcoma granulocítico aislado tienen un buen pronóstico si reciben el tratamiento actual para la LMA.[
En un estudio de 1459 niños con diagnóstico reciente de LMA, se encontró que la supervivencia de los pacientes con sarcoma granulocítico orbitario y sarcoma granulocítico en el sistema nervioso central (SNC) fue mejor que la supervivencia de los pacientes con enfermedad en la médula ósea y sarcoma granulocítico en otros sitios, y que la de los pacientes de LMA sin enfermedad extramedular.[
Profilaxis en el sistema nervioso central para la leucemia mieloide aguda
El compromiso del SNC en pacientes con LMA y su repercusión pronóstica se planteó antes en la sección Factores pronósticos en la leucemia mieloide aguda infantil. La radioterapia y la quimioterapia intratecal se han usado para el tratamiento de la leucemia en el SNC en el momento del diagnóstico y para la prevención del compromiso leucémico posterior en el SNC. El uso de radiación como profilaxis prácticamente se abandonó porque no se ha documentado beneficio y por sus secuelas a largo plazo.[
Evidencia (profilaxis en el sistema nervioso central):
- En el ensayo del COG AAML0531 (NCT00372593) se usó citarabina en monoterapia como profilaxis.[
66 ]- A diferencia de la baja tasa de recaída relacionada con la enfermedad SNC1 (3,9 %) que se observó en el 71 % de los pacientes inscritos, los pacientes con leves indicios de leucemia en el SNC en el momento del diagnóstico (SNC2 o blastocitos cuando el recuento de GB en el LCR era de <5 células/CGA; 16 % de los pacientes con diagnóstico reciente) recibieron citarabina intratecal 2 veces por semana hasta que se depuró el LCR. De los pacientes con enfermedad SNC2 (96 %) en quienes se depuró el LCR de blastocitos leucémicos (95,8 %), el 11,7 % presentó después recaída en el SNC.
- El compromiso SNC3 en el momento del diagnóstico (13 %) confirió resultados aún más precarios. A pesar de que en el 90,7 % de los niños se logró la depuración de blastocitos leucémicos, el 17,7 % presentó después recaída en el SNC. En un análisis multivariante, el compromiso SNC3 empeoró significativamente el riesgo de recaída aislada en el SNC (CRI, 7,82; P = 0,003).
- En otras metodologías se incorporan otros fármacos intratecales; por ejemplo, una estrategia triple de administración intratecal de una combinación de citarabina, hidrocortisona y metotrexato.[
67 ]- El SJCRH informó que después de cambiar de una estrategia triple (tratamiento estándar previo) a una estrategia de monoterapia de citarabina, la incidencia de recaída aislada en el SNC aumentó del 0 % (0 de 131 pacientes) al 9 % (3 de 33 pacientes), lo que llevó a volver a usar la estrategia triple, gracias a lo cual, se recuperó la incidencia de recaída en el SNC del 0 (0 de 79 pacientes).
Terapia de posremisión de la leucemia mieloide aguda
Un objetivo importante del tratamiento de los niños con leucemia mieloide aguda (LMA) es la prolongación de la remisión inicial con quimioterapia adicional o TCMH.
Las opciones de tratamiento para los niños con LMA en posremisión son las siguientes:
- Quimioterapia.
- Trasplante de células madre hematopoyéticas.
- Terapia dirigida (por ejemplo, inhibidores de FLT3).[
68 ]
Quimioterapia
La quimioterapia posremisión incluye algunos de los fármacos que se utilizan durante la inducción y también se introducen fármacos sin resistencia cruzada y, a menudo, dosis altas de citarabina. En los estudios de adultos con LMA se demostró que la consolidación con un régimen de dosis altas de citarabina mejora los desenlaces en comparación con la consolidación con un régimen de dosis estándar de citarabina, en particular, para los pacientes con los subtipos de LMA con inv(16) y t(8;21).[
Todavía no está claro el número óptimo de cursos de terapia posremisión, pero al parecer se necesitan al menos 2 o 3 cursos de terapia intensiva después de la inducción.[
Evidencia (número de cursos de quimioterapia posremisión):
- En un estudio del United Kingdom Medical Research Council (MRC), se asignó al azar a pacientes adultos y niños a recibir 4 a 5 cursos de terapia intensiva.[
6 ,16 ][Nivel de evidencia A1]- No se observó ventaja en la supervivencia sin recaída ni en la SG al usar 5 cursos.
- Teniendo en cuenta los datos del MRC, en el ensayo del COG AAML1031 (NCT01371981) los pacientes sin riesgo alto tratados sin TCMH durante la primera RC (73 de todos los pacientes) recibieron 4 ciclos de quimioterapia (2 ciclos de inducción y 2 ciclos de consolidación) en lugar de 5 ciclos (2 ciclos de inducción y 3 ciclos de consolidación); en los ensayos previos del COG AAML0531 (NCT00372593) y AAML03P1 (NCT00070174), los pacientes que no se sometieron a un TCMH, recibieron 5 ciclos de quimioterapia.[
72 ]- En un análisis retrospectivo, los pacientes de riesgo bajo tratados sin TCMH en el ensayo del COG AAML1031 (4 ciclos de quimioterapia) tuvieron desenlaces significativamente peores que aquellos que recibieron 5 ciclos de quimioterapia en el ensayo AAML0531 (desenlaces de 4 vs. 5 ciclos).
- La tasa de SG fue del 77,0 % en los pacientes que recibieron 4 ciclos de quimioterapia, en comparación con el 83,5 % en los pacientes que recibieron 5 ciclos de quimioterapia (CRI, 1,45; IC 95 %, 0,97–2,17; P = 0,068).
- La tasa de SSE fue del 56 % en los pacientes que recibieron 4 ciclos, en comparación con el 67 % en los pacientes que recibieron 5 ciclos (CRI, 1,45; IC 95 %, 1,10–1,91; P = 0,009).
- La tasa de recaída fue del 40,9 % en los pacientes que recibieron 4 ciclos, en comparación con el 31,4 % en los pacientes que recibieron 5 ciclos (CRI, 1,40; IC 95 %, 1,06–1,85; P = 0,019).
- Se encontró una excepción en el subgrupo de riesgo bajo definido según las características citogenéticas o moleculares favorables que no presentaban ERM al final del ciclo 1 de inducción. Este subconjunto de pacientes presentó desenlaces similares cuando recibieron 4 ciclos de quimioterapia (AAML1031) o 5 ciclos de quimioterapia (AAML0531).
Un estudio adicional del número de cursos de intensificación y fármacos específicos permitirá mejorar el abordaje de este problema, aunque estos datos indican que solo se deben administrar 4 cursos de quimioterapia al grupo favorable descrito antes, y que todos los otros pacientes que no se sometieron a trasplante deben recibir 5 cursos de quimioterapia.
- En un análisis retrospectivo, los pacientes de riesgo bajo tratados sin TCMH en el ensayo del COG AAML1031 (4 ciclos de quimioterapia) tuvieron desenlaces significativamente peores que aquellos que recibieron 5 ciclos de quimioterapia en el ensayo AAML0531 (desenlaces de 4 vs. 5 ciclos).
Trasplante de células madre hematopoyéticas
Desde finales de la década de 1970 se está evaluando el uso del trasplante de células madre hematopoyéticas (TCMH) durante la primera remisión; además se han publicado evaluaciones basadas en la evidencia sobre las indicaciones para los TCMH autógenos y alogénicos. En ensayos prospectivos de trasplantes en niños con LMA se indica que, en general, el 60 % al 70 % de los niños que encuentran un donante con compatibilidad de HLA y que se someten a TCMH alogénico durante la primera remisión consiguen remisiones a largo plazo,[
En los ensayos prospectivos donde se compara el TCMH alogénico con el uso de quimioterapia o un TCMH autógeno, se observó una SSE superior en adultos y niños asignados a trasplante alogénico de acuerdo con la disponibilidad de un donante emparentado con compatibilidad de HLA 6/6 o 5/6.[
El uso actual del TCMH alogénico exige la incorporación de la clasificación de riesgo para determinar si se debe realizar el trasplante durante la primera remisión. Debido a la mejora del desenlace en los pacientes con características pronósticas favorables (mutaciones citogenéticas o moleculares de riesgo bajo) tratados con regímenes quimioterapéuticos contemporáneos y a la falta de superioridad demostrable del TCMH en esta población de pacientes, este grupo de pacientes por lo general recibe un TCMH de un donante emparentado compatible (DEC) solo después de la primera recaída y en el momento de la segunda RC.[
En un análisis del Center for International Blood and Marrow Transplant Research (CIBMTR) se examinaron las variables previas al trasplante para crear un modelo que pudiera predecir la supervivencia sin leucemia (SSL) después del trasplante en pacientes pediátricos (edad <18 años). Todos los pacientes se sometían a trasplante por primera vez y recibieron acondicionamiento mielosupresor; se incluyeron todos los tipos de fuentes de células madre. En los pacientes con LMA, los factores pronósticos relacionados con una SSL inferior incluyeron edad menor a 3 años, características citogenéticas de riesgo intermedio o riesgo desfavorable, y una segunda RC o posterior, con positividad o no para la ERM en el momento de la RC. Se estableció una escala para estratificar a los pacientes según los factores de riesgo y predecir la supervivencia. La tasa de SSL a 5 años fue del 78 % en el grupo de riesgo bajo, del 53 % en el grupo de riesgo intermedio, del 40 % en el grupo de riesgo alto y del 25 % en el grupo de riesgo muy alto.[
Hay evidencia contradictoria sobre la función del TCMH alogénico en la primera remisión de pacientes con características de riesgo intermedio (sin características citogenéticas o mutaciones moleculares de riesgo bajo o riesgo alto):
Evidencia (trasplante alogénico de células madre hematopoyéticas en la primera remisión de pacientes con leucemia mieloide aguda de riesgo intermedio):
- En un estudio en el que se combinaron los resultados de los ensayos POG-8821, CCG-2891, COG-2961 (NCT00002798),y MRC-AML10, se identificó una ventaja de la SSE y la SG para el TCMH alogénico en pacientes con LMA de riesgo intermedio, pero no de riesgo bajo (inv(16) y t(8;21)) ni de riesgo alto (del(5q), monosomía 5 o 7, o más del 15 % de blastocitos después de la primera inducción para los estudios de POG/CCG); en el estudio del MRC, en la categoría de riesgo alto se incluyeron las anomalías de 3q y características citogenéticas complejas.[
74 ] Las debilidades de este estudio fueron un alto porcentaje de pacientes sin asignación de grupo de riesgo y unas tasas relativamente bajas de SSC y SG para los pacientes con LMA de riesgo intermedio asignados a quimioterapia, en comparación con los resultados observados en ensayos clínicos más recientes.[6 ,17 ] - En el ensayo clínico AML99 del Japanese Childhood AML Cooperative Study Group, se observó una diferencia significativa en la SSE de los pacientes con riesgo intermedio asignados a TCMH de un DEC, pero no hubo una diferencia significativa en la SG.[
85 ] - En el ensayo clínico AML-BFM 99, no se observaron diferencias significativas de la SSE o la SG en los pacientes de riesgo intermedio asignados a TCMH de un DEC versus los asignados a quimioterapia.[
80 ]
Dados los mejores desenlaces de los pacientes con LMA de riesgo intermedio en ensayos clínicos recientes y la carga de toxicidad aguda y crónica relacionada con el trasplante alogénico, muchos grupos de tratamiento de LMA infantil (incluso el COG) emplean quimioterapia para los pacientes de riesgo intermedio durante la primera remisión y reservan el TCMH alogénico para después de una posible recaída.[
Hay información contradictoria en relación con la función del TCMH alogénico en la primera remisión para los pacientes con enfermedad de riesgo alto, que se complica por las diversas definiciones de riesgo alto que usan los grupos de estudio.
Evidencia (trasplante alogénico de células madre hematopoyéticas en la primera remisión para pacientes con LMA de riesgo alto):
- En un análisis retrospectivo del COG y el CIBMTR se comparó la quimioterapia sola con el TCMH de donante emparentado compatible y de donante no emparentado compatible en pacientes con LMA y características citogenéticas de riesgo alto, definidas como monosomía 7/del(7q), monosomía 5/del(5q), anomalías de 3q, t(6;9) o cariotipos complejos.[
87 ]- En el análisis no se demostraron diferencias en la SG a 5 años en los 3 grupos de tratamiento.
- En un estudio de la Nordic Society for Pediatric Hematology and Oncology, se notificó que con la terapia de reinducción de cronograma intenso seguida de trasplante del mejor donante disponible para los pacientes con LMA que no respondieron a la terapia de inducción se obtuvo el 70 % de supervivencia al cabo de una mediana de seguimiento de 2,6 años.[
88 ][Nivel de evidencia B4] - En un estudio retrospectivo de una sola institución, se incluyó a 36 pacientes consecutivos (edad 0–30 años) con LMA de riesgo alto (mutaciones FLT3 ITD, reordenamientos de 11q23 en KMT2A, presencia de anomalías en los cromosomas 5 o 7, fracaso de la inducción o enfermedad persistente) que estaban en una primera remisión morfológica antes del trasplante alogénico.[
89 ]- Los investigadores informaron sobre una tasa de SG a 5 años del 72 % y una tasa SSL (desde el momento del trasplante) del 69 % con el uso de un régimen de acondicionamiento mielosupresor.
- También notificaron una tasa de mortalidad relacionada con el tratamiento del 17 %.
- Estos desenlaces fueron similares en 14 pacientes con LMA de riesgo estándar que recibieron un trasplante durante el mismo período.
- En un análisis de un subgrupo del ensayo clínico AML-BFM 98, se demostraron mejores tasas de supervivencia en los pacientes con anomalías de 11q23 asignados a TCMH alogénico, pero no en aquellos sin anomalías de 11q23.[
80 ] - Entre los niños con mutaciones FLT3 ITD (proporción alélica alta), aquellos que se sometieron a TCMH de DEC (n = 6) tuvieron tasas de SG más altas que quienes recibieron quimioterapia estándar (n = 28); sin embargo, el número de casos estudiados limitó la capacidad de llegar a conclusiones.[
90 ] - En un informe retrospectivo posterior de 3 ensayos consecutivos de adultos jóvenes con LMA, se encontró que los pacientes con una proporción alélica alta de mutaciones FLT3 ITD se beneficiaron del TCMH alogénico (P =0,03); no así los pacientes con proporción alélica baja (P = 0,64).[
91 ] - En un análisis de subgrupos de un ensayo de fase III del COG, se evaluó el uso de gemtuzumab ozogamicina durante la terapia de inducción en niños con diagnóstico reciente de LMA.[
25 ]- En los pacientes con proporción alélica alta de mutaciones FLT3 ITD sometidos a TCMH, se observó una tasa de recaída más baja que en los pacientes que también recibieron gemtuzumab ozogamicina (15 vs. 53 %, P = 0,007).
- Por el contrario, los pacientes que recibieron gemtuzumab ozogamicina exhibieron tasas de mortalidad relacionada con el tratamiento más altas (19 vs. 7 %, P = 0,08), lo que se tradujo en una mejora general de la SSE (65 vs. 40 %, P = 0,08).
Muchos de los grupos de ensayos clínicos pediátricos, aunque no todos, ordenan el TCMH alogénico para los pacientes de riesgo alto durante la primera remisión.[
Puesto que las definiciones de LMA de riesgo alto, intermedio y bajo continúan en evolución debido a la relación permanente de las características moleculares del tumor con el desenlace (por ejemplo, mutaciones FLT3 ITD, mutaciones en WT1 y mutaciones en NPM1) y la respuesta al tratamiento (por ejemplo, evaluaciones de la ERM después de la terapia de posinducción), se necesitan más análisis de subpoblaciones de pacientes tratados con TCMH alogénico en ensayos clínicos actuales y futuros.
Si se decide realizar un trasplante durante la primera RC, todavía no se han determinado el régimen preparatorio ni la fuente donante de células óptimos, aunque se estudian otros tipos de donantes, como los donantes haploidénticos.[
Evidencia (régimen mielosupresor):
- En un ensayo aleatorizado en el que se comparó busulfano y fludarabina con busulfano y ciclofosfamida como régimen de preparación para la LMA durante la primera RC, se demostró que el primer régimen produjo menos toxicidad, así como SSE y SG comparables.[
96 ] - Además, en un estudio prospectivo grande de cohortes del CIBMTR de niños y adultos con LMA, síndromes mielodisplásicos (SMD) y leucemia mielógena crónica (LMC), se observó una supervivencia superior en los pacientes con enfermedad en estadio temprano (LMC en fase crónica, LMA en primera RC y SMD con anemia resistente al tratamiento) cuando se usaron regímenes a base de busulfano en comparación con la ICT.[
97 ] - En un estudio del CIBMTR en 624 niños con diagnóstico nuevo de LMA que recibieron un trasplante entre 2008 y 2016, los pacientes se trataron con un régimen a base de ICT (n = 199) o un régimen que no incluía la ICT (n = 425) y se observaron los siguientes resultados:[
98 ]- Los pacientes que recibieron ICT presentaron una mortalidad no relacionada con recidiva más alta (P < 0,0001), pero tasas de recidiva más bajas (P < 0,0001), lo que se tradujo en tasas de SSL y SG equivalentes.
- Los pacientes que recibieron ICT presentaron más EICH aguda de grados 2 y 3 (56 vs. 27 %; P < 0,0001), pero una incidencia de EICH crónica equivalente.
- Los sobrevivientes que recibieron ICT exhibieron una incidencia más alta de insuficiencia gonadal o del crecimiento (24 vs. 8 %; P < 0,0001), pero no se observaron diferencias en el compromiso pulmonar, cardiaco o renal.
Para los subtipos diferentes a la LPA, no hay datos que demuestren que la terapia de mantenimiento administrada después de la terapia intensiva de posremisión prolongue de manera significativa la duración de la remisión. En 2 estudios aleatorizados en los que se usó terapia intensiva de consolidación contemporánea, no se demostró beneficio de la quimioterapia de mantenimiento;[
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Leucemia mieloide aguda recidivante o resistente al tratamiento y otras neoplasias malignas mieloides
El diagnóstico de la leucemia mieloide aguda (LMA) recidivante o en recaída, de acuerdo con los criterios del COG, es esencialmente el mismo que se indica en los criterios para establecer el diagnóstico de la LMA. Por lo general, se determina este diagnóstico en pacientes que tienen más del 5 % de blastocitos en la médula ósea y que lograron una remisión previa después del tratamiento por un diagnóstico inicial de LMA según los criterios de clasificación de la Organización Mundial de la Salud (OMS).[
A pesar de que se induce una segunda remisión en más de la mitad de los niños con LMA tratados con fármacos similares a los empleados en la terapia de inducción inicial, el pronóstico de los niños con LMA recidivante o progresiva suele ser adverso.[
Leucemia mieloide aguda infantil recidivante
Alrededor del 50 % al 60 % de las recaídas se presentan durante el primer año del diagnóstico; la mayoría de las recaídas ocurren al cabo de 4 años del diagnóstico.[
Pronóstico y factores pronósticos
Los factores que afectan la capacidad de lograr una segunda remisión son los siguientes:
- Duración de la primera remisión. La duración de la primera remisión es un factor importante que afecta la capacidad de lograr una segunda remisión; los niños con una primera remisión de menos de 1 año tienen tasas mucho más bajas de segunda remisión (50–60 %) que los niños cuya primera remisión dura más de 1 año (70–90 %).[
60 ,103 ,104 ] La supervivencia de los niños que tienen primeras remisiones cortas es mucho menor (cerca de 10 %) que la de los niños con una primera remisión que supera 1 año (cerca de 40 %).[60 ,103 ,104 ,105 ] El Therapeutic Advances in Childhood Leucemia and Lymphoma Consortium también identificó la duración de la remisión anterior como un factor pronóstico potente, con tasas de SG a 5 años del 54 % ± 10 % en los pacientes con una duración de la primera remisión superior a 12 meses y del 19 % ± 6 % en pacientes con períodos más cortos de primera remisión.[106 ] En este mismo análisis, los desenlaces, sobre todo en los pacientes con recaída temprana, se deterioraron con cada intento de reinducir la remisión (56 ± 5 %, 25 ± 8 %, y 17 ± 7 % para cada intento consecutivo). - Alteraciones moleculares. Además, se notificó que determinadas alteraciones moleculares en el momento de la recaída afectan la supervivencia posterior. Por ejemplo, la presencia de mutaciones en WT1 o mutaciones FLT3 ITD en la primera recaída se relaciona, como factor independiente de riesgo, con una SG más precaria en los pacientes que alcanzaron una segunda remisión.[
107 ]
En los estudios que se mencionan a continuación se identificaron otros factores pronósticos:
- En un informe de 379 niños con LMA que recidivaron luego del tratamiento inicial en los protocolos del grupo alemán BFM, la tasa de segunda remisión completa fue del 63 % y la tasa de SG fue del 23 %.[
108 ][Nivel de evidencia C1] Los factores pronósticos más importantes relacionados con un desenlace favorable luego de la recaída fueron el logro de una segunda remisión completa, la recaída más allá de los 12 meses desde el diagnóstico inicial, la ausencia de trasplante alogénico de médula ósea durante la primera remisión y la presencia de características citogenéticas favorables (t(8;21), t(15;17) e inv(16)). - En el ensayo internacional Relapsed AML 2001/01 (NCT00186966), también se observó que la respuesta temprana a la terapia de rescate tuvo gran importancia pronóstica.[
109 ][Nivel de evidencia C2] - En un estudio retrospectivo de Japón con 71 pacientes de LMA en recaída, se notificó una tasa de SG a 5 años del 37 %. Los pacientes que presentaron una recaída temprana tuvieron una tasa de segunda remisión del 27 %, en comparación con el 88 % de los pacientes con recaída tardía. La SG a 5 años fue más alta en los pacientes que se sometieron a un TCMH después de alcanzar una segunda remisión completa (66 %) que en los pacientes sin remisión (17 %).[
105 ] - Se analizó la supervivencia de los pacientes en recaída en 2 ensayos de LMA consecutivos de la Nordic Society of Pediatric Hematology and Oncology (NOPHO) realizados entre 1993 y 2012 (de 543 niños tratados originalmente, 208 pacientes recayeron). De estos niños, 146 (70 %) obtuvieron segundas remisiones después de una variedad de regímenes de reinducción. La tasa de SG a 5 años fue del 39 %. Los factores de pronóstico favorable fueron la recaída tardía (≥1 año desde el diagnóstico), ausencia de TCMH durante la primera remisión y un subtipo de LMA con factor de unión nuclear. Para los niños en segunda remisión sometidos a TCMH, la tasa de SG a 5 años fue del 61 %, en contraste a la tasa de SG a 5 años del 18 % para aquellos que no recibieron un TCMH como parte de su tratamiento (P < 0,001).[
110 ]
Tratamiento de la leucemia mieloide aguda recidivante
Las opciones de tratamiento para los niños con leucemia mieloide aguda (LMA) recidivante son las siguientes:
- Quimioterapia.
- Abordajes inmunoterapéuticos.
- Terapia dirigida (inhibidores de FLT3).
- Trasplante de células madre hematopoyéticas.
- Segundo trasplante después de la recaída posterior a un primer trasplante.
Quimioterapia
Los regímenes que se han usado con éxito para inducir la remisión en niños con LMA recidivante por lo común incluyen dosis altas de citarabina administradas en combinación con los siguientes fármacos:
- Mitoxantrona.[
60 ] - Fludarabina e idarrubicina.[
111 ] - L-asparaginasa.[
112 ] - Etopósido.
- Daunorrubicina liposomal. En un estudio del grupo internacional BFM, se compararon la fludarabina, la citarabina y el G-CSF (FLAG) con FLAG y daunorrubicina liposomal. La tasa de SG a 4 años fue del 38 %, sin diferencia en la supervivencia para todo el grupo; sin embargo, la adición de la daunorrubicina liposomal aumentó la probabilidad de lograr una remisión y condujo a una mejora significativa de la SG en los pacientes con mutaciones del factor de unión nuclear (82 %, FLAG y daunorrubicina liposomal vs. 58 %, FLAG; P = 0,04).[
113 ][Nivel de evidencia A1] - CPX-351. El CPX-351 es un medicamento de combinación liposomal fija de los fármacos daunorrubicina y citarabina, que se evaluó en el ensayo de fase I/II del COG AAML1421 (NCT02642965) para niños con LMA en recaída. Se administró CPX-351 (135 unidades/m2 /día que contiene 60 mg/m2 de daunorrubicina) sin dexrazoxano durante el ciclo 1 en los días 1, 3, 5 seguido de un ciclo de FLAG. El CPX-351 se toleró bien y no se observó toxicidad inesperada; un caso de toxicidad que limitó la dosis (disminución de la fracción de eyección de grado 3 que se resolvió) y ningún caso de mortalidad por toxicidad. De los pacientes, el 40 % presentó exantema maculopapular. De 37 pacientes evaluables, el 75,7 % tuvo una remisión completa (RC), RC con recuperación incompleta de plaquetas (RCp) o RC con recuperación hematológica incompleta (RCi) después del ciclo de CPX-351; 21 de 25 pacientes en RC o RCp no presentaron enfermedad residual mínima (ERM) después del ciclo 2, y 20 de 25 pacientes no tuvieron ERM antes del trasplante de células madre hematopoyéticas (TCMH).[
114 ][Nivel de evidencia B4] - Venetoclax. En el ensayo del St. Jude Children's Research Hospital VENAML(NCT03194932) se evaluó el venetoclax, un inhibidor selectivo de BCL-2, en combinación con citarabina e idarrubicina o sin esta, en pacientes pediátricos con LMA en recaída o resistente al tratamiento. La combinación se toleró bien; los efectos adversos más comunes de grado 3 y 4 fueron neutropenia febril (66 % de los pacientes), infecciones del torrente sanguíneo (16 % de los pacientes) e infecciones fúngicas invasivas (16 % de los pacientes). De 20 pacientes tratados con la dosis recomendada de la fase II, 14 pacientes (70 %) lograron una respuesta completa con recuperación hematológica o sin esta, y 2 pacientes (10 %) lograron una respuesta parcial.
Se han usado regímenes a base de clofarabina y [
Los regímenes de dosis estándar de citarabina en el estudio MRC AML10 del Reino Unido para niños con diagnóstico reciente de LMA (citarabina y daunorrubicina con etopósido o tioguanina) produjeron tasas similares de remisión a las tasas de los regímenes de dosis altas de citarabina cuando se usaron en un entorno de recaída.[
Abordajes inmunoterapéuticos
Antes de que la FDA aprobara su uso en niños con LMA de novo en 2020, el gemtuzumab ozogamicina estaba aprobado para niños mayores de 2 años con LMA en recaída o resistente al tratamiento.
- En el ensayo del COG AAML00P2 (NCT00028899) se estableció que la dosis máxima tolerada (DMT) de gemtuzumab ozogamicina, cuando se combina con mitoxantrona y dosis altas de citarabina, era de 3 mg/m2; la DMT de gemtuzumab ozogamicina, cuando se combina con el régimen Capizzi II a base de dosis altas de citarabina, fue de 2 mg/m2. [
61 ]- Estos regímenes produjeron una tasa general de respuesta de remisión del 45 % (±15 %), una tasa de SSC a un año del 38 % (±14 %) y una tasa de SG a 1 año del 53 % (±15 %).
- Durante el ciclo que contenía gemtuzumab ozogamicina, se observó el síndrome de oclusión sinusoidal en un paciente con un trasplante de células madre (TCM) anterior y en 4 de 28 pacientes durante TCM posteriores (2 pacientes con grado 1, 1 paciente con grado 3, 1 paciente con grado 4); todos ellos se recuperaron.
- En la sección de aumento gradual de la dosis del estudio AML15 (NCT00006122) del U.K. Medical Research Council en adultos, la dosis de gemtuzumab ozogamicina se aumentó por encima de los 3 mg/m2 por dosis (la misma DMT que se determinó en el estudio AAML00P2) y se administró con una quimioterapia de base intensiva convencional; este régimen no fue factible debido a hepatotoxicidad y retraso en la recuperación hematopoyética. Tampoco se toleró bien el gemtuzumab ozogamicina de 3 mg/m2 por dosis, cuando se administró con cursos consecutivos de quimioterapia intensiva.[
121 ]
- El estudio Relapsed AML 2001/02 fue un ensayo de grupo único para niños (n = 30) con LMA en segunda recaída o resistente al tratamiento después de que no respondieran a un segundo régimen de inducción. El gemtuzumab ozogamicina en monoterapia se administró en dosis de 7,5 mg/dosis (los niños menores de 3 años recibieron 0,25 mg/kg) cada 14 días con un total de 2 dosis. Se observó RC o RCp en el 37 % de los pacientes; 9 pacientes se sometieron a TCM posteriores y 3 de esos pacientes se mantuvieron en RC continua. Todos los pacientes recibieron defibrotida profiláctica durante el TCM y no presentaron síndrome de oclusión sinusoidal (SOS).[
122 ]- En un estudio anterior de niños que recibieron monoterapia de gemtuzumab ozogamicina en una dosificación de 6 a 9 mg/m2 por dosis, los pacientes no recibieron profilaxis con defibrotida durante los TCM posteriores. En estos estudios se demostró un aumento de riesgo de SOS, sobre todo en los pacientes que se sometieron a TCM menos de 3,5 meses después de la última dosis de gemtuzumab ozogamicina.[
123 ]
- En un estudio anterior de niños que recibieron monoterapia de gemtuzumab ozogamicina en una dosificación de 6 a 9 mg/m2 por dosis, los pacientes no recibieron profilaxis con defibrotida durante los TCM posteriores. En estos estudios se demostró un aumento de riesgo de SOS, sobre todo en los pacientes que se sometieron a TCM menos de 3,5 meses después de la última dosis de gemtuzumab ozogamicina.[
- En 2 estudios prospectivos del grupo Acute Leukemia French Association (ALFA) se evaluó el gemtuzumab ozogamicina fraccionado (3 mg/m2 /dosis en los días 1, 4, y 7) en el entorno de adultos con LMA en recaída.
- En el ensayo MYLOFRANCE 1 se evaluó la dosis fraccionada de monoterapia en 57 adultos con LMA en primera recaída, y se obtuvo una tasa de RC del 26 % y una tasa de RCp del 7 %. No se presentó síndrome de oclusión sinusoidal durante el tratamiento ni durante los TCM posteriores.[
124 ] - Posteriormente, el ensayo MYLOFRANCE 2, fue un estudio de fase I/II (n = 20) en el que se combinó la misma dosis fraccionada de gemtuzumab ozogamicina con daunomicina y citarabina en un diseño de búsqueda de dosis recomendada para el tratamiento de base. De los pacientes, 9 lograron una RC y 2 lograron una RCp. Se encontró que la dosis recomendada de la fase II era 60 mg/m2 por día durante 3 días para la daunomicina y 200 mg/m2 por día durante 7 días para la citarabina. No se presentó síndrome de oclusión sinusoidal.[
125 ] Se ha demostrado que la dosis fraccionada de gemtuzumab ozogamicina es inocua y eficaz en adultos con LMA de novo;[27 ] ahora se está evaluando en el estudio MyeChild01 (NCT02724163) de fase III en el Reino Unido en pacientes pediátricos con LMA de novo.
- En el ensayo MYLOFRANCE 1 se evaluó la dosis fraccionada de monoterapia en 57 adultos con LMA en primera recaída, y se obtuvo una tasa de RC del 26 % y una tasa de RCp del 7 %. No se presentó síndrome de oclusión sinusoidal durante el tratamiento ni durante los TCM posteriores.[
Terapia dirigida (inhibidores de FLT3)
Midostaurina Hay poca experiencia con midostaurina en pacientes pediátricos con LMA.
- Se notificó un ensayo de fase I/II de aumento gradual de la dosis en monoterapia, de 22 niños con LMA en recaída o resistente al tratamiento (9 de ellos con mutaciones en FLT3). De los pacientes, 7 recibieron la dosis de 30 mg/m2 2 veces al día, y 15 pacientes recibieron la dosis más alta, correspondiente a 60 mg/m2 2 veces al día, con una mediana de duración de la dosis de 16 días.[
35 ]- En general, el 72,7 % de los pacientes tuvieron efectos adversos relacionados con el tratamiento; solo un paciente presentó toxicidad que limitó la dosis (elevación de ALT de grado 3–4).
- De los pacientes de LMA con mutaciones en FLT3, el 55,5 % (21,2–86,3 %) tuvo alguna respuesta clínica al cabo de una mediana de 14 días (intervalo, 8–22 días), además, 1 paciente logró remisión completa con recuperación incompleta del recuento y pudo recibir un TCM; este paciente fue el único sobreviviente a largo plazo del estudio.
- En Europa está en curso un ensayo de fase II que comenzó con dosis de 30 mg/m2 2 veces al día (NCT03591510).
Gilteritinib. Al igual que en la LMA de novo, la mayoría del centro de atención y la experiencia publicada con inhibidores de FLT3 está puesta en adultos con LMA y esto es así también en el entorno de las recaídas y la resistencia al tratamiento. Para la LMA en recaída o resistente al tratamiento, el gilterinib, un inhibidor de tipo 1 de FLT3 con actividad contra las mutaciones en FLT3 (ITD y D835/I836 del dominio de tirosina–cinasa [TKD]), es el primer y único inhibidor de FLT3 que fue aprobado por la FDA para su uso en adultos como régimen de monoterapia a partir del ensayo ADMIRAL (NCT02421939).[
- En el ensayo ADMIRAL de fase III en adultos (edad, 18 años o más) con LMA en recaída o resistente al tratamiento que albergaba la mutación en FLT3, 247 pacientes se asignaron al azar a recibir gilteritinib en monoterapia (120 mg/día una vez por día) o 1 de 4 regímenes quimioterapéuticos de rescate.[
126 ]- La mediana de SG fue significativamente mejor en los pacientes que recibieron gilteritinib (9,3 meses vs. 5,6 meses; CRI, 0,64; IC 95 %, 0,49–0,83; P < 0,001); el 37,1 % versus el 16,7 % de los pacientes estaban vivos al cabo de un año.
- Es importante destacar que, dado que el TCM es esencial para la supervivencia a largo plazo en pacientes con LMA que alberga la mutación en FLT3, un porcentaje más alto de pacientes que recibían gilteritinib se sometió a un TCM (25,5 vs. 15,3 %). Y tuvo la misma eficacia en ambas cohortes de LMA (FLT3 ITD y FLT3 TKD).
- Se presentaron menos efectos adversos en los pacientes que recibieron gilteritinib que en aquellos que recibieron los regímenes quimioterapéuticos de rescate; sin embargo, algunos pacientes que recibieron gilteritinib tuvieron concentraciones elevadas de transaminasas hepáticas. El principal efecto tóxico fue la mielodepresión.
En el ensayo AAML1831 (NCT04293562) del COG está en estudio el uso de gilteritinib en niños con LMA de novo que alberga la mutación en FLT3.
Sorafenib. El sorafenib se ha evaluado en pacientes pediátricos con LMA en recaída o resistente al tratamiento.
- En un ensayo de fase I con disminución gradual de la dosis de sorafenib oral en pacientes pediátricos con leucemia aguda en recaída o resistente al tratamiento, el sorafenib se administró en monoterapia los días 1 al 7, y luego en combinación con clofarabina y dosis altas de citarabina durante 5 días, seguido de sorafenib en monoterapia hasta el día 28.[
127 ]- Se determinó que la dosis recomendada de sorafenib para la fase II era de 150 mg/m2 por dosis (dosis máxima, 300 mg) 2 veces al día (n = 6) después de que los pacientes tuvieran reacciones cutáneas significativas en las manos y en los pies (grados 2–3 en 4 de 4 pacientes; toxicidad limitante de la dosis [TLD] de grado 3 en 2 de 4 pacientes) con los primeros 200 mg/m2 por dosis, 2 veces al día (n = 4).
- El día 8, se observó una reducción blástica medular en 10 de un total de 12 pacientes (4 de 5 pacientes con LMA positiva para FLT3 ITD).
- De los 11 pacientes con LMA, 6 pacientes alcanzaron la remisión completa (RC), 2 alcanzaron la remisión completa con recuperación incompleta del recuento sanguíneo (RCi), y 1 paciente alcanzó la remisión parcial (RP) el día 22 o más tarde.
- Los 5 pacientes con mutación FLT3 ITD alcanzaron la RC o la RCi.
- En un análisis retrospectivo se examinó a 15 niños con LMA que recibieron sorafenib para profilaxis (n = 6) o por recaída (n = 9) después de un TCM. Las dosis de sorafenib variaron de 75 a 340 mg/m2 por día (mediana de la dosis, 230 mg/m2) y se administró en forma de monoterapia en 11 de 15 pacientes.[
128 ]- Se observaron efectos tóxicos en 11 pacientes, 7 de los cuales recibieron dosis mayores a 200 mg/m2; los eventos adversos incluyeron disminución del recuento (n = 6), reacciones en la piel de las manos o de los pies (n = 6), disfunción cardíaca (n = 2), entre otros.
- De los 7 pacientes que presentaron TLD, 6 pudieron reiniciar o continuar el tratamiento con sorafenib, después de ajustar la dosis.
- El sorafenib presentó la mayor eficacia en pacientes con ERM, previa a un TCM o posterior a este (5 de 5 pacientes permanecieron libres de enfermedad), mientras que solo 1 de los 6 pacientes que comenzaron el tratamiento con sorafenib por recidiva morfológica, permaneció en RC.
- La enfermedad de injerto contra huésped no se agravó con el tratamiento con sorafenib.
Trasplante de células madre hematopoyéticas
La selección de otros tratamientos luego de alcanzar una segunda remisión completa depende del tratamiento previo, así como de consideraciones individuales. De manera clásica, se recomienda la quimioterapia de consolidación seguida de trasplante de células madre hematopoyéticas (TCMH), aunque no hay datos prospectivos controlados sobre la contribución de cursos adicionales de terapia una vez que se logra la segunda remisión completa.[
Evidencia (TCMH después de una segunda remisión completa):
- El grupo BFM examinó los desenlaces de niños con LMA durante un período de 35 años y encontró que la mayor mejoría en los desenlaces generales se observó para la supervivencia posterior a la recaída. Esta mejoría de la SSC después de la recaída o la enfermedad resistente al tratamiento solo se observó en pacientes que recibieron un trasplante de células madre como parte de la terapia de rescate.[
129 ] - Se notificó que los TCMH de donante no emparentado resultan en probabilidades de SSL a 5 años del 45 %, el 20 % y el 12 % en los pacientes de LMA que recibieron trasplante durante la segunda remisión completa, una recaída manifiesta y un fracaso de la inducción primaria, respectivamente.[
130 ][Nivel de evidencia C1] - En varios estudios, incluso en un estudio prospectivo numeroso del CIBMTR de cohortes de niños y adultos con enfermedades mieloides, se observó una supervivencia similar o superior con regímenes de busulfano en comparación con la ICT.[
97 ,98 ,131 ,132 ] - El trasplante de donante fraterno compatible por lo general produjo los mejores desenlaces, pero el empleo de donantes emparentados con incompatibilidad de un solo antígeno o donantes no emparentados compatibles produce una supervivencia muy similar a expensas de tasas elevadas de enfermedad de injerto contra receptor (EICH) y mortalidad no relacionada con una recaída.[
133 ] El uso de sangre del cordón umbilical produce desenlaces similares a los de otros donantes no emparentados, pero cuando la compatibilidad de los pacientes se determina como mínimo en 7/8 alelos (HLA A, B, C, DRB1) se presenta una mortalidad no relacionada con una recaída más baja.[134 ] Los abordajes haploidénticos se usan cada vez más y producen desenlaces comparables a otras fuentes de células madre en el ámbito pediátrico.[135 ] No se han hecho comparaciones directas de donantes haploidénticos y otros donantes no emparentados en el ámbito pediátrico, pero en los estudios de adultos se observaron desenlaces similares.[136 ] - Los abordajes de intensidad reducida se han usado con éxito en el entorno pediátrico, sobre todo en niños que no son aptos para someterse a abordajes mielosupresores.[
137 ] En un ensayo aleatorizado de adultos, se observaron desenlaces superiores con los abordajes mielosupresores en comparación con los regímenes de intensidad reducida.[138 ]
Segundo trasplante después de la recaída posterior a un primer trasplante
Hay indicios de que se puede lograr una supervivencia a largo plazo en un grupo de pacientes pediátricos que reciben un segundo trasplante por una recaída después de un primer trasplante mielosupresor. La supervivencia se relacionó con recaída tardía (>6–12 meses desde el primer trasplante), obtención de una respuesta completa antes del segundo procedimiento, y uso de un segundo régimen mielosupresor, si fuera posible.[
Recaída en el sistema nervioso central
La recaída aislada en el SNC se presenta en el 3 % al 6 % de los pacientes con LMA infantil.[
- Edad menor de 2 años en el momento del diagnóstico inicial.
- Leucemia M5.
- Anomalías 11q23.
- Compromiso SNC2 o SNC3 en el momento del diagnóstico inicial.[
66 ]
El riesgo de recaída en el SNC aumenta a medida que aumenta el compromiso leucémico del SNC en el momento del diagnóstico inicial de la LMA (SNC1: 0,6 %, SNC2: 2,6 %, SNC3: 5,8 % de incidencia de recaída aislada en el SNC, P < 0,001; CRI multivariante de SNC3: 7,82, P = 0,0003).[
Leucemia mieloide aguda infantil resistente al tratamiento (fracaso de la inducción)
Las opciones de tratamiento para los niños con leucemia mieloide aguda (LMA) resistente al tratamiento son las siguientes:
- Quimioterapia.
- Gemtuzumab ozogamicina.
Al igual que los pacientes con recaída de una LMA, los pacientes cuya inducción fracasa por lo general se derivan a trasplante de células madre hematopoyéticas (TCMH) cuando logran la remisión porque en los estudios se indica una mejor supervivencia sin complicaciones (SSC) que para los pacientes tratados con quimioterapia sola (31,2 vs. 5 %, P < 0,0001). El logro de una remisión completa (RC) morfológica en estos pacientes es un factor pronóstico significativo de supervivencia sin enfermedad (SSE) después de un TCMH (46 vs. 0 %; P = 0,02), con un fracaso que resulta en primer lugar de una recaída (riesgo de recaída, 53,9 vs. 88,9 %; P = 0,02).[
Evidencia (tratamiento de la LMA infantil resistente al tratamiento con gemtuzumab ozogamicina):
- En el ensayo del SJCRH AML02 (NCT00136084), se administró gemtuzumab ozogamicina solo (n = 17), por lo general cuando la ERM era baja pero detectable (0,1–5,6 %), o en combinación con quimioterapia (n = 29) en aquellos pacientes con ERM alta (1–97 %) después del primer ciclo de inducción.[
146 ]- Cuando el fármaco se administró solo, en 13 de 17 pacientes despareció la ERM.
- Cuando se administró en combinación con quimioterapia, en 13 de 29 pacientes desapareció la ERM y en 28 de 29 pacientes se redujo la ERM.
- En una comparación con una cohorte no aleatorizada de pacientes con un 1–25 % de ERM después de la inducción 1, se encontró que agregar gemtuzumab ozogamicina a la quimioterapia versus la quimioterapia sola produjo diferencias significativas en la ERM (P = 0,03); la ERM desapareció o se redujo en todos los pacientes que recibieron gemtuzumab ozogamicina versus solo en el 82 % de los pacientes que no lo recibieron. Esto se observó a pesar de grados de ERM más altos después de la inducción 1 en la cohorte de pacientes que recibió gemtuzumab ozogamicina (mediana, 9,5 vs. 2,9 % en el grupo sin gemtuzumab ozogamicina, P < 0,01). Se produjo una mejora sin significación estadística en las tasas de SG a 5 años (55 ± 13,9 % vs. 36,4 ± 9,7 %, P = 0,28) y en las tasas de SSC a 5 años (50 ± 9,3 % vs. 31,8 ± 13,4 %, P = 0,28).
- No se observó efecto del TCMH en la mortalidad relacionada con el tratamiento.
- En un ensayo de fase II de gemtuzumab ozogamicina solo, para niños con LMA en recaída o resistente al tratamiento que no reaccionaron bien a intentos de reinducción previos, 11 de 30 pacientes lograron una RC o RC parcial, con una tasa de SG a 3 años del 27 % versus el 0 % (P = 0,001) para quienes respondieron al tratamiento versus aquellos que no respondieron.[
122 ]
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de ensayo clínico nacional o institucional en curso:
- AAML1831 (NCT04293562) (A Study to Compare Standard Chemotherapy to Therapy With CPX-351 and/or Gilteritinib for Patients With Newly Diagnosed AML With or Without FLT3 Mutations): este estudio conjunto de la industria y el COG es un ensayo aleatorizado que evalúa si el medicamento liposomal CPX-351, que contiene los fármacos daunomicina y citarabina, mejora la SSC en comparación con la daunomicina y la citarabina estándar. En un grupo adicional para pacientes de LMA con mutaciones FLT3 ITD sin características citomoleculares favorables (NPM1 o CEBPA) se evalúa el efecto de gilteritinib, un inhibidor selectivo de FLT3.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
- Ries LAG, Melbert D, Krapcho M, et al.: SEER Cancer Statistics Review, 1975-2005. National Cancer Institute, 2007. Also available online. Last accessed April 7, 2022.
- Gibson BE, Wheatley K, Hann IM, et al.: Treatment strategy and long-term results in paediatric patients treated in consecutive UK AML trials. Leukemia 19 (12): 2130-8, 2005.
- Lange BJ, Smith FO, Feusner J, et al.: Outcomes in CCG-2961, a children's oncology group phase 3 trial for untreated pediatric acute myeloid leukemia: a report from the children's oncology group. Blood 111 (3): 1044-53, 2008.
- Creutzig U, Büchner T, Sauerland MC, et al.: Significance of age in acute myeloid leukemia patients younger than 30 years: a common analysis of the pediatric trials AML-BFM 93/98 and the adult trials AMLCG 92/99 and AMLSG HD93/98A. Cancer 112 (3): 562-71, 2008.
- Kaspers GJ, Creutzig U: Pediatric acute myeloid leukemia: international progress and future directions. Leukemia 19 (12): 2025-9, 2005.
- Gibson BE, Webb DK, Howman AJ, et al.: Results of a randomized trial in children with Acute Myeloid Leukaemia: medical research council AML12 trial. Br J Haematol 155 (3): 366-76, 2011.
- Creutzig U, Zimmermann M, Lehrnbecher T, et al.: Less toxicity by optimizing chemotherapy, but not by addition of granulocyte colony-stimulating factor in children and adolescents with acute myeloid leukemia: results of AML-BFM 98. J Clin Oncol 24 (27): 4499-506, 2006.
- Cooper TM, Franklin J, Gerbing RB, et al.: AAML03P1, a pilot study of the safety of gemtuzumab ozogamicin in combination with chemotherapy for newly diagnosed childhood acute myeloid leukemia: a report from the Children's Oncology Group. Cancer 118 (3): 761-9, 2012.
- Gamis AS, Alonzo TA, Meshinchi S, et al.: Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children's Oncology Group trial AAML0531. J Clin Oncol 32 (27): 3021-32, 2014.
- Stevens RF, Hann IM, Wheatley K, et al.: Marked improvements in outcome with chemotherapy alone in paediatric acute myeloid leukemia: results of the United Kingdom Medical Research Council's 10th AML trial. MRC Childhood Leukaemia Working Party. Br J Haematol 101 (1): 130-40, 1998.
- Creutzig U, Ritter J, Zimmermann M, et al.: Improved treatment results in high-risk pediatric acute myeloid leukemia patients after intensification with high-dose cytarabine and mitoxantrone: results of Study Acute Myeloid Leukemia-Berlin-Frankfurt-Münster 93. J Clin Oncol 19 (10): 2705-13, 2001.
- Hann IM, Stevens RF, Goldstone AH, et al.: Randomized comparison of DAT versus ADE as induction chemotherapy in children and younger adults with acute myeloid leukemia. Results of the Medical Research Council's 10th AML trial (MRC AML10). Adult and Childhood Leukaemia Working Parties of the Medical Research Council. Blood 89 (7): 2311-8, 1997.
- Burnett AK, Russell NH, Hills RK, et al.: Optimization of chemotherapy for younger patients with acute myeloid leukemia: results of the medical research council AML15 trial. J Clin Oncol 31 (27): 3360-8, 2013.
- Creutzig U, Ritter J, Zimmermann M, et al.: Idarubicin improves blast cell clearance during induction therapy in children with AML: results of study AML-BFM 93. AML-BFM Study Group. Leukemia 15 (3): 348-54, 2001.
- Pession A, Masetti R, Rizzari C, et al.: Results of the AIEOP AML 2002/01 multicenter prospective trial for the treatment of children with acute myeloid leukemia. Blood 122 (2): 170-8, 2013.
- Burnett AK, Hills RK, Milligan DW, et al.: Attempts to optimize induction and consolidation treatment in acute myeloid leukemia: results of the MRC AML12 trial. J Clin Oncol 28 (4): 586-95, 2010.
- Creutzig U, Zimmermann M, Bourquin JP, et al.: Randomized trial comparing liposomal daunorubicin with idarubicin as induction for pediatric acute myeloid leukemia: results from Study AML-BFM 2004. Blood 122 (1): 37-43, 2013.
- Elgarten CW, Wood AC, Li Y, et al.: Outcomes of intensification of induction chemotherapy for children with high-risk acute myeloid leukemia: A report from the Children's Oncology Group. Pediatr Blood Cancer 68 (12): e29281, 2021.
- Rubnitz JE, Lacayo NJ, Inaba H, et al.: Clofarabine Can Replace Anthracyclines and Etoposide in Remission Induction Therapy for Childhood Acute Myeloid Leukemia: The AML08 Multicenter, Randomized Phase III Trial. J Clin Oncol 37 (23): 2072-2081, 2019.
- Woods WG, Kobrinsky N, Buckley JD, et al.: Timed-sequential induction therapy improves postremission outcome in acute myeloid leukemia: a report from the Children's Cancer Group. Blood 87 (12): 4979-89, 1996.
- Bishop JF, Matthews JP, Young GA, et al.: A randomized study of high-dose cytarabine in induction in acute myeloid leukemia. Blood 87 (5): 1710-7, 1996.
- Becton D, Dahl GV, Ravindranath Y, et al.: Randomized use of cyclosporin A (CsA) to modulate P-glycoprotein in children with AML in remission: Pediatric Oncology Group Study 9421. Blood 107 (4): 1315-24, 2006.
- Rubnitz JE, Inaba H, Dahl G, et al.: Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol 11 (6): 543-52, 2010.
- Pollard JA, Guest E, Alonzo TA, et al.: Gemtuzumab Ozogamicin Improves Event-Free Survival and Reduces Relapse in Pediatric KMT2A-Rearranged AML: Results From the Phase III Children's Oncology Group Trial AAML0531. J Clin Oncol 39 (28): 3149-3160, 2021.
- Tarlock K, Alonzo TA, Gerbing RB, et al.: Gemtuzumab Ozogamicin Reduces Relapse Risk in FLT3/ITD Acute Myeloid Leukemia: A Report from the Children's Oncology Group. Clin Cancer Res 22 (8): 1951-7, 2016.
- Guest EM, Aplenc R, Sung L, et al.: Gemtuzumab ozogamicin in infants with AML: results from the Children's Oncology Group trials AAML03P1 and AAML0531. Blood 130 (7): 943-945, 2017.
- Castaigne S, Pautas C, Terré C, et al.: Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 379 (9825): 1508-16, 2012.
- Pollard JA, Loken M, Gerbing RB, et al.: CD33 Expression and Its Association With Gemtuzumab Ozogamicin Response: Results From the Randomized Phase III Children's Oncology Group Trial AAML0531. J Clin Oncol 34 (7): 747-55, 2016.
- Olombel G, Guerin E, Guy J, et al.: The level of blast CD33 expression positively impacts the effect of gemtuzumab ozogamicin in patients with acute myeloid leukemia. Blood 127 (17): 2157-60, 2016.
- Lamba JK, Chauhan L, Shin M, et al.: CD33 Splicing Polymorphism Determines Gemtuzumab Ozogamicin Response in De Novo Acute Myeloid Leukemia: Report From Randomized Phase III Children's Oncology Group Trial AAML0531. J Clin Oncol 35 (23): 2674-2682, 2017.
- Hills RK, Castaigne S, Appelbaum FR, et al.: Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol 15 (9): 986-96, 2014.
- Perl AE: Availability of FLT3 inhibitors: how do we use them? Blood 134 (9): 741-745, 2019.
- Stone RM, Mandrekar SJ, Sanford BL, et al.: Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 377 (5): 454-464, 2017.
- Schlenk RF, Weber D, Fiedler W, et al.: Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood 133 (8): 840-851, 2019.
- Zwaan CM, Söderhäll S, Brethon B, et al.: A phase 1/2, open-label, dose-escalation study of midostaurin in children with relapsed or refractory acute leukaemia. Br J Haematol 185 (3): 623-627, 2019.
- Pollard JA, Alonzo TA, Gerbing R, et al.: Sorafenib in Combination With Standard Chemotherapy for Children With High Allelic Ratio FLT3/ITD+ Acute Myeloid Leukemia: A Report From the Children's Oncology Group Protocol AAML1031. J Clin Oncol 40 (18): 2023-2035, 2022.
- Sung L, Gamis A, Alonzo TA, et al.: Infections and association with different intensity of chemotherapy in children with acute myeloid leukemia. Cancer 115 (5): 1100-8, 2009.
- Kaya Z, Gursel T, Kocak U, et al.: Invasive fungal infections in pediatric leukemia patients receiving fluconazole prophylaxis. Pediatr Blood Cancer 52 (4): 470-5, 2009.
- Kobayashi R, Kaneda M, Sato T, et al.: The clinical feature of invasive fungal infection in pediatric patients with hematologic and malignant diseases: a 10-year analysis at a single institution at Japan. J Pediatr Hematol Oncol 30 (12): 886-90, 2008.
- Ozer H, Armitage JO, Bennett CL, et al.: 2000 update of recommendations for the use of hematopoietic colony-stimulating factors: evidence-based, clinical practice guidelines. American Society of Clinical Oncology Growth Factors Expert Panel. J Clin Oncol 18 (20): 3558-85, 2000.
- Lehrnbecher T, Zimmermann M, Reinhardt D, et al.: Prophylactic human granulocyte colony-stimulating factor after induction therapy in pediatric acute myeloid leukemia. Blood 109 (3): 936-43, 2007.
- Ehlers S, Herbst C, Zimmermann M, et al.: Granulocyte colony-stimulating factor (G-CSF) treatment of childhood acute myeloid leukemias that overexpress the differentiation-defective G-CSF receptor isoform IV is associated with a higher incidence of relapse. J Clin Oncol 28 (15): 2591-7, 2010.
- Kurt B, Flynn P, Shenep JL, et al.: Prophylactic antibiotics reduce morbidity due to septicemia during intensive treatment for pediatric acute myeloid leukemia. Cancer 113 (2): 376-82, 2008.
- Inaba H, Gaur AH, Cao X, et al.: Feasibility, efficacy, and adverse effects of outpatient antibacterial prophylaxis in children with acute myeloid leukemia. Cancer 120 (13): 1985-92, 2014.
- Sung L, Aplenc R, Alonzo TA, et al.: Effectiveness of supportive care measures to reduce infections in pediatric AML: a report from the Children's Oncology Group. Blood 121 (18): 3573-7, 2013.
- Yeh TC, Liu HC, Hou JY, et al.: Severe infections in children with acute leukemia undergoing intensive chemotherapy can successfully be prevented by ciprofloxacin, voriconazole, or micafungin prophylaxis. Cancer 120 (8): 1255-62, 2014.
- Alexander S, Fisher BT, Gaur AH, et al.: Effect of Levofloxacin Prophylaxis on Bacteremia in Children With Acute Leukemia or Undergoing Hematopoietic Stem Cell Transplantation: A Randomized Clinical Trial. JAMA 320 (10): 995-1004, 2018.
- Taplitz RA, Kennedy EB, Bow EJ, et al.: Antimicrobial Prophylaxis for Adult Patients With Cancer-Related Immunosuppression: ASCO and IDSA Clinical Practice Guideline Update. J Clin Oncol 36 (30): 3043-3054, 2018.
- Ethier MC, Science M, Beyene J, et al.: Mould-active compared with fluconazole prophylaxis to prevent invasive fungal diseases in cancer patients receiving chemotherapy or haematopoietic stem-cell transplantation: a systematic review and meta-analysis of randomised controlled trials. Br J Cancer 106 (10): 1626-37, 2012.
- Robenshtok E, Gafter-Gvili A, Goldberg E, et al.: Antifungal prophylaxis in cancer patients after chemotherapy or hematopoietic stem-cell transplantation: systematic review and meta-analysis. J Clin Oncol 25 (34): 5471-89, 2007.
- Mandhaniya S, Swaroop C, Thulkar S, et al.: Oral voriconazole versus intravenous low dose amphotericin B for primary antifungal prophylaxis in pediatric acute leukemia induction: a prospective, randomized, clinical study. J Pediatr Hematol Oncol 33 (8): e333-41, 2011.
- Mattiuzzi GN, Kantarjian H, Faderl S, et al.: Amphotericin B lipid complex as prophylaxis of invasive fungal infections in patients with acute myelogenous leukemia and myelodysplastic syndrome undergoing induction chemotherapy. Cancer 100 (3): 581-9, 2004.
- Mattiuzzi GN, Kantarjian H, O'Brien S, et al.: Intravenous itraconazole for prophylaxis of systemic fungal infections in patients with acute myelogenous leukemia and high-risk myelodysplastic syndrome undergoing induction chemotherapy. Cancer 100 (3): 568-73, 2004.
- Tacke D, Buchheidt D, Karthaus M, et al.: Primary prophylaxis of invasive fungal infections in patients with haematologic malignancies. 2014 update of the recommendations of the Infectious Diseases Working Party of the German Society for Haematology and Oncology. Ann Hematol 93 (9): 1449-56, 2014.
- Grau S, de la Cámara R, Sabater FJ, et al.: Cost-effectiveness of posaconazole versus fluconazole or itraconazole in the prevention of invasive fungal infections among high-risk neutropenic patients in Spain. BMC Infect Dis 12: 83, 2012.
- Fisher BT, Zaoutis T, Dvorak CC, et al.: Effect of Caspofungin vs Fluconazole Prophylaxis on Invasive Fungal Disease Among Children and Young Adults With Acute Myeloid Leukemia: A Randomized Clinical Trial. JAMA 322 (17): 1673-1681, 2019.
- Getz KD, Sung L, Ky B, et al.: Occurrence of Treatment-Related Cardiotoxicity and Its Impact on Outcomes Among Children Treated in the AAML0531 Clinical Trial: A Report From the Children's Oncology Group. J Clin Oncol 37 (1): 12-21, 2019.
- Feijen EAM, Leisenring WM, Stratton KL, et al.: Derivation of Anthracycline and Anthraquinone Equivalence Ratios to Doxorubicin for Late-Onset Cardiotoxicity. JAMA Oncol 5 (6): 864-871, 2019.
- Getz KD, Sung L, Alonzo TA, et al.: Effect of Dexrazoxane on Left Ventricular Systolic Function and Treatment Outcomes in Patients With Acute Myeloid Leukemia: A Report From the Children's Oncology Group. J Clin Oncol 38 (21): 2398-2406, 2020.
- Wells RJ, Adams MT, Alonzo TA, et al.: Mitoxantrone and cytarabine induction, high-dose cytarabine, and etoposide intensification for pediatric patients with relapsed or refractory acute myeloid leukemia: Children's Cancer Group Study 2951. J Clin Oncol 21 (15): 2940-7, 2003.
- Aplenc R, Alonzo TA, Gerbing RB, et al.: Safety and efficacy of gemtuzumab ozogamicin in combination with chemotherapy for pediatric acute myeloid leukemia: a report from the Children's Oncology Group. J Clin Oncol 26 (14): 2390-3295, 2008.
- Dusenbery KE, Howells WB, Arthur DC, et al.: Extramedullary leukemia in children with newly diagnosed acute myeloid leukemia: a report from the Children's Cancer Group. J Pediatr Hematol Oncol 25 (10): 760-8, 2003.
- Støve HK, Sandahl JD, Abrahamsson J, et al.: Extramedullary leukemia in children with acute myeloid leukemia: A population-based cohort study from the Nordic Society of Pediatric Hematology and Oncology (NOPHO). Pediatr Blood Cancer 64 (12): , 2017.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Superior outcome of pediatric acute myeloid leukemia patients with orbital and CNS myeloid sarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 58 (4): 519-24, 2012.
- Creutzig U, Zimmermann M, Bourquin JP, et al.: CNS irradiation in pediatric acute myleoid leukemia: equal results by 12 or 18 Gy in studies AML-BFM98 and 2004. Pediatr Blood Cancer 57 (6): 986-92, 2011.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Central nervous system disease in pediatric acute myeloid leukemia: A report from the Children's Oncology Group. Pediatr Blood Cancer 64 (12): , 2017.
- Pui CH, Howard SC: Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol 9 (3): 257-68, 2008.
- Pollard JA, Alonzo TA, Brown PA, et al.: Sorafenib in combination with standard chemotherapy for children with high allelic ratio FLT3/ITD+ AML improves event-free survival and reduces relapse risk: a report from the Children's Oncology Group protocol AAML1031. [Abstract] Blood 134 (Suppl 1): A-613, 292, 2019. Also available online. Last accessed April 7, 2022.
- Mayer RJ, Davis RB, Schiffer CA, et al.: Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N Engl J Med 331 (14): 896-903, 1994.
- Wells RJ, Woods WG, Buckley JD, et al.: Treatment of newly diagnosed children and adolescents with acute myeloid leukemia: a Childrens Cancer Group study. J Clin Oncol 12 (11): 2367-77, 1994.
- Wells RJ, Woods WG, Lampkin BC, et al.: Impact of high-dose cytarabine and asparaginase intensification on childhood acute myeloid leukemia: a report from the Childrens Cancer Group. J Clin Oncol 11 (3): 538-45, 1993.
- Getz KD, Alonzo TA, Sung L, et al.: Cytarabine dose reduction in patients with low-risk acute myeloid leukemia: A report from the Children's Oncology Group. Pediatr Blood Cancer 69 (1): e29313, 2022.
- Woods WG, Neudorf S, Gold S, et al.: A comparison of allogeneic bone marrow transplantation, autologous bone marrow transplantation, and aggressive chemotherapy in children with acute myeloid leukemia in remission. Blood 97 (1): 56-62, 2001.
- Horan JT, Alonzo TA, Lyman GH, et al.: Impact of disease risk on efficacy of matched related bone marrow transplantation for pediatric acute myeloid leukemia: the Children's Oncology Group. J Clin Oncol 26 (35): 5797-801, 2008.
- Ravindranath Y, Yeager AM, Chang MN, et al.: Autologous bone marrow transplantation versus intensive consolidation chemotherapy for acute myeloid leukemia in childhood. Pediatric Oncology Group. N Engl J Med 334 (22): 1428-34, 1996.
- Feig SA, Lampkin B, Nesbit ME, et al.: Outcome of BMT during first complete remission of AML: a comparison of two sequential studies by the Children's Cancer Group. Bone Marrow Transplant 12 (1): 65-71, 1993.
- Amadori S, Testi AM, Aricò M, et al.: Prospective comparative study of bone marrow transplantation and postremission chemotherapy for childhood acute myelogenous leukemia. The Associazione Italiana Ematologia ed Oncologia Pediatrica Cooperative Group. J Clin Oncol 11 (6): 1046-54, 1993.
- Bleakley M, Lau L, Shaw PJ, et al.: Bone marrow transplantation for paediatric AML in first remission: a systematic review and meta-analysis. Bone Marrow Transplant 29 (10): 843-52, 2002.
- Koreth J, Schlenk R, Kopecky KJ, et al.: Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA 301 (22): 2349-61, 2009.
- Klusmann JH, Reinhardt D, Zimmermann M, et al.: The role of matched sibling donor allogeneic stem cell transplantation in pediatric high-risk acute myeloid leukemia: results from the AML-BFM 98 study. Haematologica 97 (1): 21-9, 2012.
- Creutzig U, Reinhardt D: Current controversies: which patients with acute myeloid leukaemia should receive a bone marrow transplantation?--a European view. Br J Haematol 118 (2): 365-77, 2002.
- Oliansky DM, Rizzo JD, Aplan PD, et al.: The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute myeloid leukemia in children: an evidence-based review. Biol Blood Marrow Transplant 13 (1): 1-25, 2007.
- Niewerth D, Creutzig U, Bierings MB, et al.: A review on allogeneic stem cell transplantation for newly diagnosed pediatric acute myeloid leukemia. Blood 116 (13): 2205-14, 2010.
- Qayed M, Ahn KW, Kitko CL, et al.: A validated pediatric disease risk index for allogeneic hematopoietic cell transplantation. Blood 137 (7): 983-993, 2021.
- Tsukimoto I, Tawa A, Horibe K, et al.: Risk-stratified therapy and the intensive use of cytarabine improves the outcome in childhood acute myeloid leukemia: the AML99 trial from the Japanese Childhood AML Cooperative Study Group. J Clin Oncol 27 (24): 4007-13, 2009.
- Abrahamsson J, Forestier E, Heldrup J, et al.: Response-guided induction therapy in pediatric acute myeloid leukemia with excellent remission rate. J Clin Oncol 29 (3): 310-5, 2011.
- Kelly MJ, Horan JT, Alonzo TA, et al.: Comparable survival for pediatric acute myeloid leukemia with poor-risk cytogenetics following chemotherapy, matched related donor, or unrelated donor transplantation. Pediatr Blood Cancer 61 (2): 269-75, 2014.
- Wareham NE, Heilmann C, Abrahamsson J, et al.: Outcome of poor response paediatric AML using early SCT. Eur J Haematol 90 (3): 187-94, 2013.
- Burke MJ, Wagner JE, Cao Q, et al.: Allogeneic hematopoietic cell transplantation in first remission abrogates poor outcomes associated with high-risk pediatric acute myeloid leukemia. Biol Blood Marrow Transplant 19 (7): 1021-5, 2013.
- Meshinchi S, Alonzo TA, Stirewalt DL, et al.: Clinical implications of FLT3 mutations in pediatric AML. Blood 108 (12): 3654-61, 2006.
- Schlenk RF, Kayser S, Bullinger L, et al.: Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood 124 (23): 3441-9, 2014.
- Beier R, Albert MH, Bader P, et al.: Allo-SCT using BU, CY and melphalan for children with AML in second CR. Bone Marrow Transplant 48 (5): 651-6, 2013.
- Liu DH, Xu LP, Liu KY, et al.: Long-term outcomes of unmanipulated haploidentical HSCT for paediatric patients with acute leukaemia. Bone Marrow Transplant 48 (12): 1519-24, 2013.
- Locatelli F, Masetti R, Rondelli R, et al.: Outcome of children with high-risk acute myeloid leukemia given autologous or allogeneic hematopoietic cell transplantation in the aieop AML-2002/01 study. Bone Marrow Transplant 50 (2): 181-8, 2015.
- Nemecek ER, Hilger RA, Adams A, et al.: Treosulfan, Fludarabine, and Low-Dose Total Body Irradiation for Children and Young Adults with Acute Myeloid Leukemia or Myelodysplastic Syndrome Undergoing Allogeneic Hematopoietic Cell Transplantation: Prospective Phase II Trial of the Pediatric Blood and Marrow Transplant Consortium. Biol Blood Marrow Transplant 24 (8): 1651-1656, 2018.
- Liu H, Zhai X, Song Z, et al.: Busulfan plus fludarabine as a myeloablative conditioning regimen compared with busulfan plus cyclophosphamide for acute myeloid leukemia in first complete remission undergoing allogeneic hematopoietic stem cell transplantation: a prospective and multicenter study. J Hematol Oncol 6: 15, 2013.
- Bredeson C, LeRademacher J, Kato K, et al.: Prospective cohort study comparing intravenous busulfan to total body irradiation in hematopoietic cell transplantation. Blood 122 (24): 3871-8, 2013.
- Dandoy CE, Davies SM, Woo Ahn K, et al.: Comparison of total body irradiation versus non-total body irradiation containing regimens for de novo acute myeloid leukemia in children. Haematologica 106 (7): 1839-1845, 2021.
- Perel Y, Auvrignon A, Leblanc T, et al.: Treatment of childhood acute myeloblastic leukemia: dose intensification improves outcome and maintenance therapy is of no benefit--multicenter studies of the French LAME (Leucémie Aiguë Myéloblastique Enfant) Cooperative Group. Leukemia 19 (12): 2082-9, 2005.
- Arber DA, Vardiman JW, Brunning RD: Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 110-23.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Webb DK: Management of relapsed acute myeloid leukaemia. Br J Haematol 106 (4): 851-9, 1999.
- Stahnke K, Boos J, Bender-Götze C, et al.: Duration of first remission predicts remission rates and long-term survival in children with relapsed acute myelogenous leukemia. Leukemia 12 (10): 1534-8, 1998.
- Webb DK, Wheatley K, Harrison G, et al.: Outcome for children with relapsed acute myeloid leukaemia following initial therapy in the Medical Research Council (MRC) AML 10 trial. MRC Childhood Leukaemia Working Party. Leukemia 13 (1): 25-31, 1999.
- Nakayama H, Tabuchi K, Tawa A, et al.: Outcome of children with relapsed acute myeloid leukemia following initial therapy under the AML99 protocol. Int J Hematol 100 (2): 171-9, 2014.
- Gorman MF, Ji L, Ko RH, et al.: Outcome for children treated for relapsed or refractory acute myelogenous leukemia (rAML): a Therapeutic Advances in Childhood Leukemia (TACL) Consortium study. Pediatr Blood Cancer 55 (3): 421-9, 2010.
- Bachas C, Schuurhuis GJ, Reinhardt D, et al.: Clinical relevance of molecular aberrations in paediatric acute myeloid leukaemia at first relapse. Br J Haematol 166 (6): 902-10, 2014.
- Sander A, Zimmermann M, Dworzak M, et al.: Consequent and intensified relapse therapy improved survival in pediatric AML: results of relapse treatment in 379 patients of three consecutive AML-BFM trials. Leukemia 24 (8): 1422-8, 2010.
- Creutzig U, Zimmermann M, Dworzak MN, et al.: The prognostic significance of early treatment response in pediatric relapsed acute myeloid leukemia: results of the international study Relapsed AML 2001/01. Haematologica 99 (9): 1472-8, 2014.
- Karlsson L, Forestier E, Hasle H, et al.: Outcome after intensive reinduction therapy and allogeneic stem cell transplant in paediatric relapsed acute myeloid leukaemia. Br J Haematol 178 (4): 592-602, 2017.
- Tavil B, Aytac S, Balci YI, et al.: Fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin (FLAG-IDA) for the treatment of children with poor-prognosis acute leukemia: the Hacettepe experience. Pediatr Hematol Oncol 27 (7): 517-28, 2010.
- Capizzi RL, Davis R, Powell B, et al.: Synergy between high-dose cytarabine and asparaginase in the treatment of adults with refractory and relapsed acute myelogenous leukemia--a Cancer and Leukemia Group B Study. J Clin Oncol 6 (3): 499-508, 1988.
- Kaspers GJ, Zimmermann M, Reinhardt D, et al.: Improved outcome in pediatric relapsed acute myeloid leukemia: results of a randomized trial on liposomal daunorubicin by the International BFM Study Group. J Clin Oncol 31 (5): 599-607, 2013.
- Cooper TM, Absalon MJ, Alonzo TA, et al.: Phase I/II Study of CPX-351 Followed by Fludarabine, Cytarabine, and Granulocyte-Colony Stimulating Factor for Children With Relapsed Acute Myeloid Leukemia: A Report From the Children's Oncology Group. J Clin Oncol 38 (19): 2170-2177, 2020.
- Hijiya N, Gaynon P, Barry E, et al.: A multi-center phase I study of clofarabine, etoposide and cyclophosphamide in combination in pediatric patients with refractory or relapsed acute leukemia. Leukemia 23 (12): 2259-64, 2009.
- Jeha S, Razzouk B, Rytting M, et al.: Phase II study of clofarabine in pediatric patients with refractory or relapsed acute myeloid leukemia. J Clin Oncol 27 (26): 4392-7, 2009.
- Shukla N, Kobos R, Renaud T, et al.: Phase II trial of clofarabine with topotecan, vinorelbine, and thiotepa in pediatric patients with relapsed or refractory acute leukemia. Pediatr Blood Cancer 61 (3): 431-5, 2014.
- Chaleff S, Hurwitz CA, Chang M, et al.: Phase II study of 2-chlorodeoxyadenosine plus idarubicin for children with acute myeloid leukaemia in first relapse: a paediatric oncology group study. Br J Haematol 156 (5): 649-55, 2012.
- Cooper TM, Alonzo TA, Gerbing RB, et al.: AAML0523: a report from the Children's Oncology Group on the efficacy of clofarabine in combination with cytarabine in pediatric patients with recurrent acute myeloid leukemia. Cancer 120 (16): 2482-9, 2014.
- Horton TM, Perentesis JP, Gamis AS, et al.: A Phase 2 study of bortezomib combined with either idarubicin/cytarabine or cytarabine/etoposide in children with relapsed, refractory or secondary acute myeloid leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 61 (10): 1754-60, 2014.
- Kell WJ, Burnett AK, Chopra R, et al.: A feasibility study of simultaneous administration of gemtuzumab ozogamicin with intensive chemotherapy in induction and consolidation in younger patients with acute myeloid leukemia. Blood 102 (13): 4277-83, 2003.
- Zwaan CM, Reinhardt D, Zimmerman M, et al.: Salvage treatment for children with refractory first or second relapse of acute myeloid leukaemia with gemtuzumab ozogamicin: results of a phase II study. Br J Haematol 148 (5): 768-76, 2010.
- Arceci RJ, Sande J, Lange B, et al.: Safety and efficacy of gemtuzumab ozogamicin in pediatric patients with advanced CD33+ acute myeloid leukemia. Blood 106 (4): 1183-8, 2005.
- Taksin AL, Legrand O, Raffoux E, et al.: High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia 21 (1): 66-71, 2007.
- Farhat H, Reman O, Raffoux E, et al.: Fractionated doses of gemtuzumab ozogamicin with escalated doses of daunorubicin and cytarabine as first acute myeloid leukemia salvage in patients aged 50-70-year old: a phase 1/2 study of the acute leukemia French association. Am J Hematol 87 (1): 62-5, 2012.
- Perl AE, Martinelli G, Cortes JE, et al.: Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N Engl J Med 381 (18): 1728-1740, 2019.
- Inaba H, Rubnitz JE, Coustan-Smith E, et al.: Phase I pharmacokinetic and pharmacodynamic study of the multikinase inhibitor sorafenib in combination with clofarabine and cytarabine in pediatric relapsed/refractory leukemia. J Clin Oncol 29 (24): 3293-300, 2011.
- Tarlock K, Chang B, Cooper T, et al.: Sorafenib treatment following hematopoietic stem cell transplant in pediatric FLT3/ITD acute myeloid leukemia. Pediatr Blood Cancer 62 (6): 1048-54, 2015.
- Rasche M, Zimmermann M, Borschel L, et al.: Successes and challenges in the treatment of pediatric acute myeloid leukemia: a retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia 32 (10): 2167-2177, 2018.
- Bunin NJ, Davies SM, Aplenc R, et al.: Unrelated donor bone marrow transplantation for children with acute myeloid leukemia beyond first remission or refractory to chemotherapy. J Clin Oncol 26 (26): 4326-32, 2008.
- Woodard P, Carpenter PA, Davies SM, et al.: Unrelated donor bone marrow transplantation for myelodysplastic syndrome in children. Biol Blood Marrow Transplant 17 (5): 723-8, 2011.
- Uberti JP, Agovi MA, Tarima S, et al.: Comparative analysis of BU and CY versus CY and TBI in full intensity unrelated marrow donor transplantation for AML, CML and myelodysplasia. Bone Marrow Transplant 46 (1): 34-43, 2011.
- Shaw PJ, Kan F, Woo Ahn K, et al.: Outcomes of pediatric bone marrow transplantation for leukemia and myelodysplasia using matched sibling, mismatched related, or matched unrelated donors. Blood 116 (19): 4007-15, 2010.
- Eapen M, Klein JP, Ruggeri A, et al.: Impact of allele-level HLA matching on outcomes after myeloablative single unit umbilical cord blood transplantation for hematologic malignancy. Blood 123 (1): 133-40, 2014.
- Locatelli F, Merli P, Pagliara D, et al.: Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood 130 (5): 677-685, 2017.
- Rashidi A, DiPersio JF, Westervelt P, et al.: Comparison of Outcomes after Peripheral Blood Haploidentical versus Matched Unrelated Donor Allogeneic Hematopoietic Cell Transplantation in Patients with Acute Myeloid Leukemia: A Retrospective Single-Center Review. Biol Blood Marrow Transplant 22 (9): 1696-1701, 2016.
- Pulsipher MA, Boucher KM, Wall D, et al.: Reduced-intensity allogeneic transplantation in pediatric patients ineligible for myeloablative therapy: results of the Pediatric Blood and Marrow Transplant Consortium Study ONC0313. Blood 114 (7): 1429-36, 2009.
- Scott BL, Pasquini MC, Logan BR, et al.: Myeloablative Versus Reduced-Intensity Hematopoietic Cell Transplantation for Acute Myeloid Leukemia and Myelodysplastic Syndromes. J Clin Oncol 35 (11): 1154-1161, 2017.
- Meshinchi S, Leisenring WM, Carpenter PA, et al.: Survival after second hematopoietic stem cell transplantation for recurrent pediatric acute myeloid leukemia. Biol Blood Marrow Transplant 9 (11): 706-13, 2003.
- Nishikawa T, Inagaki J, Nagatoshi Y, et al.: The second therapeutic trial for children with hematological malignancies who relapsed after their first allogeneic SCT: long-term outcomes. Pediatr Transplant 16 (7): 722-8, 2012.
- Yaniv I, Krauss AC, Beohou E, et al.: Second Hematopoietic Stem Cell Transplantation for Post-Transplantation Relapsed Acute Leukemia in Children: A Retrospective EBMT-PDWP Study. Biol Blood Marrow Transplant 24 (8): 1629-1642, 2018.
- Uden T, Bertaina A, Abrahamsson J, et al.: Outcome of children relapsing after first allogeneic haematopoietic stem cell transplantation for acute myeloid leukaemia: a retrospective I-BFM analysis of 333 children. Br J Haematol 189 (4): 745-750, 2020.
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Risk factors and therapy for isolated central nervous system relapse of pediatric acute myeloid leukemia. J Clin Oncol 23 (36): 9172-8, 2005.
- Abbott BL, Rubnitz JE, Tong X, et al.: Clinical significance of central nervous system involvement at diagnosis of pediatric acute myeloid leukemia: a single institution's experience. Leukemia 17 (11): 2090-6, 2003.
- Quarello P, Fagioli F, Basso G, et al.: Outcome of children with acute myeloid leukaemia (AML) experiencing primary induction failure in the AIEOP AML 2002/01 clinical trial. Br J Haematol 171 (4): 566-73, 2015.
- O'Hear C, Inaba H, Pounds S, et al.: Gemtuzumab ozogamicin can reduce minimal residual disease in patients with childhood acute myeloid leukemia. Cancer 119 (22): 4036-43, 2013.
Leucemia promielocítica aguda
La leucemia promielocítica aguda (LPA) es un subtipo de leucemia mieloide aguda (LMA) diferenciado por múltiples factores, como los siguientes:
- Cuadro clínico inicial de coagulopatía universal (coagulación intravascular diseminada) y características morfológicas únicas (French-American-British [FAB] M3 o sus variantes).
- Características etiológicas moleculares únicas por compromiso del oncogén RARA.
- Sensibilidad única al diferenciador tretinoína y al proapoptótico trióxido de arsénico.[
1 ]
Estas características únicas de la LPA exigen un índice alto de sospecha en el momento del diagnóstico para que se puedan iniciar las medidas de atención complementaria apropiadas con el fin de evitar complicaciones por coagulopatía durante los primeros días del tratamiento. También es importante instituir un régimen de inducción diferente con el fin de reducir el riesgo de complicaciones por coagulopatías y ofrecer una supervivencia sin recaída a largo plazo y una supervivencia general (SG) mejoradas en comparación con las obtenidas con los abordajes previos para la LPA y que son comparables con los desenlaces para los pacientes con otras formas de LMA.[
Anomalías moleculares
La anomalía cromosómica característica relacionada con la LPA es la t(15;17). Esta translocación afecta un punto de ruptura que incluye el receptor del ácido retinoico y lleva a la producción de la proteína de fusión de la leucemia promielocítica (PML)::receptor de ácido retinoico (RARA).[
Se confirma el diagnóstico de los pacientes en quienes se sospecha una LPA cuando se detecta la proteína de fusión PML::RARA (por ejemplo, mediante hibridación fluorescente in situ [FISH], reacción en cadena de la polimerasa con retrotranscripción [RCP-RT] o análisis citogenéticos convencionales). Se puede establecer con rapidez la presencia de la proteína de fusión PML::RARA usando un método de inmunofluorescencia con un anticuerpo monoclonal anti-PML porque se analiza el patrón de distribución característico de la PML en presencia de la proteína de fusión.[
Cuadro clínico inicial
Desde el punto de vista clínico, la LPA se caracteriza por coagulopatía grave que a menudo se presenta en el momento del diagnóstico.[
La terapia con tretinoína se inicia tan pronto como se sospeche LPA a partir de las manifestaciones clínicas y morfológicas iniciales,[
En general, la LPA en niños es similar a la LPA en adultos, aunque los niños tienen una incidencia más alta de hiperleucocitosis (definida como un recuento de GB mayor de 10 × 109 /l) y mayor incidencia del subtipo morfológico microgranular.[
Clasificación de riesgo para la estratificación del tratamiento
La importancia pronóstica del recuento de GB sirve para definir las poblaciones de riesgo alto y riesgo bajo, y asignar el tratamiento posinducción: los pacientes de riesgo alto se definen de manera más común por un GB de 10 × 109 /l o más alto.[
En el ensayo del COG AAML0631 (NCT00866918), que incluyó el tratamiento con quimioterapia, tretinoína y trióxido de arsénico, la clasificación de riesgo definió principalmente el riesgo de muerte prematura más que el riesgo de recaída (riesgo estándar, 0 de 66 pacientes vs. riesgo alto, 4 de 35 pacientes).[
El sistema nervioso central y la leucemia promielocítica aguda
En la mayoría de los pacientes de leucemia promielocítica aguda (LPA) no se corrobora el compromiso del sistema nervioso central (SNC) en el momento del diagnóstico debido a una coagulación intravascular diseminada. Entre 101 niños inscritos en el ensayo del COG AAML0631 (NCT00866918), se identificaron 28 niños que tenían exámenes del líquido cefalorraquídeo (LCR) en el momento del diagnóstico, y en 7 de esos niños se detectaron blastocitos en punciones atraumáticas.[
En general, las recaídas en el SNC son infrecuentes en los pacientes con LPA, en particular en aquellos que tienen un recuento de GB inferior a 10 × 109 /l.[
Tratamiento de la leucemia promielocítica aguda
Los programas de tratamiento contemporáneos para la LPA se basan en la sensibilidad de las células leucémicas de los pacientes con LPA a los efectos de inducción de diferenciación, y los efectos apoptóticos de la tretinoína y el trióxido de arsénico. La adición de tretinoína a la quimioterapia fue la primera diferencia del tratamiento de la LPA en relación con el tratamiento de otros subtipos de LMA diferentes a la LPA. Con la incorporación del trióxido de arsénico en los regímenes terapéuticos modernos, el uso de la quimioterapia tradicional en adultos y niños se restringe cada vez más a la fase de inducción en pacientes de riesgo alto.[
Las opciones de tratamiento para los niños con LPA son las siguientes:
- Tretinoína.
- Trióxido de arsénico.
- Quimioterapia.
- Cuidados médicos de apoyo.
Debido al alto índice de actividad de la combinación de trióxido de arsénico y tretinoína en adultos con LPA,[
Antes de que se descubriera este abordaje, la quimioterapia se utilizaba en todas o en la mayoría de las fases del tratamiento, incluyendo la inducción, la consolidación y el mantenimiento en ensayos pediátricos como el AAML0631 (NCT00866918). En la actualidad, los regímenes que utilizan quimioterapia son en su mayor parte de interés histórico. También se utilizan como referencia en los casos de resistencia al tratamiento debido a los resultados obtenidos en los ensayos clínicos aleatorizados donde se compararon los regímenes con la combinación de tretinoína y trióxido de arsénico con quimioterapia o sin esta. A partir de los resultados de un ensayo finalizado del grupo cooperativo COG (AAML1331 [NCT02339740]), se verificó el beneficio del tratamiento con tretinoína y trióxido de arsénico en niños con LPA recién diagnosticada,[
Casi todos los niños con LPA tratados con tretinoína, trióxido de arsénico y los cuidados de apoyo modernos (descritos a continuación) lograron una RC sin mortalidad relacionada con coagulopatía.[
La evaluación de la respuesta a la terapia de inducción durante el primer mes de tratamiento usando criterios morfológicos y moleculares tal vez produzca resultados engañosos por la persistencia de células leucémicas en diferenciación que se pueden encontrar en pacientes que a la larga lograrán una RC.[
En los pacientes con LPA, la terapia de consolidación en ocasiones incluye ciclos repetidos de tretinoína y trióxido de arsénico, sin quimioterapia adicional, de acuerdo a la experiencia con adultos y niños.[
Es probable que no se necesite terapia de mantenimiento en los pacientes con LPA que se tratan con tretinoína y trióxido de arsénico, de acuerdo con los datos de los ensayos en adultos y el ensayo del COG AAML1331 (NCT02339740).[
El trióxido de arsénico es el fármaco más activo para el tratamiento de la LPA, y aunque antes solo se usaba para la LPA recidivante, ahora se incorporó en el tratamiento de pacientes recién diagnosticados. Los datos que respaldan el uso de trióxido de arsénico surgieron de ensayos en los que solo participaron adultos, pero de manera más reciente, se observó su eficacia en ensayos que incluyeron pacientes pediátricos.
Evidencia (terapia con trióxido de arsénico):
- En los adultos con diagnóstico reciente de LPA tratados en el ensayo CALGB-C9710 (NCT00003934), se encontró que la adición de dos cursos de consolidación con trióxido de arsénico al régimen de tratamiento estándar de la LPA produjo los siguientes resultados:[
47 ]- Una mejora significativa de las tasas de SSC (80 % vs. 63 % a los 3 años; P < 0,0001) y supervivencia sin enfermedad (SSE) (90 % vs. 70 % a los 3 años; P < 0,0001), aunque el desenlace de los pacientes que no recibieron trióxido de arsénico fue inferior a los resultados de los ensayos del Gruppo Italiano Malattie EMatologiche dell'Adulto (GIMEMA) o PETHEMA.
- En los niños y adolescentes con diagnóstico reciente de LPA tratados en el ensayo del COG AAML0631 (NCT00866918) se incorporaron dos ciclos de consolidación de trióxido de arsénico al régimen de quimioterapia con dosis acumuladas de antraciclinas más bajas en comparación con los controles históricos.[
28 ]- La tasa de SG a 3 años fue del 94 % y la tasa de SSC fue del 91 %.
- Los pacientes con LPA de riesgo estándar tuvieron una tasa de SG del 98 % y una tasa de SSC del 95 %.
- Los pacientes de riesgo alto tuvieron una tasa de SG del 86 % y una tasa de SSC del 83 %. Las muertes prematuras fueron la causa principal de esta supervivencia más baja en comparación con los pacientes de riesgo estándar.
- El riesgo de recaída después de la consolidación con trióxido de arsénico fue del 4 %, similar que para la LPA de riesgo estándar y riesgo alto.
- El uso simultáneo de trióxido de arsénico y tretinoína en pacientes con LPA recién diagnosticada produjo tasas más altas de RC.[
48 ,49 ,50 ] En las experiencias iniciales en niños con LPA recién diagnosticada se observaron tasas altas de RC al trióxido de arsénico, como monoterapia o combinado con tretinoína.[51 ][Nivel de evidencia C1] A partir de los resultados de un metanálisis de 7 estudios publicados de pacientes adultos con LPA se indicó que el uso de una combinación de trióxido de arsénico y tretinoína quizá sea más eficaz que el uso de trióxido de arsénico solo para inducir una RC.[52 ]- En ensayos tempranos en niños, el efecto del arsénico añadido a la inducción (solo o con tretinoína) sobre la SSC y la SG fue prometedor.[
51 ,53 ,54 ]
- En ensayos tempranos en niños, el efecto del arsénico añadido a la inducción (solo o con tretinoína) sobre la SSC y la SG fue prometedor.[
- El trióxido de arsénico se evaluó como un componente de la terapia de inducción con idarrubicina y tretinoína en el ensayo clínico APML4, en el que se inscribieron niños y adultos (N = 124 pacientes evaluables).[
26 ] Los pacientes recibieron 2 cursos de terapia de consolidación con trióxido de arsénico y tretinoína (sin una antraciclina) y terapia de mantenimiento con tretinoína, mercaptopurina y metotrexato.[55 ]- La tasa de ausencia de recaída a 2 años fue del 97,5 %, la tasa de supervivencia sin fracaso (SSF) fue del 88,1 % y la tasa de SG fue del 93,2 %.
- Estos resultados son superiores a los observados en los pacientes del ensayo clínico anterior (APML3) que no recibieron trióxido de arsénico.
- En un ensayo clínico alemán e italiano de fase III (APL0406 [NCT00482833]), se comparó tretinoína y quimioterapia con tretinoína y trióxido de arsénico en adultos con LPA clasificados como de riesgo bajo e intermedio (recuento de GB ≤10 × 109 /l).[
34 ] Los pacientes se asignaron al azar para recibir tretinoína y trióxido de arsénico durante la terapia de inducción y consolidación, o terapia de inducción estándar con tretinoína e idarrubicina seguida de 3 ciclos de terapia de consolidación con tretinoína y quimioterapia, además de terapia de mantenimiento con dosis bajas de quimioterapia y tretinoína.- Todos los pacientes que recibieron tretinoína y trióxido de arsénico (n = 77) alcanzaron una RC al final de la terapia de inducción, mientras que el 95 % de los pacientes que recibieron tretinoína y quimioterapia (n = 9) alcanzaron la RC.
- Las tasas de SSC fueron del 97 % en el grupo de tretinoína y trióxido de arsénico en comparación con el 86 % en el grupo de tretinoína y quimioterapia (P = 0,02).
- La probabilidad de SG a 2 años fue del 99 % (intervalo de confianza [IC] 95 %, 96–100 %) en el grupo de tretinoína y trióxido de arsénico, y del 91 % (IC 95 %, 85–97 %) en el grupo de tretinoína y quimioterapia (P = 0,02).
- En un análisis actualizado de largo plazo se demostró que, a los 50 meses, el grupo de tretinoína y trióxido de arsénico presentó una superioridad incluso mayor, con tasas de SG del 97 % versus 80 % (P < 0,001).[
34 ,35 ] - Estos resultados indican que la LPA de riesgo bajo a intermedio es curable en un porcentaje alto de pacientes sin quimioterapia convencional.
- El ensayo del COG AAML1331 (NCT02339740), de no inferioridad y con controles históricos, se realizó entre 2015 y 2019. En el estudio se incluyó a pacientes pediátricos (intervalo de edad, 1–21 años) con LPA y se examinó si añadir trióxido de arsénico a la terapia de inducción, y continuar con este tratamiento durante la consolidación, mantendría los excelentes resultados observados en el ensayo AAML0631 (NCT00866918). Además, la quimioterapia se eliminó por completo, excepto cuando los pacientes con LPA de riesgo alto recibieron ciclos cortos de idarubicina durante el terapia de inducción. Los pacientes con LPA de riesgo estándar no recibieron idarrubicina en el ciclo de inducción. En los ciclos de consolidación se eliminaron la mitoxantrona, las dosis altas de citarabina y la idarrubicina. Luego, se quitaron la mercaptopurina y el metotrexato de los ciclos de mantenimiento. Las dosis intratecales de citarabina también se eliminaron. En el estudio AAML1331 se incluyeron 158 pacientes, 98 de los cuales se clasificaron como de riesgo estándar y 56 como de riesgo alto.[
29 ]Los pacientes de riesgo estándar recibieron tretinoína y trióxido de arsénico del día 1 al 28, con la posibilidad de continuar el tratamiento hasta el día 70 para lograr una RC. Los pacientes de riesgo alto recibieron el mismo programa de terapia de inducción que los pacientes de riesgo estándar, con la adición de idarubicina en los días de inducción 1, 3, 5 y 7. Los pacientes de riesgo alto también recibieron dexametasona diaria como tratamiento profiláctico para prevenir el síndrome de diferenciación en los días 1 al 14. Todos los pacientes recibieron el mismo tratamiento de consolidación, que consistió en tretinoína del día 1 al 14 y del día 29 al 42. Los pacientes también recibieron trióxido de arsénico 5 días a la semana durante 4 semanas consecutivas en cada ciclo de 8 semanas (3 rondas). El cuarto ciclo de terapia de consolidación concluyó el día 28. No hubo ninguna fase de terapia de mantenimiento.
- La mediana de duración de la terapia de inducción en todos los pacientes (riesgo estándar y riesgo alto) fue de 47 días. La terapia de inducción incluyó un período de descanso de 14 días antes de iniciar el tratamiento de consolidación. Todos los pacientes de riesgo estándar y de riesgo alto que completaron la terapia de inducción alcanzaron una RC hematológica o una RC con recuperación hematológica incompleta antes del día 70.
- Durante la terapia de inducción, un paciente de riesgo estándar murió por complicaciones debidas a coagulopatía, síndrome de diferenciación y posterior insuficiencia orgánica. Ningún paciente de riesgo alto murió por causa de complicaciones.
- Todos los pacientes que se sometieron a la prueba de PCR cuantitativa después de completar su segunda ronda de terapia de consolidación estaban en remisión molecular.
- Ningún paciente experimentó recaída durante el tratamiento. Un paciente de riesgo estándar (1 %, recidiva en el SNC) y 2 pacientes de riesgo alto (4 %) presentaron recaídas tras la finalización del tratamiento. La terapia de rescate en estos pacientes fue exitosa.
- Se compararon los ensayos AAML1331 y AAML0631 y se notificó lo siguiente:
- Los pacientes de riesgo estándar tuvieron tasas equivalentes de SSC a 2 años (98 vs. 97 %) y de SG (99 vs. 98,5 %).
- Los pacientes de riesgo alto que se inscribieron en el ensayo AAML1331 tuvieron tasas significativamente mejores de SSC a 2 años (96,4 vs. 82,9 %, P = 0,05) y de SG (100 vs. 85,7 %, P = 0,02).
- En el ensayo AAML1331, los pacientes con síntomas en el SNC o hemorragia se examinaron y se trataron con triple quimioterapia intratecal. Mientras que en el estudio AAML0631, los pacientes de riesgo estándar recibieron 3 dosis profilácticas de quimioterapia intratecal, y los pacientes de riesgo alto recibieron 4 dosis profilácticas de quimioterapia intratecal.
- La duración del tratamiento fue significativamente menor en el ensayo AAML1331 (9 meses) que en el ensayo AAML0631 (2 años).
- Las hospitalizaciones durante la terapia de consolidación se redujeron significativamente en el ensayo AAML1331, en comparación con el ensayo AAML0631 (0 días vs. 13 días, respectivamente; P < 0,001).
- En el ensayo AAML1331, la muerte temprana fue significativamente menor en los pacientes de riesgo alto (0 vs. 4 en el ensayo AAML0631, P = 0,02), y no fue significativamente diferente en los pacientes de riesgo estándar (1 vs. 0 en el ensayo AAML0631, P = 0,16).
En resumen, en los niños con LPA, las tasas de supervivencia del 90 % se pueden lograr usando programas de tratamiento con inicio rápido de tretinoína y medidas de atención complementaria apropiadas y que combinan trióxido de arsénico con tretinoína para la terapia de inducción y consolidación.[
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
Complicaciones exclusivas del tratamiento de la leucemia promielocítica aguda
Además de la mencionada presencia universal de coagulopatía en los pacientes con LPA recién diagnosticada, los médicos deben saber que hay otras complicaciones que solo se presentan en los pacientes con LPA. Estas son dos afecciones relacionadas con la tretinoína, el pseudotumor cerebral y el síndrome de diferenciación (también llamado síndrome del ácido retinoico), y una complicación relacionada con el trióxido de arsénico, la prolongación del intervalo QT.
- Pseudotumor cerebral. El pseudotumor cerebral por lo general se manifiesta con cefalea, papiledema, parálisis del sexto nervio craneal, deterioro de los campos visuales e imágenes intracraneales normales acompañadas de una presión de apertura elevada en la punción lumbar (infrecuente en pacientes con LPA). Se sabe que el pseudotumor cerebral se relaciona con la tretinoína y se presume que tiene el mismo mecanismo que la toxicidad por vitamina A que aumenta la producción de líquido cefalorraquídeo.
Se ha informado que en ensayos pediátricos la incidencia del pseudotumor cerebral osciló entre el 1,7 % (con definiciones muy estrictas de la complicación) y el 6 % al 16 %.[
14 ,15 ,28 ,29 ,57 ] Se piensa que el pseudotumor cerebral es más prevalente en los niños que reciben tretinoína, lo que lleva a que se reduzca la dosis en los ensayos clínicos de LPA infantil contemporáneos.[14 ,29 ] El pseudotumor cerebral se presenta de manera más frecuente durante la inducción al cabo de una mediana de 15 días (intervalo, 1–35 días) después de comenzar la tretinoína, pero se sabe que también se presenta en otras fases del tratamiento.[57 ] Es posible que la incidencia y gravedad del pseudotumor cerebral aumente con el uso simultáneo de azoles debido a la inhibición del metabolismo de la tretinoína mediado por el citocromo P450.Cuando se sospecha un diagnóstico de pseudotumor cerebral, se interrumpe la administración de tretinoína hasta que los síntomas se resuelven, y luego el fármaco se aumenta hasta lograr la dosis completa según la tolerabilidad.[
57 ] - Síndrome de diferenciación. El síndrome de diferenciación (también conocido como síndrome del ácido retinoico o síndrome de la tretinoína) es un síndrome potencialmente mortal que se piensa que está mediado por una respuesta inflamatoria que se manifiesta por ganancia de peso, fiebre, edema, infiltrados pulmonares, derrames pleurales y pericárdicos, hipotensión y en los casos más graves, insuficiencia renal aguda.[
58 ] En el estudio contemporáneo del COG AAML0631 (NCT00866918), el 20 % de los pacientes presentaron este síndrome durante la inducción, y la prevalencia fue mayor en los niños de riesgo alto (31 %) que en los niños de riesgo bajo (13 %); este factor de riesgo también se observó en adultos con LPA.[28 ,59 ] Este síndrome presenta una incidencia máxima bimodal durante la primera y tercera semanas de la terapia de inducción.Dado que el síndrome de diferenciación se da con más frecuencia en pacientes de riesgo alto, se administra dexametasona con tretinoína o trióxido de arsénico para prevenir esta complicación.[
58 ] También se administra profilaxis con dexametasona e hidroxiurea (para citorreducción) en los pacientes de riesgo estándar si su recuento de GB se eleva hasta más de 10 000/µl después del inicio de la tretinoína o el arsénico. Si se presenta síndrome de diferenciación, es posible aumentar la dosis de dexametasona del paciente e interrumpir temporalmente la administración de tretinoína y trióxido de arsénico, y al igual que sucede con el pseudotumor cerebral, se reinician a dosis más bajas y se va aumentando la dosis según la tolerabilidad. Cuando se utilizó este abordaje en el ensayo del COG AAML1331 (NCT02339740), el 24,5 % de los pacientes de riesgo estándar y el 30,4 % de los pacientes de riesgo alto presentaron síndrome de diferenciación. Solo un paciente de riesgo estándar falleció por síndrome de diferenciación y coagulopatía.[29 ] - Coagulopatía. Junto con el síndrome de diferenciación, las complicaciones por coagulopatía aumentan el riesgo de muerte durante la inducción (muerte temprana en la LPA). Los blastocitos de la LPA inducen coagulopatía por activación de la cascada de coagulación (causada por la expresión del factor tisular y otros procoagulantes) con un aumento simultáneo de la fibrinólisis primaria y secundaria resultante de la expresión de anexina II en los blastocitos de la LPA. El riesgo de muerte por coagulopatía se relaciona con un recuento de GB elevado, disminución del recuento de plaquetas y estudios de coagulación anormales (fibrinógeno, tiempo de protrombina).[
12 ] En los estudios de adultos y niños se ha demostrado que los sistemas de puntuación que utilizan características clínicas y valores de laboratorio pueden ayudar a predecir el riesgo de desarrollar una coagulopatía grave o mortal.[60 ,61 ] Los cuidados médicos intensivos para corregir la coagulopatía, incluso antes de que aparezcan los signos y síntomas clínicos de hemorragia o trombosis, son importantes para prevenir la muerte prematura. - Prolongación del intervalo QT. El trióxido de arsénico se relaciona con una prolongación del intervalo QT que a veces produce arritmias potencialmente mortales (por ejemplo, torsades de pointes).[
62 ] Es fundamental vigilar de cerca los electrólitos en los pacientes que reciben trióxido de arsénico con el fin de mantener las concentraciones de potasio y magnesio dentro de intervalos normales, además se debe conocer que hay otros fármacos que se sabe que prolongan el intervalo QT.[63 ]
Vigilancia de la enfermedad mínima
En la actualidad, las terapias de inducción y consolidación que se usan, producen remisión molecular evaluada mediante RT-PCR para la proteína de fusión PML::RARA en la mayoría de los pacientes con LPA; se encuentran indicios de enfermedad molecular en el 1 % o menos al final de la terapia de consolidación.[
Los pacientes con enfermedad persistente o en recaída según la medición por RT-PCR de la proteína de fusión PML::RARA quizá obtengan beneficio de una intervención con terapias para recaídas [
Variantes moleculares de la leucemia promielocítica aguda además dePML::RARAy su efecto sobre el tratamiento
Las variantes moleculares poco frecuentes de LPA producen proteínas de fusión por la unión de parejas de genes específicos (por ejemplo, ZBTB16, NPM1, STAT5B, y NUMA1) con RARA.[
- Variante con la fusión ZBTB16::RARA. La variante con la fusión ZBTB16::RARA, caracterizada por t(11;17)(q23;q21), representa cerca del 0,8 % de los casos de LPA, expresa CD56 de superficie, y tiene gránulos diminutos en comparación con la LPA que tiene t(15;17).[
71 ,72 ,73 ] Los pacientes con LPA y fusiones ZBTB16::RARA tienen un pronóstico precario y por lo general no responden a la tretinoína o el trióxido de arsénico.[70 ,71 ,72 ,73 ] - Variantes con las fusiones NPM1::RARA o NUMA1::RARA. Es posible que las variantes infrecuentes de LPA que tienen las fusiones NPM1::RARA (t(5;17)(q35;q21)) o NUMA1::RARA (t(11;17)(q13;q21)) respondan bien a la tretinoína.[
70 ,74 ,75 ,76 ,77 ]
Tratamiento de la leucemia promielocítica aguda recidivante
En el pasado, del 10 % al 20 % de los pacientes con LPA recaían; no obstante, en estudios vigentes en los que se incorpora la terapia con trióxido de arsénico se observa una incidencia acumulada de recaída de menos del 5 %.[
En los pacientes que al inicio recibieron tratamientos con quimioterapia, la duración de la primera remisión constituyó un factor pronóstico en la LPA; los pacientes que recaen en el transcurso de 12 a 18 meses del diagnóstico inicial tienen el desenlace más precario.[
En ensayos previos, muchos niños que recayeron recibieron antraciclinas (las exposiciones oscilaron entre 400 mg/m2 y 750 mg/m2).[
Las opciones de tratamiento para los niños con LPA recidivante son las siguientes:
- Trióxido de arsénico o tretinoína.
- Gemtuzumab ozogamicina.
- Trasplante de células madre hematopoyéticas.
Trióxido de arsénico
Para los niños con LPA recidivante, se deberá considerar el uso de trióxido de arsénico en monoterapia o regímenes con tretinoína, según la terapia administrada durante la primera remisión. El trióxido de arsénico es un fármaco activo para los pacientes con LPA recidivante; cerca del 85 % al 94 % de los pacientes logran una remisión después del tratamiento con este fármaco.[
En los adultos con recaída de LPA, cerca del 85 % al 94 % logra la remisión morfológica después del tratamiento con trióxido de arsénico.[
Debido a que el trióxido de arsénico causa prolongación del intervalo QT que puede producir arritmias potencialmente mortales,[
Gemtuzumab ozogamicina
La administración en monoterapia de gemtuzumab ozogamicina, un anticuerpo monoclonal anti-CD33 y anti-caliqueamicina, produjo una remisión molecular en el 91 % (9 de 11 pacientes) después de 2 dosis y una remisión molecular en el 100 % (13 de 13 pacientes) después de 3 dosis, por lo tanto, se demostró una actividad excelente de este fármaco para la recaída de la LPA.[
Trasplante de células madre hematopoyéticas
En estudios retrospectivos del ámbito pediátrico se notificaron tasas semejantes de SSC a 5 años después de abordajes de trasplante autógeno o alogénico, de casi el 70 %.[
Evidencia (trasplante autógeno de células madre hematopoyéticas):
- En relación con el trasplante autógeno, en un estudio de pacientes adultos se demostró una mejora en la tasa de SSC a 7 años (77 % vs. 50 %) cuando en el paciente y en el producto de células madre se obtuvo un resultado negativo para el transcrito de fusión de leucemia promielocítica/receptor de ácido retinoico en una prueba de reacción en cadena de la polimerasa (remisión molecular) antes del trasplante.[
93 ] - En otro estudio, se demostró que de 7 pacientes sometidos a TCMH autógeno que obtuvieron un resultado positivo para enfermedad residual mínima (ERM) en el estudio celular, todos recayeron antes de 9 meses después del trasplante; sin embargo, solo recayó 1 paciente de los 8 que tenían células de un donante autógeno sin expresión de ERM.[
94 ] - En otro informe, se demostró que la tasa de SSC a 5 años fue del 83,3 % en los pacientes sometidos a TCMH autógeno durante la segunda remisión molecular y del 34,5 % en los pacientes que solo recibieron terapia de mantenimiento.[
95 ] - En otro informe retrospectivo se encontró que el 94 % de los pacientes pediátricos y adultos (64 de 67) con LPA en recaída, después de recibir de manera primaria trióxido de arsénico en monoterapia lograron una remisión molecular después del tratamiento con regímenes de reinducción que contienen arsénico. En los pacientes (n = 35) que recibieron consolidación posremisión con TCMH, la tasa de SG a 5 años fue del 90,3 % (± 5,3 %) y la tasa de SSC fue del 87,1 % (± 6,0 %). Estos desenlaces fueron significativamente superiores a los desenlaces de los pacientes que recibieron un régimen de mantenimiento con arsénico (n = 28); estos pacientes presentaron una tasa de SG a 5 años del 58,6 % (± 10,4 %) y una tasa de SSC del 47,7 % (± 10,3 %).[
85 ]
Estos datos respaldan el uso del trasplante autógeno en los pacientes que no presentan ERM durante la segunda RC y en cuyas extracciones de células madre no se observa ERM.
Es probable que no sea viable realizar ensayos clínicos sobre la recaída de la LPA con el fin de comparar abordajes de tratamiento porque la LPA es muy infrecuente en niños y la enfermedad tiene un desenlace favorable. No obstante, un grupo internacional de expertos emitió recomendaciones para el tratamiento de la recaída de la LPA a partir de informes de la experiencia con niños y adultos.[
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
- Melnick A, Licht JD: Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 93 (10): 3167-215, 1999.
- Sanz MA, Grimwade D, Tallman MS, et al.: Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 113 (9): 1875-91, 2009.
- Sanz MA, Lo-Coco F: Modern approaches to treating acute promyelocytic leukemia. J Clin Oncol 29 (5): 495-503, 2011.
- Falini B, Flenghi L, Fagioli M, et al.: Immunocytochemical diagnosis of acute promyelocytic leukemia (M3) with the monoclonal antibody PG-M3 (anti-PML). Blood 90 (10): 4046-53, 1997.
- Gomis F, Sanz J, Sempere A, et al.: Immunofluorescent analysis with the anti-PML monoclonal antibody PG-M3 for rapid and accurate genetic diagnosis of acute promyelocytic leukemia. Ann Hematol 83 (11): 687-90, 2004.
- Dimov ND, Medeiros LJ, Kantarjian HM, et al.: Rapid and reliable confirmation of acute promyelocytic leukemia by immunofluorescence staining with an antipromyelocytic leukemia antibody: the M. D. Anderson Cancer Center experience of 349 patients. Cancer 116 (2): 369-76, 2010.
- Tallman MS, Hakimian D, Kwaan HC, et al.: New insights into the pathogenesis of coagulation dysfunction in acute promyelocytic leukemia. Leuk Lymphoma 11 (1-2): 27-36, 1993.
- Altman JK, Rademaker A, Cull E, et al.: Administration of ATRA to newly diagnosed patients with acute promyelocytic leukemia is delayed contributing to early hemorrhagic death. Leuk Res 37 (9): 1004-9, 2013.
- Lehmann S, Ravn A, Carlsson L, et al.: Continuing high early death rate in acute promyelocytic leukemia: a population-based report from the Swedish Adult Acute Leukemia Registry. Leukemia 25 (7): 1128-34, 2011.
- Park JH, Qiao B, Panageas KS, et al.: Early death rate in acute promyelocytic leukemia remains high despite all-trans retinoic acid. Blood 118 (5): 1248-54, 2011.
- Abla O, Ribeiro RC, Testi AM, et al.: Predictors of thrombohemorrhagic early death in children and adolescents with t(15;17)-positive acute promyelocytic leukemia treated with ATRA and chemotherapy. Ann Hematol 96 (9): 1449-1456, 2017.
- Breen KA, Grimwade D, Hunt BJ: The pathogenesis and management of the coagulopathy of acute promyelocytic leukaemia. Br J Haematol 156 (1): 24-36, 2012.
- Visani G, Gugliotta L, Tosi P, et al.: All-trans retinoic acid significantly reduces the incidence of early hemorrhagic death during induction therapy of acute promyelocytic leukemia. Eur J Haematol 64 (3): 139-44, 2000.
- de Botton S, Coiteux V, Chevret S, et al.: Outcome of childhood acute promyelocytic leukemia with all-trans-retinoic acid and chemotherapy. J Clin Oncol 22 (8): 1404-12, 2004.
- Testi AM, Biondi A, Lo Coco F, et al.: GIMEMA-AIEOPAIDA protocol for the treatment of newly diagnosed acute promyelocytic leukemia (APL) in children. Blood 106 (2): 447-53, 2005.
- Ortega JJ, Madero L, Martín G, et al.: Treatment with all-trans retinoic acid and anthracycline monochemotherapy for children with acute promyelocytic leukemia: a multicenter study by the PETHEMA Group. J Clin Oncol 23 (30): 7632-40, 2005.
- Guglielmi C, Martelli MP, Diverio D, et al.: Immunophenotype of adult and childhood acute promyelocytic leukaemia: correlation with morphology, type of PML gene breakpoint and clinical outcome. A cooperative Italian study on 196 cases. Br J Haematol 102 (4): 1035-41, 1998.
- Sanz MA, Lo Coco F, Martín G, et al.: Definition of relapse risk and role of nonanthracycline drugs for consolidation in patients with acute promyelocytic leukemia: a joint study of the PETHEMA and GIMEMA cooperative groups. Blood 96 (4): 1247-53, 2000.
- Sanz MA, Martín G, González M, et al.: Risk-adapted treatment of acute promyelocytic leukemia with all-trans-retinoic acid and anthracycline monochemotherapy: a multicenter study by the PETHEMA group. Blood 103 (4): 1237-43, 2004.
- Lo-Coco F, Avvisati G, Vignetti M, et al.: Front-line treatment of acute promyelocytic leukemia with AIDA induction followed by risk-adapted consolidation for adults younger than 61 years: results of the AIDA-2000 trial of the GIMEMA Group. Blood 116 (17): 3171-9, 2010.
- Callens C, Chevret S, Cayuela JM, et al.: Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia 19 (7): 1153-60, 2005.
- Gale RE, Hills R, Pizzey AR, et al.: Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood 106 (12): 3768-76, 2005.
- Arrigoni P, Beretta C, Silvestri D, et al.: FLT3 internal tandem duplication in childhood acute myeloid leukaemia: association with hyperleucocytosis in acute promyelocytic leukaemia. Br J Haematol 120 (1): 89-92, 2003.
- Noguera NI, Breccia M, Divona M, et al.: Alterations of the FLT3 gene in acute promyelocytic leukemia: association with diagnostic characteristics and analysis of clinical outcome in patients treated with the Italian AIDA protocol. Leukemia 16 (11): 2185-9, 2002.
- Tallman MS, Kim HT, Montesinos P, et al.: Does microgranular variant morphology of acute promyelocytic leukemia independently predict a less favorable outcome compared with classical M3 APL? A joint study of the North American Intergroup and the PETHEMA Group. Blood 116 (25): 5650-9, 2010.
- Iland HJ, Bradstock K, Supple SG, et al.: All-trans-retinoic acid, idarubicin, and IV arsenic trioxide as initial therapy in acute promyelocytic leukemia (APML4). Blood 120 (8): 1570-80; quiz 1752, 2012.
- Kutny MA, Moser BK, Laumann K, et al.: FLT3 mutation status is a predictor of early death in pediatric acute promyelocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 59 (4): 662-7, 2012.
- Kutny MA, Alonzo TA, Gerbing RB, et al.: Arsenic Trioxide Consolidation Allows Anthracycline Dose Reduction for Pediatric Patients With Acute Promyelocytic Leukemia: Report From the Children's Oncology Group Phase III Historically Controlled Trial AAML0631. J Clin Oncol 35 (26): 3021-3029, 2017.
- Kutny MA, Alonzo TA, Abla O, et al.: Assessment of Arsenic Trioxide and All-trans Retinoic Acid for the Treatment of Pediatric Acute Promyelocytic Leukemia: A Report From the Children's Oncology Group AAML1331 Trial. JAMA Oncol 8 (1): 79-87, 2022.
- de Botton S, Sanz MA, Chevret S, et al.: Extramedullary relapse in acute promyelocytic leukemia treated with all-trans retinoic acid and chemotherapy. Leukemia 20 (1): 35-41, 2006.
- Montesinos P, Díaz-Mediavilla J, Debén G, et al.: Central nervous system involvement at first relapse in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline monochemotherapy without intrathecal prophylaxis. Haematologica 94 (9): 1242-9, 2009.
- Chow J, Feusner J: Isolated central nervous system recurrence of acute promyelocytic leukemia in children. Pediatr Blood Cancer 52 (1): 11-3, 2009.
- Kaspers G, Gibson B, Grimwade D, et al.: Central nervous system involvement in relapsed acute promyelocytic leukemia. Pediatr Blood Cancer 53 (2): 235-6; author reply 237, 2009.
- Lo-Coco F, Avvisati G, Vignetti M, et al.: Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 369 (2): 111-21, 2013.
- Platzbecker U, Avvisati G, Cicconi L, et al.: Improved Outcomes With Retinoic Acid and Arsenic Trioxide Compared With Retinoic Acid and Chemotherapy in Non-High-Risk Acute Promyelocytic Leukemia: Final Results of the Randomized Italian-German APL0406 Trial. J Clin Oncol 35 (6): 605-612, 2017.
- Creutzig U, Dworzak MN, Bochennek K, et al.: First experience of the AML-Berlin-Frankfurt-Münster group in pediatric patients with standard-risk acute promyelocytic leukemia treated with arsenic trioxide and all-trans retinoid acid. Pediatr Blood Cancer 64 (8): , 2017.
- Yang MH, Wan WQ, Luo JS, et al.: Multicenter randomized trial of arsenic trioxide and Realgar-Indigo naturalis formula in pediatric patients with acute promyelocytic leukemia: Interim results of the SCCLG-APL clinical study. Am J Hematol 93 (12): 1467-1473, 2018.
- Zhang L, Zou Y, Chen Y, et al.: Role of cytarabine in paediatric acute promyelocytic leukemia treated with the combination of all-trans retinoic acid and arsenic trioxide: a randomized controlled trial. BMC Cancer 18 (1): 374, 2018.
- Zheng H, Jiang H, Hu S, et al.: Arsenic Combined With All-Trans Retinoic Acid for Pediatric Acute Promyelocytic Leukemia: Report From the CCLG-APL2016 Protocol Study. J Clin Oncol 39 (28): 3161-3170, 2021.
- Altucci L, Rossin A, Raffelsberger W, et al.: Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat Med 7 (6): 680-6, 2001.
- Huang ME, Ye YC, Chen SR, et al.: Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 72 (2): 567-72, 1988.
- Castaigne S, Chomienne C, Daniel MT, et al.: All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood 76 (9): 1704-9, 1990.
- Imaizumi M, Tawa A, Hanada R, et al.: Prospective study of a therapeutic regimen with all-trans retinoic acid and anthracyclines in combination of cytarabine in children with acute promyelocytic leukaemia: the Japanese childhood acute myeloid leukaemia cooperative study. Br J Haematol 152 (1): 89-98, 2011.
- Gregory J, Kim H, Alonzo T, et al.: Treatment of children with acute promyelocytic leukemia: results of the first North American Intergroup trial INT0129. Pediatr Blood Cancer 53 (6): 1005-10, 2009.
- Testi AM, Pession A, Diverio D, et al.: Risk-adapted treatment of acute promyelocytic leukemia: results from the International Consortium for Childhood APL. Blood 132 (4): 405-412, 2018.
- Sanz MA, Montesinos P, Rayón C, et al.: Risk-adapted treatment of acute promyelocytic leukemia based on all-trans retinoic acid and anthracycline with addition of cytarabine in consolidation therapy for high-risk patients: further improvements in treatment outcome. Blood 115 (25): 5137-46, 2010.
- Powell BL, Moser B, Stock W, et al.: Arsenic trioxide improves event-free and overall survival for adults with acute promyelocytic leukemia: North American Leukemia Intergroup Study C9710. Blood 116 (19): 3751-7, 2010.
- Shen ZX, Shi ZZ, Fang J, et al.: All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A 101 (15): 5328-35, 2004.
- Ravandi F, Estey E, Jones D, et al.: Effective treatment of acute promyelocytic leukemia with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab ozogamicin. J Clin Oncol 27 (4): 504-10, 2009.
- Hu J, Liu YF, Wu CF, et al.: Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A 106 (9): 3342-7, 2009.
- Cheng Y, Zhang L, Wu J, et al.: Long-term prognosis of childhood acute promyelocytic leukaemia with arsenic trioxide administration in induction and consolidation chemotherapy phases: a single-centre experience. Eur J Haematol 91 (6): 483-9, 2013.
- Wang H, Chen XY, Wang BS, et al.: The efficacy and safety of arsenic trioxide with or without all-trans retinoic acid for the treatment of acute promyelocytic leukemia: a meta-analysis. Leuk Res 35 (9): 1170-7, 2011.
- Zhang L, Zhao H, Zhu X, et al.: Retrospective analysis of 65 Chinese children with acute promyelocytic leukemia: a single center experience. Pediatr Blood Cancer 51 (2): 210-5, 2008.
- Zhou J, Zhang Y, Li J, et al.: Single-agent arsenic trioxide in the treatment of children with newly diagnosed acute promyelocytic leukemia. Blood 115 (9): 1697-702, 2010.
- Iland HJ, Collins M, Bradstock K, et al.: Use of arsenic trioxide in remission induction and consolidation therapy for acute promyelocytic leukaemia in the Australasian Leukaemia and Lymphoma Group (ALLG) APML4 study: a non-randomised phase 2 trial. Lancet Haematol 2 (9): e357-66, 2015.
- Douer D, Zickl LN, Schiffer CA, et al.: All-trans retinoic acid and late relapses in acute promyelocytic leukemia: very long-term follow-up of the North American Intergroup Study I0129. Leuk Res 37 (7): 795-801, 2013.
- Coombs CC, DeAngelis LM, Feusner JH, et al.: Pseudotumor Cerebri in Acute Promyelocytic Leukemia Patients on Intergroup Protocol 0129: Clinical Description and Recommendations for New Diagnostic Criteria. Clin Lymphoma Myeloma Leuk 16 (3): 146-51, 2016.
- Sanz MA, Montesinos P: How we prevent and treat differentiation syndrome in patients with acute promyelocytic leukemia. Blood 123 (18): 2777-82, 2014.
- Montesinos P, Bergua JM, Vellenga E, et al.: Differentiation syndrome in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline chemotherapy: characteristics, outcome, and prognostic factors. Blood 113 (4): 775-83, 2009.
- Mitrovic M, Suvajdzic N, Bogdanovic A, et al.: International Society of Thrombosis and Hemostasis Scoring System for disseminated intravascular coagulation ≥ 6: a new predictor of hemorrhagic early death in acute promyelocytic leukemia. Med Oncol 30 (1): 478, 2013.
- Rajpurkar M, Alonzo TA, Wang YC, et al.: Risk Markers for Significant Bleeding and Thrombosis in Pediatric Acute Promyelocytic Leukemia; Report From the Children's Oncology Group Study AAML0631. J Pediatr Hematol Oncol 41 (1): 51-55, 2019.
- Unnikrishnan D, Dutcher JP, Varshneya N, et al.: Torsades de pointes in 3 patients with leukemia treated with arsenic trioxide. Blood 97 (5): 1514-6, 2001.
- Barbey JT: Cardiac toxicity of arsenic trioxide. Blood 98 (5): 1632; discussion 1633-4, 2001.
- Jurcic JG, Nimer SD, Scheinberg DA, et al.: Prognostic significance of minimal residual disease detection and PML/RAR-alpha isoform type: long-term follow-up in acute promyelocytic leukemia. Blood 98 (9): 2651-6, 2001.
- Diverio D, Rossi V, Avvisati G, et al.: Early detection of relapse by prospective reverse transcriptase-polymerase chain reaction analysis of the PML/RARalpha fusion gene in patients with acute promyelocytic leukemia enrolled in the GIMEMA-AIEOP multicenter "AIDA" trial. GIMEMA-AIEOP Multicenter "AIDA" Trial. Blood 92 (3): 784-9, 1998.
- Lo Coco F, Diverio D, Avvisati G, et al.: Therapy of molecular relapse in acute promyelocytic leukemia. Blood 94 (7): 2225-9, 1999.
- Esteve J, Escoda L, Martín G, et al.: Outcome of patients with acute promyelocytic leukemia failing to front-line treatment with all-trans retinoic acid and anthracycline-based chemotherapy (PETHEMA protocols LPA96 and LPA99): benefit of an early intervention. Leukemia 21 (3): 446-52, 2007.
- Zelent A, Guidez F, Melnick A, et al.: Translocations of the RARalpha gene in acute promyelocytic leukemia. Oncogene 20 (49): 7186-203, 2001.
- Yan W, Zhang G: Molecular Characteristics and Clinical Significance of 12 Fusion Genes in Acute Promyelocytic Leukemia: A Systematic Review. Acta Haematol 136 (1): 1-15, 2016.
- Rego EM, Ruggero D, Tribioli C, et al.: Leukemia with distinct phenotypes in transgenic mice expressing PML/RAR alpha, PLZF/RAR alpha or NPM/RAR alpha. Oncogene 25 (13): 1974-9, 2006.
- Licht JD, Chomienne C, Goy A, et al.: Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood 85 (4): 1083-94, 1995.
- Guidez F, Ivins S, Zhu J, et al.: Reduced retinoic acid-sensitivities of nuclear receptor corepressor binding to PML- and PLZF-RARalpha underlie molecular pathogenesis and treatment of acute promyelocytic leukemia. Blood 91 (8): 2634-42, 1998.
- Grimwade D, Biondi A, Mozziconacci MJ, et al.: Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Groupe Français de Cytogénétique Hématologique, Groupe de Français d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action "Molecular Cytogenetic Diagnosis in Haematological Malignancies". Blood 96 (4): 1297-308, 2000.
- Sukhai MA, Wu X, Xuan Y, et al.: Myeloid leukemia with promyelocytic features in transgenic mice expressing hCG-NuMA-RARalpha. Oncogene 23 (3): 665-78, 2004.
- Redner RL, Corey SJ, Rush EA: Differentiation of t(5;17) variant acute promyelocytic leukemic blasts by all-trans retinoic acid. Leukemia 11 (7): 1014-6, 1997.
- Wells RA, Catzavelos C, Kamel-Reid S: Fusion of retinoic acid receptor alpha to NuMA, the nuclear mitotic apparatus protein, by a variant translocation in acute promyelocytic leukaemia. Nat Genet 17 (1): 109-13, 1997.
- Wells RA, Hummel JL, De Koven A, et al.: A new variant translocation in acute promyelocytic leukaemia: molecular characterization and clinical correlation. Leukemia 10 (4): 735-40, 1996.
- Marjerrison S, Antillon F, Bonilla M, et al.: Outcome of children treated for relapsed acute myeloid leukemia in Central America. Pediatr Blood Cancer 61 (7): 1222-6, 2014.
- Lengfelder E, Lo-Coco F, Ades L, et al.: Arsenic trioxide-based therapy of relapsed acute promyelocytic leukemia: registry results from the European LeukemiaNet. Leukemia 29 (5): 1084-91, 2015.
- Holter Chakrabarty JL, Rubinger M, Le-Rademacher J, et al.: Autologous is superior to allogeneic hematopoietic cell transplantation for acute promyelocytic leukemia in second complete remission. Biol Blood Marrow Transplant 20 (7): 1021-5, 2014.
- Fox E, Razzouk BI, Widemann BC, et al.: Phase 1 trial and pharmacokinetic study of arsenic trioxide in children and adolescents with refractory or relapsed acute leukemia, including acute promyelocytic leukemia or lymphoma. Blood 111 (2): 566-73, 2008.
- Niu C, Yan H, Yu T, et al.: Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow-up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patients. Blood 94 (10): 3315-24, 1999.
- Shen ZX, Chen GQ, Ni JH, et al.: Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 89 (9): 3354-60, 1997.
- Avvisati G, Lo-Coco F, Paoloni FP, et al.: AIDA 0493 protocol for newly diagnosed acute promyelocytic leukemia: very long-term results and role of maintenance. Blood 117 (18): 4716-25, 2011.
- Fouzia NA, Sharma V, Ganesan S, et al.: Management of relapse in acute promyelocytic leukaemia treated with up-front arsenic trioxide-based regimens. Br J Haematol 192 (2): 292-299, 2021.
- Lu J, Huang X, Bao L, et al.: Treatment outcomes in relapsed acute promyelocytic leukemia patients initially treated with all-trans retinoic acid and arsenic compound-based combined therapies. Oncol Lett 7 (1): 177-182, 2014.
- Zhu HH, Qin YZ, Huang XJ: Resistance to arsenic therapy in acute promyelocytic leukemia. N Engl J Med 370 (19): 1864-6, 2014.
- Soignet SL, Maslak P, Wang ZG, et al.: Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med 339 (19): 1341-8, 1998.
- Zhang P: The use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia. J Biol Regul Homeost Agents 13 (4): 195-200, 1999 Oct-Dec.
- Lo-Coco F, Cimino G, Breccia M, et al.: Gemtuzumab ozogamicin (Mylotarg) as a single agent for molecularly relapsed acute promyelocytic leukemia. Blood 104 (7): 1995-9, 2004.
- Dvorak CC, Agarwal R, Dahl GV, et al.: Hematopoietic stem cell transplant for pediatric acute promyelocytic leukemia. Biol Blood Marrow Transplant 14 (7): 824-30, 2008.
- Bourquin JP, Thornley I, Neuberg D, et al.: Favorable outcome of allogeneic hematopoietic stem cell transplantation for relapsed or refractory acute promyelocytic leukemia in childhood. Bone Marrow Transplant 34 (9): 795-8, 2004.
- de Botton S, Fawaz A, Chevret S, et al.: Autologous and allogeneic stem-cell transplantation as salvage treatment of acute promyelocytic leukemia initially treated with all-trans-retinoic acid: a retrospective analysis of the European acute promyelocytic leukemia group. J Clin Oncol 23 (1): 120-6, 2005.
- Meloni G, Diverio D, Vignetti M, et al.: Autologous bone marrow transplantation for acute promyelocytic leukemia in second remission: prognostic relevance of pretransplant minimal residual disease assessment by reverse-transcription polymerase chain reaction of the PML/RAR alpha fusion gene. Blood 90 (3): 1321-5, 1997.
- Thirugnanam R, George B, Chendamarai E, et al.: Comparison of clinical outcomes of patients with relapsed acute promyelocytic leukemia induced with arsenic trioxide and consolidated with either an autologous stem cell transplant or an arsenic trioxide-based regimen. Biol Blood Marrow Transplant 15 (11): 1479-84, 2009.
- Abla O, Kutny MA, Testi AM, et al.: Management of relapsed and refractory childhood acute promyelocytic leukaemia: recommendations from an international expert panel. Br J Haematol 175 (4): 588-601, 2016.
Proliferaciones mieloides relacionadas con el síndrome de Down
Las leucemias mieloides que se presentan en niños con síndrome de Down, en particular en los pacientes menores de 4 años, pertenecen a un subgrupo diferente de LMA que se caracteriza por la coexistencia de trisomía 21 y mutaciones en GATA1 en los blastocitos leucémicos que con frecuencia, aunque no siempre, son megacarioblásticos. Esta leucemia específica se subdivide además en 2 versiones: una versión transitoria en recién nacidos y niños pequeños llamada mielopoyesis anormal transitoria (MAT), que remite espontáneamente con el tiempo; y una versión persistente pero quimiosensible que aparece más tarde, entre los 90 días y los 3 años de edad.[
Mielopoyesis anormal transitoria relacionada con el síndrome de Down
Alrededor del 10 % de los neonatos con síndrome de Down presentan mielopoyesis anormal transitoria (MAT) (también conocida como trastorno mieloproliferativo transitorio [TMT]).[
Aunque la MAT por lo general es una afección que resuelve por sí sola, se relaciona con morbilidad significativa y es mortal en el 10 % al 17 % de los lactantes afectados.[
A continuación se enumeran tres grupos de riesgo identificados a partir de las manifestaciones clínicas diagnósticas de hepatomegalia con síntomas potencialmente mortales o sin estos:[
- El riesgo bajo incluye los pacientes sin hepatomegalia ni síntomas potencialmente mortales (38 % de los pacientes, tasa de supervivencia general [SG] del 92 ± 8 %).
- El riesgo intermedio incluye los pacientes con hepatomegalia sola (40 % de los pacientes, y tasa de SG del 77 ± 12 %).
- El riesgo alto incluye los pacientes con hepatomegalia y síntomas potencialmente mortales (21 % de los pacientes, tasa de SG del 51 ± 19 %).
Las intervenciones terapéuticas se justifican en los pacientes con edema grave evidente o insuficiencia orgánica. Debido a que la MAT con el tiempo remite de manera espontánea, el tratamiento es corto y se orienta en primer lugar a la reducción de la carga leucémica y el alivio de los síntomas inmediatos. Se han utilizado múltiples abordajes de tratamiento, como los siguientes:
- Exanguinotransfusión.
- Leucocitaféresis.
- Dosis bajas de citarabina. De estos abordajes, se encontró que solo la citarabina disminuye de manera uniforme las complicaciones de la MAT y la mortalidad relacionada.[
5 ,8 ]; [9 ][Nivel de evidencia B4] La dosificación de citarabina oscila entre 0,4 y 1,5 mg/kg por dosis administrada por vía intravenosa (IV) o subcutánea (SC) 1 o 2 veces al día durante 4 a 12 días,[8 ] con eficacia similar y menos toxicidad que las dosis más altas administradas por infusiones durante 5 días continuos, que producen neutropenia grave prolongada.[5 ] En un ensayo prospectivo donde se utilizó la citarabina en dosis de 1,5 mg/kg por día IV o SC durante 7 días en pacientes sintomáticos, se notificó una reducción significativa de las muertes prematuras en comparación con los controles históricos similares (12 % ± 5 % vs. 33 % ± 7 %, respectivamente; P = 0,2).[9 ][Nivel de evidencia B4]
La presentación posterior de leucemia mieloide relacionada con el síndrome de Down se observa en el 10 % al 30 % de los niños que tienen remisiones espontáneas de MAT; se informó que ocurre a una media de edad de 16 meses (intervalo, 1–30 meses).[
Leucemia mieloide relacionada con el síndrome de Down
Los niños con síndrome de Down tienen un riesgo de leucemia 10 a 45 veces superior en comparación con los niños sin síndrome de Down.[
Pronóstico y tratamiento de los niños con síndrome de Down y leucemia mieloide aguda
Por lo general, el desenlace es favorable para los niños con síndrome de Down que presentan LMA (en la clasificación de la Organización Mundial de la Salud se la llama leucemia mieloide relacionada con el síndrome de Down).[
Los factores pronósticos de los niños con síndrome de Down y LMA son los siguientes:
- Edad. El pronóstico es particularmente bueno (tasas de SSC de más del 85 %) en los niños de 4 años o menos en el momento del diagnóstico; este grupo de edad representa la gran mayoría de los pacientes con síndrome de Down y LMA.[
20 ,21 ,22 ,23 ] El pronóstico de los niños con síndrome de Down que tienen más de 4 años es mucho más precario, lo que exige administrar el tratamiento que se usa en niños con LMA sin síndrome de Down, a menos que se encuentre una mutación en GATA1.[24 ] - Recuento de glóbulos blancos. En un estudio retrospectivo numeroso del Berlin-Frankfurt-Münster (BFM) de 451 niños con LMA y síndrome de Down (edad >6 meses y <5 años) se observó que la tasa de SSC a 7 años fue del 78 % y la tasa de SG a 7 años fue del 79 %. En un análisis multivariante, el recuento de GB (≥20 × 109 /l) y la edad (>3 años) fueron factores pronósticos independientes de una SSC más baja. La tasa de SSC a 7 años para la población de mayor edad (>3 años) y para la población con el mayor recuento de GB incluso superó el 60 %.[
25 ] - Cariotipo de LMA. La LMA con cariotipo normal (excepto la trisomía 21) que se observa en alrededor del 29 % de los pacientes, fue un factor pronóstico independiente de SG y SSC inferiores (tasa de SSC a 7 años del 65 % en comparación con el 82 % de los pacientes con cariotipos anómalos).[
25 ] No obstante, ello no se comprobó en un ensayo posterior.[23 ] En este mismo ensayo, se observó que la presencia de trisomía 8 afectó de manera adversa el pronóstico. En otro estudio, los cariotipos complejos (≥3 anormalidades independientes) fueron pronósticos de aumento en la incidencia acumulada de recaída a los 2 años (un 30,8 % en comparación con un 7,5 % en pacientes sin cariotipos complejos; P = 0,001).[26 ] - ERM. Se encontró que la ERM al final de la inducción 1 fue un factor pronóstico importante;[
21 ,27 ] este resultado fue compatible con el hallazgo del BFM que indica que la respuesta temprana se correlaciona con una mejora de la SG.[23 ] No obstante, una ERM negativa al final de la inducción 1 no definió un grupo de pacientes que podría recibir quimioterapia reducida.[26 ]
Entre el 29 % y 47 % de los pacientes con síndrome de Down manifiestan al inicio síndromes mielodisplásicos (SMD) (<20 % de blastocitos), pero sus desenlaces son similares a los de los pacientes con LMA.[
Las opciones de tratamiento para los niños con síndrome de Down y leucemia mieloide aguda recién diagnosticada son las siguientes:
- Quimioterapia.
El tratamiento apropiado para los niños más pequeños (edad ≤4 años) con síndrome de Down y LMA es menos intenso que el tratamiento estándar vigente para la LMA infantil. El trasplante de células madre hematopoyéticas no está indicado durante la primera remisión.[
Evidencia (quimioterapia):
- En un ensayo del Children's Oncology Group (COG) de niños con síndrome de Down y LMA recién diagnosticada (AAML0431 [NCT00369317]), se inscribieron 204 niños para recibir un régimen de dosis altas de citarabina durante el segundo de 4 ciclos de inducción (reduciendo la exposición acumulada a la antraciclina de 320 a 240 mg) en lugar del ciclo de intensificación usado en el ensayo anterior del COG A2971 (NCT00003593).[
20 ,21 ] Se redujeron las dosis intratecales de 7 a 2 inyecciones en total y la intensificación incluyó 2 ciclos de citarabina y etopósido.- Cuando se comparó este ensayo con el ensayo anterior, estos cambios provocaron una mejoría general de casi el 10 %.
- La tasa de SSC a 4 años fue del 89,9 % y la tasa de SG fue del 93 %.
- Se presentó recaída en 14 pacientes y 2 muertes relacionadas con el tratamiento, ambos casos vinculados con neumonía y no ocurrieron durante la inducción 2.
- Ningún paciente tenía compromiso del sistema nervioso central (SNC) en este ensayo ni en el ensayo anterior del COG A2971 (NCT00003593).[
20 ] - El único factor pronóstico que se identificó fue la ERM mediante citometría de flujo en el día 28 de la inducción 1. Entre los pacientes que obtuvieron un resultado negativo para ERM (≤0,01 %), la tasa de SSE fue del 92,7 %; en el 14,4 % de los pacientes que obtuvieron un resultado positivo para la ERM, la tasa de SSE fue del 76,2 % (P = 0,011).
- En un ensayo conjunto (ML-DS 2006) del BFM, el Dutch Childhood Oncology Group (DCOG) y la Nordic Society of Pediatric Hematology and Oncology (NOPHO), se inscribieron 170 niños con síndrome de Down para participar en un ensayo cuyo objetivo fue reducir el tratamiento al eliminar el etopósido durante la consolidación, disminuir el número de dosis intratecales de 11 a 4 y eliminar el mantenimiento del grupo de terapia reducida de síndrome de Down del AML-BFM 98.[
23 ] Así como en los ensayos del COG, ningún paciente tenía enfermedad en el SNC en el momento del diagnóstico.- Los desenlaces no fueron peores a pesar de la reducción de la quimioterapia. La tasa de SG fue del 89 % ± 3 % y la tasa de SSC fue del 87 % ± 3 %, similar a la que se observó en el ensayo AML-BFM 98 (SG, 90 ± 4 % [P = NS]; SSC, 89 % ± 4 % [P = NS]). La incidencia acumulada de recaída (IAR) fue del 6 % en ambos ensayos.
- Recayeron 9 pacientes, y de ellos, 7 murieron.
- Los pacientes con buena respuesta temprana (<5 % de blastocitos en la evaluación morfológica antes del ciclo de inducción 2, n = 123 [72 %]) presentaron mejores desenlaces (tasa de SG, 92 % ± 3 % vs. 57 % ± 16 %, P < 0,0001; tasa de SSC, 88 % ± 3 % vs. 58 % ± 16 %, P = 0,0008; y tasa de IAR, 3 % ± 2 % vs. 27 % ± 18 %, P = 0,003).
- Se observaron menos efectos tóxicos en este ensayo nuevo, y la mortalidad relacionada con el tratamiento permaneció baja (2,9 % vs. 5 %, P = 0,276).
Se identificaron los siguientes dos factores de pronósticos:[
23 ]- La trisomía 8 fue un factor adverso (n = 37; tasa de SG, 95 % vs. 95%, P = 0,07; tasa de SSC, 73 % ± 8 % vs. 91 % ± 4 %, P = 0,018; tasa de IAR, 16 % ± 7 % vs. 3 % ± 2 %, P = 0,02).
- Esto se confirmó en un análisis multivariante en donde la falta de respuesta temprana adecuada y la trisomía 8 mantuvieron su efecto adverso sobre la recaída, con riesgos relativos de 8,55 (intervalo de confianza [IC] del 95 % , 1,96–37,29, P = 0,004) y 4,36 (1,24–15,39, P = 0,022), respectivamente.
Los niños con mosaicismo por trisomía 21 se tratan de manera similar a los niños con síndrome de Down evidente desde el punto de vista clínico.[
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
Enfermedad resistente al tratamiento o en recaída en niños con síndrome de Down
En un número pequeño de publicaciones se abordan los desenlaces en niños con síndrome de Down que recaen luego de la terapia inicial o que tienen LMA resistente al tratamiento. En 3 ensayos prospectivos de niños con síndrome de Down y diagnóstico nuevo de LMA, los desenlaces fueron precarios en quienes recayeron (sobrevivieron 4 de 11, 2 de 9 y 2 de 12 pacientes que recayeron).[
Las opciones de tratamiento para los niños con síndrome de Down y LMA resistente al tratamiento o en recaída son las siguientes:
- Quimioterapia, que a veces se sigue de un TCMH alogénico.
Evidencia (tratamiento de los niños con síndrome de Down y leucemia mieloide aguda resistente al tratamiento o en recaída):
- El Japanese Pediatric Leukemia/Lymphoma Study Group notificó los desenlaces de 29 pacientes con síndrome de Down y leucemia mieloide aguda (LMA) en recaída (n = 26) o resistente al tratamiento (n = 3). Como era de esperarse con el síndrome de Down, los niños de esta cohorte eran muy pequeños (mediana de edad, 2 años); casi todas las recaídas fueron tempranas (mediana de 8,6 meses, 80 % <12 meses desde el diagnóstico) y el 89 % eran M7, según la clasificación French-American-British.[
29 ][Nivel de evidencia C1]- A diferencia de los excelentes resultados logrados luego de la terapia inicial, solo el 50 % de los niños lograron una segunda remisión y la tasa de SG a 3 años fue del 26 %.
- Cerca de la mitad de los niños se sometió a trasplante alogénico y no se observaron ventajas del trasplante en comparación con la quimioterapia, pero el número de pacientes fue reducido.
- En un estudio del Center for International Blood and Marrow Transplant Research de niños con síndrome de Down y LMA sometidos a TCMH, se informaron los siguientes resultados:[
30 ][Nivel de evidencia C1]- Un desenlace precario similar, con una tasa de SG a 3 años del 19 %.
- La causa principal del fracaso después del trasplante fue la recaída, que excedió el 60 %; y la mortalidad relacionada con el trasplante fue de cerca del 20 %.
- En un estudio de un registro japonés, se notificó una mejor supervivencia después del trasplante en niños con síndrome de Down cuando recibieron regímenes de acondicionamiento de intensidad reducida, en comparación con abordajes mielosupresores, pero el número de pacientes fue muy reducido (n = 5) y se necesitan más estudios sobre la eficacia de los abordajes de intensidad reducida en los niños con síndrome de Down y LMA.[
31 ][Nivel de evidencia C2]
Referencias:
- Lange B: The management of neoplastic disorders of haematopoiesis in children with Down's syndrome. Br J Haematol 110 (3): 512-24, 2000.
- Gamis AS, Smith FO: Transient myeloproliferative disorder in children with Down syndrome: clarity to this enigmatic disorder. Br J Haematol 159 (3): 277-87, 2012.
- de Rooij JD, Branstetter C, Ma J, et al.: Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet 49 (3): 451-456, 2017.
- Homans AC, Verissimo AM, Vlacha V: Transient abnormal myelopoiesis of infancy associated with trisomy 21. Am J Pediatr Hematol Oncol 15 (4): 392-9, 1993.
- Gamis AS, Alonzo TA, Gerbing RB, et al.: Natural history of transient myeloproliferative disorder clinically diagnosed in Down syndrome neonates: a report from the Children's Oncology Group Study A2971. Blood 118 (26): 6752-9; quiz 6996, 2011.
- Massey GV, Zipursky A, Chang MN, et al.: A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481. Blood 107 (12): 4606-13, 2006.
- Muramatsu H, Kato K, Watanabe N, et al.: Risk factors for early death in neonates with Down syndrome and transient leukaemia. Br J Haematol 142 (4): 610-5, 2008.
- Klusmann JH, Creutzig U, Zimmermann M, et al.: Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood 111 (6): 2991-8, 2008.
- Flasinski M, Scheibke K, Zimmermann M, et al.: Low-dose cytarabine to prevent myeloid leukemia in children with Down syndrome: TMD Prevention 2007 study. Blood Adv 2 (13): 1532-1540, 2018.
- Ravindranath Y, Abella E, Krischer JP, et al.: Acute myeloid leukemia (AML) in Down's syndrome is highly responsive to chemotherapy: experience on Pediatric Oncology Group AML Study 8498. Blood 80 (9): 2210-4, 1992.
- Marlow EC, Ducore J, Kwan ML, et al.: Leukemia Risk in a Cohort of 3.9 Million Children with and without Down Syndrome. J Pediatr 234: 172-180.e3, 2021.
- Ravindranath Y: Down syndrome and leukemia: new insights into the epidemiology, pathogenesis, and treatment. Pediatr Blood Cancer 44 (1): 1-7, 2005.
- Ross JA, Spector LG, Robison LL, et al.: Epidemiology of leukemia in children with Down syndrome. Pediatr Blood Cancer 44 (1): 8-12, 2005.
- Gamis AS: Acute myeloid leukemia and Down syndrome evolution of modern therapy--state of the art review. Pediatr Blood Cancer 44 (1): 13-20, 2005.
- Taub JW, Ge Y: Down syndrome, drug metabolism and chromosome 21. Pediatr Blood Cancer 44 (1): 33-9, 2005.
- Crispino JD: GATA1 mutations in Down syndrome: implications for biology and diagnosis of children with transient myeloproliferative disorder and acute megakaryoblastic leukemia. Pediatr Blood Cancer 44 (1): 40-4, 2005.
- Ge Y, Stout ML, Tatman DA, et al.: GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst 97 (3): 226-31, 2005.
- Kudo K, Hama A, Kojima S, et al.: Mosaic Down syndrome-associated acute myeloid leukemia does not require high-dose cytarabine treatment for induction and consolidation therapy. Int J Hematol 91 (4): 630-5, 2010.
- Lange BJ, Kobrinsky N, Barnard DR, et al.: Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: Children's Cancer Group Studies 2861 and 2891. Blood 91 (2): 608-15, 1998.
- Sorrell AD, Alonzo TA, Hilden JM, et al.: Favorable survival maintained in children who have myeloid leukemia associated with Down syndrome using reduced-dose chemotherapy on Children's Oncology Group trial A2971: a report from the Children's Oncology Group. Cancer 118 (19): 4806-14, 2012.
- Taub JW, Berman JN, Hitzler JK, et al.: Improved outcomes for myeloid leukemia of Down syndrome: a report from the Children's Oncology Group AAML0431 trial. Blood 129 (25): 3304-3313, 2017.
- Creutzig U, Reinhardt D, Diekamp S, et al.: AML patients with Down syndrome have a high cure rate with AML-BFM therapy with reduced dose intensity. Leukemia 19 (8): 1355-60, 2005.
- Uffmann M, Rasche M, Zimmermann M, et al.: Therapy reduction in patients with Down syndrome and myeloid leukemia: the international ML-DS 2006 trial. Blood 129 (25): 3314-3321, 2017.
- Gamis AS, Woods WG, Alonzo TA, et al.: Increased age at diagnosis has a significantly negative effect on outcome in children with Down syndrome and acute myeloid leukemia: a report from the Children's Cancer Group Study 2891. J Clin Oncol 21 (18): 3415-22, 2003.
- Blink M, Zimmermann M, von Neuhoff C, et al.: Normal karyotype is a poor prognostic factor in myeloid leukemia of Down syndrome: a retrospective, international study. Haematologica 99 (2): 299-307, 2014.
- Hitzler J, Alonzo T, Gerbing R, et al.: High-dose AraC is essential for the treatment of ML-DS independent of postinduction MRD: results of the COG AAML1531 trial. Blood 138 (23): 2337-2346, 2021.
- Taga T, Tanaka S, Hasegawa D, et al.: Post-induction MRD by FCM and GATA1-PCR are significant prognostic factors for myeloid leukemia of Down syndrome. Leukemia 35 (9): 2508-2516, 2021.
- Taga T, Shimomura Y, Horikoshi Y, et al.: Continuous and high-dose cytarabine combined chemotherapy in children with down syndrome and acute myeloid leukemia: Report from the Japanese children's cancer and leukemia study group (JCCLSG) AML 9805 down study. Pediatr Blood Cancer 57 (1): 36-40, 2011.
- Taga T, Saito AM, Kudo K, et al.: Clinical characteristics and outcome of refractory/relapsed myeloid leukemia in children with Down syndrome. Blood 120 (9): 1810-5, 2012.
- Hitzler JK, He W, Doyle J, et al.: Outcome of transplantation for acute myelogenous leukemia in children with Down syndrome. Biol Blood Marrow Transplant 19 (6): 893-7, 2013.
- Muramatsu H, Sakaguchi H, Taga T, et al.: Reduced intensity conditioning in allogeneic stem cell transplantation for AML with Down syndrome. Pediatr Blood Cancer 61 (5): 925-7, 2014.
Síndromes mielodisplásicos
Los síndromes mielodisplásicos (SMD) y los síndromes mieloproliferativos (SMP) representan entre el 5 % y el 10 % de todas las neoplasias mieloides malignas en niños. Son un grupo de trastornos heterogéneos; los SMD por lo general se manifiestan con citopenias y los SMP se manifiestan con recuentos periféricos altos de glóbulos blancos, glóbulos rojos o plaquetas. Los SMD se caracterizan por hematopoyesis ineficaz y aumento de la apoptosis, mientras que los SMP se relacionan con mayor proliferación y supervivencia de células progenitoras. Dado que ambos son trastornos de células madre hematopoyéticas pluripotentes muy primitivas, las estrategias terapéuticas curativas casi siempre exigen el trasplante de células madre hematopoyéticas (TCMH) alogénico.
Factores de riesgo
Los pacientes que tienen las siguientes mutaciones de la línea germinal o trastornos hereditarios tienen un aumento significativo de riesgo de presentar un SMD:
- Anemia de Fanconi: se debe a mutaciones de la línea germinal en los genes de reparación del DNA.
- Disqueratosis congénita: se debe a mutaciones en los genes que regulan la longitud del telómero. Los genes mutados en la disqueratosis congénita son: ACD, CTC1, DKC1, NHP2, NOP10, PARN, RTEL1, TERC, TERT, TINF2 y WRAP53.
- Síndrome de Shwachman-Diamond, anemia de Diamond-Blackfan y otros síndromes de insuficiencia de la médula ósea: se deben a mutaciones en los genes que codifican las proteínas relacionadas con los ribosomas.[
1 ,2 ] Las mutaciones en GATA1 se relacionaron con la anemia de Diamond-Blackfan y la predisposición a los SMD.[3 ] - Neutropenia congénita grave: se debe a mutaciones en el gen que codifica la elastasa. Se calculó que el riesgo acumulado a 15 años de SMD en los pacientes con neutropenia congénita grave, también conocida como síndrome de Kostmann, es del 15 %, y el riesgo anual de SMD o leucemia mieloide aguda (LMA) es del 2 % al 3 %. No se conoce el modo en que las mutaciones que afectan esta proteína y la exposición crónica al factor estimulante de colonias de granulocitos (G-CSF) contribuyen a la presentación de un SMD.[
4 ] - Síndrome de trisomía 21: casi siempre hay mutaciones en GATA1 en la leucemia transitoria relacionada con la trisomía 21 y en el SMD en niños con síndrome de Down menores de 3 años.[
5 ] - Trombocitopenia amegacariocítica congénita (TAMC): las mutaciones hereditarias en los genes RUNX1 o CEPBA se relacionan con la TAMC.[
6 ,7 ] Las mutaciones en el gen MPL son la causa genética subyacente de la TAMC; los pacientes con TAMC tienen un riesgo de menos del 10 % de presentar SMD o LMA.[8 ] - Mutaciones en GATA2: en pacientes con SMD/LMA se notificaron mutaciones de la línea germinal en GATA2 junto con monocitopenia, deficiencia de linfocitos B y linfocitos citolíticos naturales, proteinosis alveolar pulmonar y susceptibilidad a infecciones oportunistas.[
9 ,10 ] - Mutaciones en RUNX1 o CEPBA: las mutaciones hereditarias en los genes RUNX1 o CEPBA se relacionan con SMD o LMA familiar.[
6 ,7 ] - Mutaciones en SAMD9 y SAMD9L: las mutaciones hereditarias en los genes SAMD9 y SAMD9 se relacionan con SMD familiar.[
11 ,12 ,13 ,14 ,15 ,16 ]
Un análisis retrospectivo en el que se usó un ensayo de captura para detectar mutaciones conocidas por predisponer a la insuficiencia de la médula ósea y SMD, se realizó en DNA genómico de muestras de células mononucleares de sangre periférica de pacientes sometidos a trasplante de células madre por SMD y anemia aplásica. Entre 46 niños de hasta 18 años con SMD, 10 pacientes (22 %) albergaban mutaciones genética constitucionales predisponentes (5 en GATA2, 1 en MPL, 1 en RTEL1, 1 en SBDS, 1 en TINF2 y 1 en TP53), de las que 2 se sospechaban antes del trasplante. Esto se considera una incidencia alta de mutaciones génicas en comparación con solo el 8 % (4 de 64) en pacientes de 18 a 40 años.[
Cuadro clínico inicial
Los pacientes a menudo manifiestan signos de citopenias, como palidez, infecciones o hematomas.
La médula ósea se suele caracterizar por hipercelularidad y cambios displásicos en los precursores mieloides. La evolución clonal con el tiempo puede llevar a la LMA. El porcentaje de blastocitos anómalos es inferior al 20 % y no hay anormalidades citogenéticas recurrentes comunes de la LMA (t(8;21), inv(16), t(15;17), ni translocaciones de KMT2A).
El SMD hipocelular, que es menos común, se puede diferenciar de la anemia aplásica, en parte, por su displasia marcada, naturaleza clonal y mayor porcentaje de precursores positivos para CD34.[
Anomalías moleculares
Características moleculares de los síndromes mielodisplásicos
Los síndromes mielodisplásicos (SMD) infantiles, si los comparamos con los SMD que se presentan en adultos, se relacionan con un conjunto característico de alteraciones genéticas. En adultos, los SMD a menudo surgen a partir de una hematopoyesis clonal y se caracterizan por la presencia de mutaciones en TET2, DNMT3A y TP53. Por el contrario, las mutaciones en estos genes son poco frecuentes en los SMD que se presentan durante la niñez, aunque en subgrupos de casos de SMD infantiles se observan mutaciones en los genes GATA2, SAMD9, SAMD9L, SETBP1, ASXL1 y de la vía RAS/MAPK.[
En un informe del panorama genómico de los SMD infantiles se describieron los resultados de la secuenciación del exoma completo de 32 pacientes pediátricos con SMD primarios infantiles y de la secuenciación dirigida de otros 14 casos.[
- Se observaron mutaciones en los genes de la vía RAS/MAPK en el 43 % de los casos de SMD primarios, las más comunes afectaban los genes PTPN11 y NRAS, pero también se observaron mutaciones en otros genes de la vía (por ejemplo, BRAF [diferentes a la mutación V600E en BRAF], CBL y KRAS). Las mutaciones en la vía RAS/MAPK fueron más comunes en pacientes con SMD-EB (65 %) que en los pacientes con citopenia refractaria infantil (17 %).
- Se observaron variantes de la línea germinal en SAMD9 (n = 4) o en SAMD9L (n = 4) en el 17 % de los pacientes de SMD primarios, donde 7 de las 8 mutaciones ocurrieron en pacientes de citopenia refractaria infantil. En todos estos casos se observó una pérdida de material en el cromosoma 7. Alrededor del 40 % de los pacientes con deleciones de parte o de la totalidad del cromosoma 7 presentaron variantes de la línea germinal en SAMD9 o en SAMD9L.
- Se observaron mutaciones en GATA2 en 3 casos (7 %), y en todos ellos se confirmó o se sospechó que eran de la línea germinal.
- Las alteraciones en el número de copias más comunes fueron las deleciones en el cromosoma 7, que se observaron en 41 % de los casos. La pérdida parcial o total del cromosoma 7 fue más frecuente en los casos con alteraciones en los genes SAMD9 y SAMD9L (100 %) y en pacientes de SMD-EB con mutación en la vía RAS/MAPK (71 %).
- Otros genes que estaban mutados en más de 1 de los 46 casos estudiados fueron SETBP1, ETV6 y TP53.
En un segundo informe se describió la utilización de un perfil de secuenciación dirigida de 105 genes en 50 pacientes pediátricos de SMD (citopenia refractaria infantil = 31 y SMD-EB = 19) que incluyó abundantes casos con monosomía 7 (48 %).[
- Se observaron mutaciones de la línea germinal en GATA2 en el 30 % de los pacientes y mutaciones en RUNX1 en el 6 % de los pacientes.
- Se observaron mutaciones somáticas en el 34 % de los pacientes y fueron más comunes en pacientes con SMD-EB que en pacientes con citopenia refractaria infantil (68 vs. 13 %).
- El gen que mutó con más frecuencia fue SETBP1 (18 %); los genes que mutaron con menor frecuencia fueron ASXL1, RUNX1 y los genes de la vía RAS/MAPK (PTPN11, NRAS, KRAS, NF1). Se encontraron mutaciones en los genes de la vía RAS/MAPK en el 12 % de los casos.
Los pacientes con mutaciones de la línea germinal en GATA2, además de SMD, exhiben un amplio abanico de defectos hematopoyéticos e inmunológicos así como manifestaciones no hematopoyéticas.[
- En el 7 % de los pacientes pediátricos con SMD primarios se identificaron mutaciones de la línea germinal en GATA2. Si bien la mediana de edad de los pacientes con mutaciones en GATA2 fue de 12,3 años en la población pediátrica del EWOG-MDS, la mayor parte de los casos de neoplasias mieloides relacionadas con la línea germinal en GATA2 se presentan durante la edad adulta.[
23 ] - Las mutaciones en GATA2 fueron más comunes en los pacientes con SMD-EB (15 %) que en los pacientes de citopenia refractaria infantil (4 %).
- Entre los pacientes con mutaciones en GATA2, el 46 % presentaba SDM-EB y el 70 % exhibió monosomía 7.
- Se identificó SMD/LMA familiar en 12 de los 53 pacientes con una mutación en GATA2 para los que se disponía de una historia familiar detallada.
- Los fenotipos no hematológicos de la deficiencia de GATA2 se presentaron en el 51 % de los pacientes de SMD con una mutación en GATA2 e incluyeron hipoacusia (9 %), linfedema o hidrocele (23 %) e inmunodeficiencia (39 %).
Las mutaciones de la línea germinal en SAMD9 y SAMD9L se relacionan con casos de SMD con pérdida adicional, total o parcial, del cromosoma 7.[
En 2016, se identificó el gen SAMD9 como la causa del síndrome MIRAGE (mielodisplasia, infección, restricción del crecimiento, hipoplasia suprarrenal, fenotipos genitales y enteropatía) que se relaciona con los SMD de aparición temprana con monosomía 7.[
- Las mutaciones causales en SAMD9 y SAMD9L son mutaciones de ganancia de función que intensifican la actividad supresora del crecimiento de SAMD9 o SAMD9L.[
14 ,16 ] - Los genes SAMD9 y SAMD9L están en el cromosoma 7q21.2. Los casos de SMD en pacientes con mutaciones en SAMD9 o SAMD9L a menudo exhiben monosomía 7 y el otro cromosoma 7 tiene SAMD9 y SAMD9Lnaturales. Esto lleva a que se pierda el efecto intensificador del gen mutado en la actividad supresora del crecimiento.
- Los pacientes con fenotipo normal, mutaciones en SAMD9 o SAMD9L y monosomía 7 a veces presentan SMD o LMA, o por el contrario, pierden la monosomía 7 y su hematopoyesis se normaliza.[
16 ] El primer caso se relaciona con la aparición de mutaciones en los genes vinculados con los SMD o la LMA (por ejemplo, ETV6 o SETBP1), mientras que el segundo caso se relaciona con modificaciones genéticas (por ejemplo, mutaciones inversas o pérdida de la heterocigosidad sin cambio en el número de copias con retención del alelo natural) que llevan a la normalización de la actividad de SAMD9 y SAMD9L. Estas observaciones indican que el seguimiento de los pacientes con monosomía 7 relacionada con mutaciones en SAMD9 o SAMD9L mediante la secuenciación clínica de las mutaciones adquiridas en genes vinculados con la formación de la LMA, permitiría identificar a los pacientes con riesgo alto de transformación leucémica que se beneficiarían más de un trasplante de células madre hematopoyéticas.[16 ]
La presencia de una monosomía 7 aislada es la anomalía citogenética más común, si bien no parece augurar un pronóstico adverso en comparación con su presencia en la LMA manifiesta. No obstante, la presencia de monosomía 7 en combinación con otras anomalías citogenéticas se relaciona con un pronóstico precario.[
Para obtener más información sobre el sistema de clasificación de los SMD de la Organización Mundial de la Salud (OMS), consultar la sección Clasificación de la Organización Mundial de la Salud sobre los hallazgos en la médula ósea y la sangre periférica de los síndromes mielodisplásicos.
Clasificación de los síndromes mielodisplásicos
Los sistemas de clasificación de los síndromes mielodisplásicos (SMD) y los síndromes mieloproliferativos (SMP) del French-American-British (FAB) y de la OMS son difíciles de usar en los pacientes pediátricos. Se han propuesto otros sistemas de clasificación para niños, pero ninguno se ha adoptado de manera uniforme, excepto el sistema de clasificación de la OMS modificado en 2008.[
El subtipo de citopenia refractaria representa alrededor del 50 % de todos los casos de SMD infantil. La presencia de una monosomía 7 aislada es la anomalía citogenética más común, si bien no parece augurar un pronóstico adverso en comparación con su presencia en la LMA manifiesta. No obstante, la presencia de monosomía 7 en combinación con otras anomalías citogenéticas se relaciona con un pronóstico precario.[
Los grupos pronóstico R-IPSS y las anomalías citogenéticas relacionadas son los siguientes:[
- Grupo de pronóstico muy bueno: -Y; del(11q).
- Grupo de pronóstico bueno: normal; del(5q); del(20q); del(12p); doble que incluye del(5q).
- Grupo de pronóstico intermedio: del(7q); +8; i(17q); +19; cualquier otro clon independiente simple o doble.
- Grupo de pronóstico precario: -7; inv(3)/t(3q)/del(3q); doble que incluye -7/del(7q); complejo, es decir 3 anomalías.
- Grupo de pronóstico muy precario: complejo, es decir >3 anomalías.
El IPSS puede ayudar a diferenciar el SMD de riesgo bajo y de riesgo alto, aunque su utilidad en niños con SMD es más limitada que en los adultos porque muchas características difieren entre niños y adultos.[
Tratamiento de los síndromes mielodisplásicos infantiles
Las opciones de tratamiento de los síndromes mielodisplásicos (SMD) infantiles son las siguientes:
- Trasplante de células madre hematopoyéticas.
- Otras terapias.
Trasplante de células madre hematopoyéticas
El SMD y los trastornos relacionados por lo general afectan una célula madre hematopoyética primitiva. Por lo tanto, el trasplante de células madre hematopoyéticas (TCMH) alogénico se considera el abordaje óptimo de tratamiento para los pacientes pediátricos con SMD. Aunque se prefiere el trasplante de un hermano compatible, se encontró una supervivencia similar con abordajes haploidénticos y de sangre de cordón de donantes no emparentados con compatibilidad buena.[
Cuando se toman decisiones sobre el tratamiento, se debe tener en cuenta cierta información. Por ejemplo, se notificaron tasas de supervivencia de hasta el 80 % en pacientes con SMD en estadio temprano que procedieron a recibir un trasplante unos pocos meses después del diagnóstico. Asimismo, un trasplante temprano y la ausencia de quimioterapia pretrasplante se relacionaron con una mejora de la supervivencia en los niños con SMD.[
Se examinó la pregunta de si se debe usar quimioterapia para el SMD de riesgo alto.
Evidencia (trasplante de células madre hematopoyéticas):
- En un análisis de 37 niños con SMD tratados en los protocolos AML 83, 87, y 93 del grupo Berlin-Frankfurt-Münster, se confirmó una respuesta a la inducción del 74 % en pacientes con anemia resistente al tratamiento y exceso de blastocitos en transformación, y se indicó que el trasplante fue beneficioso.[
53 ] - En otro estudio del mismo grupo se observó que, al usar abordajes actuales de TCMH, más del 60 % de los niños con SMD en estadio avanzado sobrevivió; los desenlaces para los pacientes que recibieron células de donantes no emparentados fueron similares a los desenlaces de los pacientes que recibieron células de donantes emparentados compatibles (DEC).[
54 ] - En el ensayo 2891 del Children Cancer Group, se inscribieron pacientes entre 1989 y 1995, incluso niños con SMD.[
44 ] Participaron 77 pacientes con anemia resistente al tratamiento (n = 2), anemia resistente al tratamiento con exceso de blastocitos (n = 33), anemia resistente al tratamiento con exceso de blastocitos en transformación (n = 26) o LMA con antecedente de SMD (n = 16). Los pacientes se asignaron al azar a recibir inducción de cronograma estándar o intenso. Más adelante, los pacientes se sometieron a un TCMH alogénico en el caso de contar con un donante emparentado adecuado, o se asignaron al azar para recibir un TCMH autógeno o quimioterapia.- Los pacientes con anemia resistente al tratamiento o anemia resistente al tratamiento con exceso de blastocitos tuvieron una tasa de remisión más baja (45 %), en tanto que los pacientes con anemia resistente al tratamiento con exceso de blastocitos en transformación (69 %) o LMA con antecedente de SMD (81 %) tuvieron tasas de remisión similares comparables a las tasas de los desenlaces de LMA de novo (77 %).
- Las tasas de supervivencia a 6 años fueron precarias en los pacientes con anemia resistente al tratamiento o anemia resistente al tratamiento con exceso de blastocitos (28 %), y en aquellos con anemia resistente al tratamiento con exceso de blastocitos en transformación (30 %).
- Los pacientes con LMA y antecedente de SMD tuvieron un desenlace similar al de los pacientes con LMA de novo (supervivencia del 50 % en comparación con el 45 %).
- El TCMH alogénico mejoró la supervivencia (P = 0,08).
Al analizar estos resultados es importante tener en cuenta que el subtipo de anemia resistente al tratamiento con exceso de blastocitos en transformación es probable que represente a pacientes con LMA manifiesta, mientras que la anemia resistente al tratamiento y la anemia resistente al tratamiento con exceso de blastocitos representan un SDM. En la clasificación de la OMS ahora se omite la categoría de anemia resistente al tratamiento con exceso de blastocitos en trasformación y se concluyó que esta entidad era en esencia una LMA.
Debido a que la supervivencia después un TCMH mejoró en los niños con formas tempranas de SMD (anemia resistente al tratamiento), se debe considerar el trasplante antes de la progresión a SMD tardía o LMA. El TCMH se debe considerar en especial cuando se necesitan transfusiones u otros tratamientos, lo que es usual en los pacientes con citopenias sintomáticas graves.[
Dado que los SMD infantiles se suelen relacionar con síndromes de predisposición hereditarios, se han documentado trasplantes en muy pocos casos de pacientes con estos trastornos. Por ejemplo, en los pacientes con anemia de Fanconi y LMA o SMD en estadio avanzado, se notificó una tasa de supervivencia general (SG) a 5 años del 33 % al 55 %.[
Mientras que algunos pacientes con síndromes de predisposición hereditarios requieren una modificación significativa de sus abordajes de trasplante debido a un exceso de toxicidad (por ejemplo, anemia de Fanconi), otros síndromes no tienen una toxicidad excesiva detectable relacionada con el proceso de trasplante. La herencia de GATA2 es un buen ejemplo de esto último. En un estudio se compararon los resultados del TCMH en 65 niños con mutaciones en la línea germinal de GATA2 y SMD, con los resultados de 404 niños con SMD y línea germinal de GATA2 natural. La supervivencia sin enfermedad, la recaída y la mortalidad sin recaída fueron similares en las 2 poblaciones.[
Para los pacientes con citopenias de importancia clínica, se considera que los cuidados médicos de apoyo como transfusiones y antibióticos profilácticos son parte del tratamiento de referencia. El empleo de factores de crecimiento hematopoyético puede mejorar el estado de la hematopoyesis, pero aún preocupa que dicho tratamiento acelere la conversión a una LMA.[
Otras terapias
Otras terapias complementarias en estudio son las siguientes:
- Se ha usado terapia con corticoesteroides, como glucocorticoides y andrógenos, con resultados variados.[
61 ] - Los tratamientos dirigidos a la captación de radicales libres de oxígeno (antioxidantes) con amifostina [
62 ,63 ] o el uso de retinoides promotores de la diferenciación,[64 ] los inhibidores de la metilación de DNA (por ejemplo, azacitidina y decitabina), y los inhibidores de la histona–desacetilasa, demostraron en conjunto alguna respuesta, pero no hay informes de ensayos definitivos en niños con SMD. La Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) aprobó la azacitidina para el tratamiento del SMD en adultos a partir de los ensayos aleatorizados.[65 ] Para obtener más información, consultar la sección Modificadores de la enfermedad en el sumario Tratamiento de los síndromes mielodisplásicos. - Se han probado fármacos como la lenalidomida, un análogo de la talidomida, a partir de resultados que demostraron aumento de la actividad en la médula ósea de pacientes con SMD. La lenalidomida demostró ser más eficaz en los pacientes con síndrome 5q-, sobre todo en aquellos con trombocitosis; en la actualidad la FDA autoriza su uso en adultos con estas características.[
66 ] - También se notificó inmunodepresión en adultos que recibieron globulina antitimocítica o ciclosporina.[
66 ,67 ]
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
- Alter BP, Giri N, Savage SA, et al.: Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol 150 (2): 179-88, 2010.
- Rosenberg PS, Huang Y, Alter BP: Individualized risks of first adverse events in patients with Fanconi anemia. Blood 104 (2): 350-5, 2004.
- Ludwig LS, Gazda HT, Eng JC, et al.: Altered translation of GATA1 in Diamond-Blackfan anemia. Nat Med 20 (7): 748-53, 2014.
- Rosenberg PS, Zeidler C, Bolyard AA, et al.: Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br J Haematol 150 (2): 196-9, 2010.
- Wechsler J, Greene M, McDevitt MA, et al.: Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet 32 (1): 148-52, 2002.
- Liew E, Owen C: Familial myelodysplastic syndromes: a review of the literature. Haematologica 96 (10): 1536-42, 2011.
- Owen C, Barnett M, Fitzgibbon J: Familial myelodysplasia and acute myeloid leukaemia--a review. Br J Haematol 140 (2): 123-32, 2008.
- Ghauri RI, Naveed M, Mannan J: Congenital amegakaryocytic thrombocytopenic purpura (CAMT). J Coll Physicians Surg Pak 24 (4): 285-7, 2014.
- Auer PL, Teumer A, Schick U, et al.: Rare and low-frequency coding variants in CXCR2 and other genes are associated with hematological traits. Nat Genet 46 (6): 629-34, 2014.
- Vinh DC, Patel SY, Uzel G, et al.: Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood 115 (8): 1519-29, 2010.
- Schwartz JR, Ma J, Lamprecht T, et al.: The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun 8 (1): 1557, 2017.
- Schwartz JR, Wang S, Ma J, et al.: Germline SAMD9 mutation in siblings with monosomy 7 and myelodysplastic syndrome. Leukemia 31 (8): 1827-1830, 2017.
- Davidsson J, Puschmann A, Tedgård U, et al.: SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 32 (5): 1106-1115, 2018.
- Narumi S, Amano N, Ishii T, et al.: SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet 48 (7): 792-7, 2016.
- Chen DH, Below JE, Shimamura A, et al.: Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am J Hum Genet 98 (6): 1146-1158, 2016.
- Wong JC, Bryant V, Lamprecht T, et al.: Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight 3 (14): , 2018.
- Keel SB, Scott A, Sanchez-Bonilla M, et al.: Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients. Haematologica 101 (11): 1343-1350, 2016.
- Kasahara S, Hara T, Itoh H, et al.: Hypoplastic myelodysplastic syndromes can be distinguished from acquired aplastic anaemia by bone marrow stem cell expression of the tumour necrosis factor receptor. Br J Haematol 118 (1): 181-8, 2002.
- Orazi A: Histopathology in the diagnosis and classification of acute myeloid leukemia, myelodysplastic syndromes, and myelodysplastic/myeloproliferative diseases. Pathobiology 74 (2): 97-114, 2007.
- Pastor V, Hirabayashi S, Karow A, et al.: Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia 31 (3): 759-762, 2017.
- Collin M, Dickinson R, Bigley V: Haematopoietic and immune defects associated with GATA2 mutation. Br J Haematol 169 (2): 173-87, 2015.
- Wlodarski MW, Hirabayashi S, Pastor V, et al.: Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 127 (11): 1387-97; quiz 1518, 2016.
- Wlodarski MW, Collin M, Horwitz MS: GATA2 deficiency and related myeloid neoplasms. Semin Hematol 54 (2): 81-86, 2017.
- Göhring G, Michalova K, Beverloo HB, et al.: Complex karyotype newly defined: the strongest prognostic factor in advanced childhood myelodysplastic syndrome. Blood 116 (19): 3766-9, 2010.
- Haase D, Germing U, Schanz J, et al.: New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood 110 (13): 4385-95, 2007.
- Arber DA, Vardiman JW, Brunning RD: Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. International Agency for Research on Cancer, 2008, pp 110-23.
- Occhipinti E, Correa H, Yu L, et al.: Comparison of two new classifications for pediatric myelodysplastic and myeloproliferative disorders. Pediatr Blood Cancer 44 (3): 240-4, 2005.
- Niemeyer CM, Baumann I: Myelodysplastic syndrome in children and adolescents. Semin Hematol 45 (1): 60-70, 2008.
- Niemeyer CM, Kratz CP: Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia: molecular classification and treatment options. Br J Haematol 140 (6): 610-24, 2008.
- Hasle H: Myelodysplastic and myeloproliferative disorders in children. Curr Opin Pediatr 19 (1): 1-8, 2007.
- Mandel K, Dror Y, Poon A, et al.: A practical, comprehensive classification for pediatric myelodysplastic syndromes: the CCC system. J Pediatr Hematol Oncol 24 (7): 596-605, 2002.
- Nösslinger T, Reisner R, Koller E, et al.: Myelodysplastic syndromes, from French-American-British to World Health Organization: comparison of classifications on 431 unselected patients from a single institution. Blood 98 (10): 2935-41, 2001.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Yamamoto S, Kato M, Watanabe K, et al.: Prognostic value of the revised International Prognostic Scoring System five-group cytogenetic abnormality classification for the outcome prediction of hematopoietic stem cell transplantation in pediatric myelodysplastic syndrome. Bone Marrow Transplant 56 (12): 3016-3023, 2021.
- Cutler CS, Lee SJ, Greenberg P, et al.: A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood 104 (2): 579-85, 2004.
- Hasle H, Baumann I, Bergsträsser E, et al.: The International Prognostic Scoring System (IPSS) for childhood myelodysplastic syndrome (MDS) and juvenile myelomonocytic leukemia (JMML). Leukemia 18 (12): 2008-14, 2004.
- Hasle H, Niemeyer CM: Advances in the prognostication and management of advanced MDS in children. Br J Haematol 154 (2): 185-95, 2011.
- Uberti JP, Agovi MA, Tarima S, et al.: Comparative analysis of BU and CY versus CY and TBI in full intensity unrelated marrow donor transplantation for AML, CML and myelodysplasia. Bone Marrow Transplant 46 (1): 34-43, 2011.
- Nemecek ER, Guthrie KA, Sorror ML, et al.: Conditioning with treosulfan and fludarabine followed by allogeneic hematopoietic cell transplantation for high-risk hematologic malignancies. Biol Blood Marrow Transplant 17 (3): 341-50, 2011.
- Shaw PJ, Kan F, Woo Ahn K, et al.: Outcomes of pediatric bone marrow transplantation for leukemia and myelodysplasia using matched sibling, mismatched related, or matched unrelated donors. Blood 116 (19): 4007-15, 2010.
- Parikh SH, Mendizabal A, Martin PL, et al.: Unrelated donor umbilical cord blood transplantation in pediatric myelodysplastic syndrome: a single-center experience. Biol Blood Marrow Transplant 15 (8): 948-55, 2009.
- Locatelli F, Merli P, Pagliara D, et al.: Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood 130 (5): 677-685, 2017.
- Smith AR, Christiansen EC, Wagner JE, et al.: Early hematopoietic stem cell transplant is associated with favorable outcomes in children with MDS. Pediatr Blood Cancer 60 (4): 705-10, 2013.
- Woods WG, Barnard DR, Alonzo TA, et al.: Prospective study of 90 children requiring treatment for juvenile myelomonocytic leukemia or myelodysplastic syndrome: a report from the Children's Cancer Group. J Clin Oncol 20 (2): 434-40, 2002.
- Andolina JR, Kletzel M, Tse WT, et al.: Allogeneic hematopoetic stem cell transplantation in pediatric myelodysplastic syndromes: improved outcomes for de novo disease. Pediatr Transplant 15 (3): 334-43, 2011.
- Al-Seraihy A, Ayas M, Al-Nounou R, et al.: Outcome of allogeneic stem cell transplantation with a conditioning regimen of busulfan, cyclophosphamide and low-dose etoposide for children with myelodysplastic syndrome. Hematol Oncol Stem Cell Ther 4 (3): 121-5, 2011.
- Woodard P, Carpenter PA, Davies SM, et al.: Unrelated donor bone marrow transplantation for myelodysplastic syndrome in children. Biol Blood Marrow Transplant 17 (5): 723-8, 2011.
- Champlin R: Hematopoietic stem cell transplantation for treatment of myleodysplastic syndromes. Biol Blood Marrow Transplant 17 (1 Suppl): S6-8, 2011.
- Nelson RP, Yu M, Schwartz JE, et al.: Long-term disease-free survival after nonmyeloablative cyclophosphamide/fludarabine conditioning and related/unrelated allotransplantation for acute myeloid leukemia/myelodysplasia. Bone Marrow Transplant 45 (8): 1300-8, 2010.
- Warlick ED: Optimizing stem cell transplantation in myelodysplastic syndromes: unresolved questions. Curr Opin Oncol 22 (2): 150-4, 2010.
- Pulsipher MA, Boucher KM, Wall D, et al.: Reduced-intensity allogeneic transplantation in pediatric patients ineligible for myeloablative therapy: results of the Pediatric Blood and Marrow Transplant Consortium Study ONC0313. Blood 114 (7): 1429-36, 2009.
- Gao L, Gao L, Gong Y, et al.: Reduced-intensity conditioning therapy with fludarabine, idarubicin, busulfan and cytarabine for allogeneic hematopoietic stem cell transplantation in acute myeloid leukemia and myelodysplastic syndrome. Leuk Res 37 (11): 1482-7, 2013.
- Creutzig U, Bender-Götze C, Ritter J, et al.: The role of intensive AML-specific therapy in treatment of children with RAEB and RAEB-t. Leukemia 12 (5): 652-9, 1998.
- Strahm B, Nöllke P, Zecca M, et al.: Hematopoietic stem cell transplantation for advanced myelodysplastic syndrome in children: results of the EWOG-MDS 98 study. Leukemia 25 (3): 455-62, 2011.
- Madureira AB, Eapen M, Locatelli F, et al.: Analysis of risk factors influencing outcome in children with myelodysplastic syndrome after unrelated cord blood transplantation. Leukemia 25 (3): 449-54, 2011.
- Mitchell R, Wagner JE, Hirsch B, et al.: Haematopoietic cell transplantation for acute leukaemia and advanced myelodysplastic syndrome in Fanconi anaemia. Br J Haematol 164 (3): 384-95, 2014.
- Ayas M, Saber W, Davies SM, et al.: Allogeneic hematopoietic cell transplantation for fanconi anemia in patients with pretransplantation cytogenetic abnormalities, myelodysplastic syndrome, or acute leukemia. J Clin Oncol 31 (13): 1669-76, 2013.
- Kato M, Yoshida N, Inagaki J, et al.: Salvage allogeneic stem cell transplantation in patients with pediatric myelodysplastic syndrome and myeloproliferative neoplasms. Pediatr Blood Cancer 61 (10): 1860-6, 2014.
- Bortnick R, Wlodarski M, de Haas V, et al.: Hematopoietic stem cell transplantation in children and adolescents with GATA2-related myelodysplastic syndrome. Bone Marrow Transplant 56 (11): 2732-2741, 2021.
- Zwierzina H, Suciu S, Loeffler-Ragg J, et al.: Low-dose cytosine arabinoside (LD-AraC) vs LD-AraC plus granulocyte/macrophage colony stimulating factor vs LD-AraC plus Interleukin-3 for myelodysplastic syndrome patients with a high risk of developing acute leukemia: final results of a randomized phase III study (06903) of the EORTC Leukemia Cooperative Group. Leukemia 19 (11): 1929-33, 2005.
- Chan G, DiVenuti G, Miller K: Danazol for the treatment of thrombocytopenia in patients with myelodysplastic syndrome. Am J Hematol 71 (3): 166-71, 2002.
- Mathew P, Gerbing R, Alonzo TA, et al.: A phase II study of amifostine in children with myelodysplastic syndrome: a report from the Children's Oncology Group study (AAML0121). Pediatr Blood Cancer 57 (7): 1230-2, 2011.
- Schanz J, Jung H, Wörmann B, et al.: Amifostine has the potential to induce haematologic responses and decelerate disease progression in individual patients with low- and intermediate-1-risk myelodysplastic syndromes. Leuk Res 33 (9): 1183-8, 2009.
- Sadek I, Zayed E, Hayne O, et al.: Prolonged complete remission of myelodysplastic syndrome treated with danazol, retinoic acid and low-dose prednisone. Am J Hematol 64 (4): 306-10, 2000.
- Silverman LR, Demakos EP, Peterson BL, et al.: Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 20 (10): 2429-40, 2002.
- Yazji S, Giles FJ, Tsimberidou AM, et al.: Antithymocyte globulin (ATG)-based therapy in patients with myelodysplastic syndromes. Leukemia 17 (11): 2101-6, 2003.
- Yoshimi A, Baumann I, Führer M, et al.: Immunosuppressive therapy with anti-thymocyte globulin and cyclosporine A in selected children with hypoplastic refractory cytopenia. Haematologica 92 (3): 397-400, 2007.
Leucemia mieloide aguda o síndromes mielodisplásicos relacionados con el tratamiento
Patogenia
La presentación de una leucemia mieloide aguda (LMA) o de un síndrome mielodisplásico (SMD) luego del tratamiento con radiación ionizante o quimioterapia, en particular, con alquilantes e inhibidores de la topoisomerasa, se denomina LMA relacionada con el tratamiento (LMA-t) o SMD relacionado con el tratamiento (SMD-t). Además de las exposiciones genotóxicas, es posible que las susceptibilidades por predisposición genética (como los polimorfismos en los componentes de desintoxicación farmacológica y de las vías de reparación del DNA) contribuyan a la presentación de una LMA o un SMD secundarios.[
El riesgo de LMA-t o SMD-t depende del régimen y, a menudo, se relaciona con la dosis acumulada de fármacos quimioterapéuticos, así como la dosis y el campo de radiación utilizados.[
Las LMA-t o los SMD-t debidos a epipodofilotoxinas y otros inhibidores de la topoisomerasa II (por ejemplo, antraciclinas) por lo general se presentan dentro de los 2 años siguientes a la exposición y es frecuente que se relacionen con anomalías en el cromosoma 11q23,[
Tratamiento de la leucemia mieloide aguda y el síndrome mielodisplásico relacionados con el tratamiento
La opción de tratamiento de la leucemia mieloide aguda (LMA) y el síndrome mielodisplásico (SMD) relacionados con el tratamiento es la siguiente:
- Trasplante de células madre hematopoyéticas.
La meta del tratamiento es lograr una remisión completa (RC) inicial con el uso de regímenes dirigidos a la leucemia mieloide aguda (LMA) y luego, por lo general, proceder directamente a un trasplante de células madre hematopoyéticas (TCMH) usando el mejor donante disponible. Sin embargo, el tratamiento es complejo debido a los siguientes aspectos:[
- Tasas crecientes de características citogenéticas adversas y fracaso subsiguiente para lograr la remisión con quimioterapia.
- Comorbilidades o limitaciones relacionadas con la quimioterapia por una neoplasia maligna previa.
En consecuencia, las tasas de RC y de supervivencia general (SG) por lo general son más bajas para los pacientes de LMA-t en comparación con los pacientes de LMA de novo.[
Los pacientes con SMD-t y anemia resistente al tratamiento a menudo no necesitan quimioterapia de inducción antes del trasplante; la función de la terapia de inducción antes del trasplante es polémica para los pacientes con anemia resistente al tratamiento con exceso de blastocitos-1.
Solo en unos pocos informes se describen los desenlaces de niños sometidos a TCMH para la LMA-t.
Evidencia (trasplante de células madre hematopoyéticas para la leucemia mieloide aguda o el síndrome mielodisplásico relacionados con el tratamiento):
- En un estudio se describieron los desenlaces de 27 niños con LMA-t que recibieron TCMH de un donante emparentado o no emparentado.[
14 ]- Las tasas de SG a 3 años fueron del 18,5 % ± 7,5 % y las tasas de supervivencia sin complicaciones (SSC) fueron del 18,7 % ± 7,5 %.
- La supervivencia precaria se debió sobre todo a la mortalidad muy alta relacionada con el trasplante (59,6 ± 8,4 %).
- En otro estudio se informó sobre una segunda experiencia retrospectiva de un solo centro con 14 pacientes con LMA-t o SMD-t que recibieron trasplantes entre 1975 y 2007.[
11 ]- La supervivencia fue del 29 %, pero en esta revisión, solo el 63 % de los pacientes con diagnóstico de LMA-t o SMD-t se sometieron a TCMH.
- En un estudio multicéntrico (CCG-2891), se examinaron los desenlaces de 24 niños con LMA-t o SMD-t en comparación con otros niños que participaron en el estudio y que tenían LMA de novo (n = 898) o SMD (n = 62). Los niños con LMA-t o SMD-t eran más mayores y casi ninguno exhibía características citogenéticas de riesgo bajo.[
15 ]- Aunque las tasas de logro de RC y SG a 3 años fueron peores en el grupo de LMA-t/SMD-t (RC, 50 % vs. 72 %; P = 0,016; tasa de SG, 26 vs. 47 %; P = 0,007), la supervivencia fue similar (tas de SG, 45 vs. 53 %; P = 0,87) cuando los pacientes lograron una RC.
- La importancia de la remisión en la supervivencia de estos pacientes se describió más en otro informe de un solo centro sobre 21 niños sometidos a TCMH por LMA-t o SMD-t entre 1994 y 2009. De estos 21 niños, 12 tenían LMA-t (11 de ellos en RC en el momento del trasplante), 7 tenían anemia resistente al tratamiento (para quienes no se administró inducción), y 2 presentaban anemia resistente al tratamiento con exceso de blastocitos.[
16 ]- La tasa de SG a 10 años para toda la cohorte fue del 61 %. Los pacientes en remisión o con anemia resistente al tratamiento tuvieron una tasa de supervivencia sin enfermedad del 66 %, y, para los 3 pacientes con más del 5 % de blastocitos en el momento del TCMH la tasa de supervivencia fue del 0 % (P = 0,015).
Debido a que la LMA-t es poco frecuente en los niños, no se sabe si la disminución significativa de la mortalidad relacionada con un trasplante después del TCMH de donante no emparentado que se observó durante los últimos años se traduzca en una mejora de la supervivencia en esta población. Los pacientes se deben someter a una evaluación cuidadosa del riesgo de morbilidad antes del TCMH ocasionada por tratamientos anteriores, y los abordajes se deben adaptar a fin de proveer la intensidad adecuada a la vez que se disminuye la mortalidad relacionada con el trasplante.
Referencias:
- Leone G, Fianchi L, Voso MT: Therapy-related myeloid neoplasms. Curr Opin Oncol 23 (6): 672-80, 2011.
- Bolufer P, Collado M, Barragan E, et al.: Profile of polymorphisms of drug-metabolising enzymes and the risk of therapy-related leukaemia. Br J Haematol 136 (4): 590-6, 2007.
- Ezoe S: Secondary leukemia associated with the anti-cancer agent, etoposide, a topoisomerase II inhibitor. Int J Environ Res Public Health 9 (7): 2444-53, 2012.
- Ding Y, Sun CL, Li L, et al.: Genetic susceptibility to therapy-related leukemia after Hodgkin lymphoma or non-Hodgkin lymphoma: role of drug metabolism, apoptosis and DNA repair. Blood Cancer J 2 (3): e58, 2012.
- Leone G, Mele L, Pulsoni A, et al.: The incidence of secondary leukemias. Haematologica 84 (10): 937-45, 1999.
- Pui CH, Ribeiro RC, Hancock ML, et al.: Acute myeloid leukemia in children treated with epipodophyllotoxins for acute lymphoblastic leukemia. N Engl J Med 325 (24): 1682-7, 1991.
- Andersen MK, Johansson B, Larsen SO, et al.: Chromosomal abnormalities in secondary MDS and AML. Relationship to drugs and radiation with specific emphasis on the balanced rearrangements. Haematologica 83 (6): 483-8, 1998.
- Ogami A, Morimoto A, Hibi S, et al.: Secondary acute promyelocytic leukemia following chemotherapy for non-Hodgkin's lymphoma in a child. J Pediatr Hematol Oncol 26 (7): 427-30, 2004.
- Okamoto T, Okada M, Wakae T, et al.: Secondary acute promyelocytic leukemia in a patient with non-Hodgkin's lymphoma treated with VP-16 and MST-16. Int J Hematol 75 (1): 107-8, 2002.
- Larson RA: Etiology and management of therapy-related myeloid leukemia. Hematology Am Soc Hematol Educ Program : 453-9, 2007.
- Aguilera DG, Vaklavas C, Tsimberidou AM, et al.: Pediatric therapy-related myelodysplastic syndrome/acute myeloid leukemia: the MD Anderson Cancer Center experience. J Pediatr Hematol Oncol 31 (11): 803-11, 2009.
- Yokoyama H, Mori S, Kobayashi Y, et al.: Hematopoietic stem cell transplantation for therapy-related myelodysplastic syndrome and acute leukemia: a single-center analysis of 47 patients. Int J Hematol 92 (2): 334-41, 2010.
- Xavier AC, Kutny M, Costa LJ: Incidence and outcomes of paediatric myelodysplastic syndrome in the United States. Br J Haematol 180 (6): 898-901, 2018.
- Woodard P, Barfield R, Hale G, et al.: Outcome of hematopoietic stem cell transplantation for pediatric patients with therapy-related acute myeloid leukemia or myelodysplastic syndrome. Pediatr Blood Cancer 47 (7): 931-5, 2006.
- Barnard DR, Lange B, Alonzo TA, et al.: Acute myeloid leukemia and myelodysplastic syndrome in children treated for cancer: comparison with primary presentation. Blood 100 (2): 427-34, 2002.
- Kobos R, Steinherz PG, Kernan NA, et al.: Allogeneic hematopoietic stem cell transplantation for pediatric patients with treatment-related myelodysplastic syndrome or acute myelogenous leukemia. Biol Blood Marrow Transplant 18 (3): 473-80, 2012.
Leucemia mielomonocítica juvenil
Incidencia
La leucemia mielomonocítica juvenil (LMMJ) es una leucemia rara, alrededor de 10 veces menos frecuente que la leucemia mieloide aguda (LMA) en niños; la incidencia anual es de 1 a 2 casos por millón de personas.[
Cuadro clínico inicial y criterios diagnósticos
Las manifestaciones clínicas comunes en el momento del diagnóstico son las siguientes:[
- Hepatoesplenomegalia (97 %).
- Linfadenopatía (76 %).
- Palidez (64 %).
- Fiebre (54 %).
- Erupción cutánea (36 %).
En el Cuadro se describen los criterios que se utilizan en la actualidad para el diagnóstico definitivo de niños que presentan las manifestaciones clínicas indicativas de LMMJ 8.[
| Categoría 1 (se necesitan todos) | Categoría 2 (un criterio es suficiente)a | Categoría 3 (los pacientes sin características genéticas deben manifestar lo siguiente además de los criterios de la categoría 1b) |
|---|---|---|
| Criterios clínicos y hematológicos | Estudios genéticos | Otras características |
| GM-CSF = factor estimulante de colonias de granulocitos y macrófagos; NF1 = neurofibromatosis de tipo 1. | ||
| a Los pacientes que tienen una lesión de categoría 2 deben satisfacer los criterios de la categoría 1, pero no los de la categoría 3. Los pacientes que no tienen una lesión de categoría 2 deben satisfacer los criterios de las categorías 1 y 3. | ||
| b Cabe destacar que sólo el 7 % de los pacientes con LMMJ no presentarán esplenomegalia al inicio, pero prácticamente todos los pacientes presentarán esplenomegalia varias semanas o meses después del cuadro clínico inicial. | ||
| Ausencia de la fusión génicaBCR::ABL1 | Mutación somática enKRAS,NRASoPTPN11(se deben excluir las mutaciones de la línea germinal) | Monosomía 7 u otras anormalidades cromosómicas, o por lo menos 2 de los criterios descritos a continuación: |
| >1 × 109 /l de monocitos circulantes | Diagnóstico clínico de NF1 o mutación en el genNF1 | — Precursores mieloides o eritroides circulantes |
| <20 % de blastocitos en la sangre periférica y médula ósea | Mutación de la línea germinal enCBLy pérdida de heterocigosis deCBL | — Aumento de la hemoglobina F según la edad |
| Esplenomegalia | — Hiperfosforilación de STAT5 | |
| — Hipersensibilidad a GM-CSF | ||
Patogenia y síndromes relacionados
La patogenia de la LMMJ se vinculó con la activación de la vía del oncogén RAS, así como con síndromes relacionados (consultar la Figura 1).[
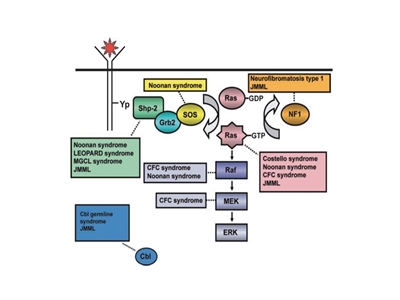
Los niños con neurofibromatosis de tipo 1 (NF1) y síndrome de Noonan tienen un aumento del riesgo de presentar LMMJ:[
- NF1. Hasta el 14 % de los casos de LMMJ se presentan en niños con NF1.[
2 ] - Síndrome de Noonan. El síndrome de Noonan a menudo se hereda como una enfermedad autosómica dominante, pero también surge de manera espontánea. Se caracteriza por dismorfia facial, estatura baja, cuello alado, anomalías neurocognitivas y cardíacas. Se observaron mutaciones de la línea germinal en PTPN11 en niños con síndrome de Noonan y en niños con LMMJ.[
10 ,11 ,12 ]Cabe destacar que algunos niños con síndrome de Noonan tienen un cuadro hematológico indistinguible de la LMMJ que remite de forma espontánea durante la lactancia de manera similar a lo que ocurre en los niños con síndrome de Down y trastorno mieloproliferativo transitorio.[
5 ,12 ]En una cohorte prospectiva numerosa de 641 pacientes con síndrome de Noonan y mutación de la línea germinal en PTPN11, se encontró que 36 pacientes (~6 %) exhibían características mieloproliferativas y 20 pacientes (~3 %) cumplieron los criterios de diagnóstico de consenso de la LMMJ.[
12 ] De los 20 pacientes que cumplían con los criterios de LMMJ, 12 pacientes tenían manifestaciones neonatales graves (por ejemplo, complicaciones potencialmente mortales relacionadas con anomalías cardíacas congénitas, derrames pleurales, infiltrados leucémicos o trombocitopenia), y 10 de los 20 pacientes murieron en el primer mes de vida. De los 8 pacientes restantes, ninguno necesitó tratamiento intensivo en el momento del diagnóstico o durante el seguimiento. Al cabo de una mediana de seguimiento de 3 años, los 16 pacientes con características mieloproliferativas que no cumplían con los criterios de LMMJ estaban vivos, y ninguno de ellos recibió quimioterapia.
Las mutaciones en el gen CBL, que da origen a la proteína ligasa de ubiquitina E3 , que participa en la selección de proteínas, en especial de tirosinas–cinasas, para la degradación en los proteasomas, se presenta en el 10 % al 15 % de los casos de LMMJ;[
Características genómicas de la leucemia mielomonocítica juvenil
Características moleculares de la leucemia mielomonocítica juvenil
El panorama genómico de la leucemia mielomonocítica juvenil (LMMJ) se caracteriza por mutaciones en 1 de los 5 genes de la vía RAS: NF1, NRAS, KRAS, PTPN11 y CBL.[
La tasa de mutaciones de las células leucémicas en la JMML es muy baja, pero se observan mutaciones adicionales en genes diferentes a los 5 genes de la vía RAS descritos antes.[
En un informe en el que se describe el panorama genómico de la LMMJ se encontró que 16 de 150 pacientes (11 %) carecían de mutaciones canónicas de la vía RAS. De esos 16 pacientes, 3 presentaban fusiones en el marco de lectura que afectaban receptores tirosina–cinasas (fusiones génicas DCTN1::ALK, RANBP2::ALK y TBL1XR1::ROS1). Todos estos pacientes presentaban monosomía 7 y tenían 56 meses o más. Un paciente con la fusión de ALK se trató con crizotinib y quimioterapia convencional, logró una remisión molecular completa, luego se sometió a un trasplante alogénico de médula ósea.[
Pronóstico (factores genómicos y moleculares)
Varios factores genómicos afectan el pronóstico de los pacientes con LMMJ; entre ellos, los siguientes:
- Número de mutaciones fuera de la vía RAS. Un factor de pronóstico de los niños con LMMJ es el número de mutaciones diferentes de las mutaciones en la vía RAS que son definitorias de enfermedad.[
18 ,19 ]- En un estudio se observó que, en el momento del diagnóstico, se encontraron entre 0 a 1 alteraciones somáticas (mutación patógena o monosomía 7) en 64 pacientes (65,3 %), mientras que se encontraron 2 o más alteraciones en 34 pacientes (34,7 %).[
19 ] En el análisis multivariante, el número de mutaciones (2 o más vs. 0 a 1) conservó la significación como factor de pronóstico de supervivencia sin complicaciones (SSC) y supervivencia general (SG) más precarias. Una mayor proporción de pacientes con diagnóstico de 2 o más alteraciones tenían más edad y eran varones; estos pacientes también exhibieron una tasa más alta de monosomía 7 o mutaciones somáticas en NF1.[19 ] - En otro estudio se observó que alrededor del 60 % de los pacientes tenían 1 o más mutaciones adicionales a la mutación en la vía RAS que son definitorias de la enfermedad. Estos pacientes tenían una SG inferior en comparación con los pacientes que no tenían mutaciones adicionales (tasa de SG a 3 años, 61 vs 85 %, respectivamente).[
18 ] - En un tercer estudio se observó una tendencia hacia una SG inferior para los pacientes con 2 mutaciones o más en comparación con los pacientes con 1 o ninguna mutación.[
20 ]
- En un estudio se observó que, en el momento del diagnóstico, se encontraron entre 0 a 1 alteraciones somáticas (mutación patógena o monosomía 7) en 64 pacientes (65,3 %), mientras que se encontraron 2 o más alteraciones en 34 pacientes (34,7 %).[
- Mutaciones dobles de la vía RAS. A pesar de que las mutaciones en los 5 genes canónicos de la vía RAS relacionados con la LMMJ (NF1, NRAS, KRAS, PTPN11, y CBL) son por lo general mutuamente excluyentes, el 4 % al 17 % de los casos presentan mutaciones en 2 de estos genes de la vía RAS,[
18 ,19 ] que es un hallazgo que se relacionó con un pronóstico más precario.[18 ,19 ]- En un informe se identificaron 2 mutaciones en la vía RAS en el 11 % de los pacientes de LMMJ, y estos pacientes tuvieron una tasa de SSC significativamente inferior (14 %) en comparación con la de los pacientes que tenían una sola mutación en la vía RAS (62 %). Los pacientes con el síndrome de Noonan se excluyeron del análisis.[
19 ] - Se informaron resultados similares en un segundo estudio en el que se observó que los pacientes con mutaciones dobles en la vía RAS (15 de 96 pacientes) tenían tasas de supervivencia más bajas que los pacientes sin mutaciones adicionales o con mutaciones adicionales a la mutación de la vía RAS.[
18 ]
- En un informe se identificaron 2 mutaciones en la vía RAS en el 11 % de los pacientes de LMMJ, y estos pacientes tuvieron una tasa de SSC significativamente inferior (14 %) en comparación con la de los pacientes que tenían una sola mutación en la vía RAS (62 %). Los pacientes con el síndrome de Noonan se excluyeron del análisis.[
- Perfil de metilación del DNA.
- En un estudio se aplicó un perfil de metilación del DNA a una cohorte de descubrimiento de 39 pacientes de LMMJ y a una cohorte de validación de 40 pacientes. En ambas cohortes se observaron subgrupos de LMMJ característicos con grados de metilación alto, medio o bajo. Los pacientes con los grados de metilación más bajos tuvieron las tasas de supervivencia más altas, y en la cohorte de metilación baja todos menos 1 de los 15 pacientes presentaron resolución espontánea. El estado de metilación alta se relacionó con tasas más bajas de SSC.[
22 ] - En otro estudio se aplicó un perfil de metilación del DNA a una cohorte de 106 pacientes de LMMJ y se observó un subgrupo de pacientes con un perfil de hipermetilación y un subgrupo de pacientes con un perfil de hipometilación. Los pacientes del grupo de hipermetilación tuvieron una SG significativamente más baja que la de los pacientes del grupo de hipometilación (tasa de SG a 5 años, 46 vs. 73 %, respectivamente). Los pacientes en el grupo de hipermetilación también tuvieron una tasa de supervivencia sin trasplante a 5 años significativamente más precaria que la de los pacientes del grupo de hipometilación (2,2 %, IC 95 %, 0,2–10,1 vs. 41,2 %; IC 95 %, 27,1–54,8 %). El estado de hipermetilación se relacionó con 2 o más mutaciones, concentraciones más altas de hemoglobina fetal, mayor edad y recuento de plaquetas más bajo en el momento del diagnóstico. Todos los pacientes con síndrome de Noonan estaban en el grupo de hipometilación.[
20 ] - En un estudio se examinaron 33 pacientes de LMMJ que tenían mutaciones en CBL y se identificaron 31 pacientes con metilación baja y 2 pacientes con metilación intermedia. Los 2 niños con metilación intermedia recayeron después de someterse a TCMH. Debido a que el tratamiento, que incluyó la observación sola, varió entre los 31 pacientes con metilación baja, no se pudo valorar por completo el efecto del perfil de metilación en las decisiones terapéuticas y los desenlaces. Sin embargo, el estado de metilación no fue pronóstico de resolución espontánea.[
17 ]
- En un estudio se aplicó un perfil de metilación del DNA a una cohorte de descubrimiento de 39 pacientes de LMMJ y a una cohorte de validación de 40 pacientes. En ambas cohortes se observaron subgrupos de LMMJ característicos con grados de metilación alto, medio o bajo. Los pacientes con los grados de metilación más bajos tuvieron las tasas de supervivencia más altas, y en la cohorte de metilación baja todos menos 1 de los 15 pacientes presentaron resolución espontánea. El estado de metilación alta se relacionó con tasas más bajas de SSC.[
- Sobreexpresión de LIN28B. La sobreexpresión de LIN28B se presenta en casi la mitad de los niños con LMMJ e identifica un subgrupo de LMMJ distintivo desde el punto de vista biológico. La LIN28B es una proteína de unión al RNA que regula la renovación de células madre.[
23 ]- La sobreexpresión de LIN28B se correlacionó de forma directa con las concentraciones sanguíneas altas de hemoglobina fetal y la edad (ambos relacionados con un pronóstico más precario), y se correlacionó de forma inversa con monosomía 7 (también relacionada con un pronóstico más precario). Aunque la sobreexpresión de LIN28B permite identificar un subconjunto de pacientes con aumento de riesgo de fracaso del tratamiento, se encontró que no era un factor de pronóstico independiente cuando se consideran otros factores como la edad o la monosomía 7.[
23 ] - En otro estudio también se observó un subgrupo de pacientes de LMMJ con aumento de la expresión de LIN28B y se identificó a LIN28B como el gen cuya expresión se relacionó de manera más contundente con el estado de hipermetilación.[
20 ]
- La sobreexpresión de LIN28B se correlacionó de forma directa con las concentraciones sanguíneas altas de hemoglobina fetal y la edad (ambos relacionados con un pronóstico más precario), y se correlacionó de forma inversa con monosomía 7 (también relacionada con un pronóstico más precario). Aunque la sobreexpresión de LIN28B permite identificar un subconjunto de pacientes con aumento de riesgo de fracaso del tratamiento, se encontró que no era un factor de pronóstico independiente cuando se consideran otros factores como la edad o la monosomía 7.[
Pronóstico (factores pronósticos)
Edad, recuento de plaquetas y concentración de hemoglobina fetal después de cualquier tratamiento. En el pasado, más del 90 % de los pacientes con leucemia mielomonocítica juvenil (LMMJ) morían a pesar de la quimioterapia,[
- Enfermedad rápidamente progresiva y deceso temprano.
- Enfermedad estable transitoria seguida de progresión y muerte.
- Mejoría clínica que dura hasta 9 años antes de la progresión o, de manera infrecuente, supervivencia a largo plazo.
Los factores pronósticos favorables para la supervivencia después de cualquier terapia son: edad menor de 2 años, recuento de plaquetas mayor a 33 × 109 /l, y concentraciones bajas de hemoglobina fetal ajustadas según la edad.[
Tratamiento de la leucemia mielomonocítica juvenil
Las opciones de tratamiento de la leucemia mielomonocítica juvenil (LMMJ) son las siguientes:
- Trasplante de células madre hematopoyéticas (TCMH).
La función de la terapia antileucémica convencional en el tratamiento de la LMMJ no está definida. La falta de criterios consensuados de respuesta para la LMMJ complica la determinación de la función de fármacos específicos para el tratamiento de la LMMJ.[
En la actualidad, el TCMH ofrece la mejor probabilidad de cura de la LMMJ.[
Evidencia (trasplante de células madre hematopoyéticas):
- En un informe reciente del European Working Group on Childhood Myelodysplastic Syndromes llevado a cabo en múltiples centros, se incluyeron 100 receptores de trasplantes tratados con un régimen preparatorio común de busulfano, ciclofosfamida y melfalán, con globulina antitimocítica o sin esta. Los receptores de trasplantes se habían tratado con quimioterapia pretrasplante de diversa intensidad o fármacos diferenciadores, y algunos pacientes se habían sometido a esplenectomía.[
25 ]- La tasa de supervivencia sin complicaciones a 5 años fue del 55 % para los niños con LMMJ que recibieron un TCMH de donante emparentado compatible con HLA idéntico y del 49 % en los niños con LMMJ que recibieron TCMH de donantes no emparentados.
- En el análisis multivariante no se observaron efectos en la supervivencia de la quimioterapia similar a la utilizada para la leucemia mieloide aguda versus la quimioterapia de dosis baja o la ausencia de quimioterapia.
- No se observaron efectos en la supervivencia con una esplenectomía anterior al trasplante o diferencias en el tamaño del bazo.
- Tampoco se encontraron diferencias en la comparación de los desenlaces a partir de donantes emparentados versus no emparentados.
- Solo factores como una edad de más de 4 años y el sexo mostraron ser factores de pronóstico adverso del desenlace y de presentar mayor riesgo de recaída (riesgo relativo [RR], 2,24 [1,07–4,69]; P = 0,032 para mayor edad; RR, 2,22 [1,09–4,50]; P = 0,028 para las mujeres).[
25 ]
- El trasplante de sangre de cordón umbilical produce una tasa de supervivencia sin enfermedad a 5 años del 44 %, con mejores desenlaces en los niños menores de 1,4 años en el momento del diagnóstico, en aquellos con un cariotipo sin monosomía 7 y en quienes reciben unidades de sangre de cordón con compatibilidad de HLA 5/6 a 6/6.[
36 ][Nivel de evidencia C2] Esto indica que la sangre de cordón umbilical puede proporcionar una fuente adicional de donantes para este grupo de niños. - En un pequeño número de pacientes también se notificó el uso de regímenes preparatorios de intensidad reducida para disminuir los efectos adversos del trasplante; en general se usa en pacientes que no son aptos para el TCMH mielosupresor.[
37 ,38 ]- El COG condujo un ensayo aleatorizado en niños con LMMJ en el que se comparó el régimen preparatorio de intensidad estándar (busulfano, ciclofosfamida y melfalán) con un régimen de intensidad reducida (busulfano y fludarabina).[
39 ]- Se interrumpió temprano la inscripción en el ensayo cuando en un análisis intermedio se observó una frecuencia más alta de recaída o persistencia de la enfermedad (7 de 9 pacientes) en los niños que recibieron el régimen de intensidad reducida que en aquellos que recibieron el régimen de intensidad estándar (1 de 6 pacientes).
- El COG condujo un ensayo aleatorizado en niños con LMMJ en el que se comparó el régimen preparatorio de intensidad estándar (busulfano, ciclofosfamida y melfalán) con un régimen de intensidad reducida (busulfano y fludarabina).[
La recidiva de la enfermedad es la causa primaria de fracaso del tratamiento para los niños con LMMJ después de un TCMH y se presenta en el 30 % al 40 % de los casos.[
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de ensayo clínico nacional o institucional en curso:
- COG-ADVL1521 (NCT03190915) (Trametinib in Treating Patients With Relapsed or Refractory JMML): en este ensayo se evalúa la actividad del trametinib (inhibidor de MEK1/2, que está secuencia abajo de las vías de señalización RAS/MAPK) en pacientes con LMMJ pediátrico en recaída o resistente al tratamiento. El fundamento del estudio de este fármaco se basa en el hallazgo de que casi todas las mutaciones genéticas que se encuentran en la LMMJ producen una vía de señalización RAS anormal. Son pacientes aptos para participar en el ensayo quienes tienen enfermedad en recaída o que persiste después de recibir quimioterapia intravenosa (como fludarabina o citarabina) o un trasplante de células madre hematopoyéticas, pero no quienes recibieron dosis bajas de quimioterapia oral (como mercaptopurina). La meta principal del ensayo es determinar la tasa de respuesta al trametinib oral administrado diariamente en ciclos de 28 días.
Referencias:
- Passmore SJ, Chessells JM, Kempski H, et al.: Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia in the UK: a population-based study of incidence and survival. Br J Haematol 121 (5): 758-67, 2003.
- Niemeyer CM, Arico M, Basso G, et al.: Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89 (10): 3534-43, 1997.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Chan RJ, Cooper T, Kratz CP, et al.: Juvenile myelomonocytic leukemia: a report from the 2nd International JMML Symposium. Leuk Res 33 (3): 355-62, 2009.
- Loh ML: Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol 152 (6): 677-87, 2011.
- Bresolin S, Zecca M, Flotho C, et al.: Gene expression-based classification as an independent predictor of clinical outcome in juvenile myelomonocytic leukemia. J Clin Oncol 28 (11): 1919-27, 2010.
- Olk-Batz C, Poetsch AR, Nöllke P, et al.: Aberrant DNA methylation characterizes juvenile myelomonocytic leukemia with poor outcome. Blood 117 (18): 4871-80, 2011.
- Stiller CA, Chessells JM, Fitchett M: Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer 70 (5): 969-72, 1994.
- Choong K, Freedman MH, Chitayat D, et al.: Juvenile myelomonocytic leukemia and Noonan syndrome. J Pediatr Hematol Oncol 21 (6): 523-7, 1999 Nov-Dec.
- Tartaglia M, Niemeyer CM, Fragale A, et al.: Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34 (2): 148-50, 2003.
- Kratz CP, Niemeyer CM, Castleberry RP, et al.: The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 106 (6): 2183-5, 2005.
- Strullu M, Caye A, Lachenaud J, et al.: Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet 51 (10): 689-97, 2014.
- Loh ML, Sakai DS, Flotho C, et al.: Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 114 (9): 1859-63, 2009.
- Muramatsu H, Makishima H, Jankowska AM, et al.: Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood 115 (10): 1969-75, 2010.
- Niemeyer CM, Kang MW, Shin DH, et al.: Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet 42 (9): 794-800, 2010.
- Pérez B, Mechinaud F, Galambrun C, et al.: Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet 47 (10): 686-91, 2010.
- Hecht A, Meyer JA, Behnert A, et al.: Molecular and phenotypic diversity of CBL-mutated juvenile myelomonocytic leukemia. Haematologica 107 (1): 178-186, 2022.
- Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 (11): 1334-40, 2015.
- Stieglitz E, Taylor-Weiner AN, Chang TY, et al.: The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47 (11): 1326-33, 2015.
- Murakami N, Okuno Y, Yoshida K, et al.: Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 131 (14): 1576-1586, 2018.
- Sakaguchi H, Okuno Y, Muramatsu H, et al.: Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet 45 (8): 937-41, 2013.
- Stieglitz E, Mazor T, Olshen AB, et al.: Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat Commun 8 (1): 2127, 2017.
- Helsmoortel HH, Bresolin S, Lammens T, et al.: LIN28B overexpression defines a novel fetal-like subgroup of juvenile myelomonocytic leukemia. Blood 127 (9): 1163-72, 2016.
- Freedman MH, Estrov Z, Chan HS: Juvenile chronic myelogenous leukemia. Am J Pediatr Hematol Oncol 10 (3): 261-7, 1988 Fall.
- Locatelli F, Nöllke P, Zecca M, et al.: Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood 105 (1): 410-9, 2005.
- Bergstraesser E, Hasle H, Rogge T, et al.: Non-hematopoietic stem cell transplantation treatment of juvenile myelomonocytic leukemia: a retrospective analysis and definition of response criteria. Pediatr Blood Cancer 49 (5): 629-33, 2007.
- Castleberry RP, Emanuel PD, Zuckerman KS, et al.: A pilot study of isotretinoin in the treatment of juvenile chronic myelogenous leukemia. N Engl J Med 331 (25): 1680-4, 1994.
- Woods WG, Barnard DR, Alonzo TA, et al.: Prospective study of 90 children requiring treatment for juvenile myelomonocytic leukemia or myelodysplastic syndrome: a report from the Children's Cancer Group. J Clin Oncol 20 (2): 434-40, 2002.
- Loh ML: Childhood myelodysplastic syndrome: focus on the approach to diagnosis and treatment of juvenile myelomonocytic leukemia. Hematology Am Soc Hematol Educ Program 2010: 357-62, 2010.
- Hasle H: Myelodysplastic and myeloproliferative disorders in children. Curr Opin Pediatr 19 (1): 1-8, 2007.
- Stieglitz E, Ward AF, Gerbing RB, et al.: Phase II/III trial of a pre-transplant farnesyl transferase inhibitor in juvenile myelomonocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (4): 629-36, 2015.
- Smith FO, King R, Nelson G, et al.: Unrelated donor bone marrow transplantation for children with juvenile myelomonocytic leukaemia. Br J Haematol 116 (3): 716-24, 2002.
- Yusuf U, Frangoul HA, Gooley TA, et al.: Allogeneic bone marrow transplantation in children with myelodysplastic syndrome or juvenile myelomonocytic leukemia: the Seattle experience. Bone Marrow Transplant 33 (8): 805-14, 2004.
- Baker D, Cole C, Price J, et al.: Allogeneic bone marrow transplantation in juvenile myelomonocytic leukemia without total body irradiation. J Pediatr Hematol Oncol 26 (3): 200-3, 2004.
- Locatelli F, Niemeyer CM: How I treat juvenile myelomonocytic leukemia. Blood 125 (7): 1083-90, 2015.
- Locatelli F, Crotta A, Ruggeri A, et al.: Analysis of risk factors influencing outcomes after cord blood transplantation in children with juvenile myelomonocytic leukemia: a EUROCORD, EBMT, EWOG-MDS, CIBMTR study. Blood 122 (12): 2135-41, 2013.
- Yabe M, Sako M, Yabe H, et al.: A conditioning regimen of busulfan, fludarabine, and melphalan for allogeneic stem cell transplantation in children with juvenile myelomonocytic leukemia. Pediatr Transplant 12 (8): 862-7, 2008.
- Koyama M, Nakano T, Takeshita Y, et al.: Successful treatment of JMML with related bone marrow transplantation after reduced-intensity conditioning. Bone Marrow Transplant 36 (5): 453-4; author reply 454, 2005.
- Dvorak CC, Satwani P, Stieglitz E, et al.: Disease burden and conditioning regimens in ASCT1221, a randomized phase II trial in children with juvenile myelomonocytic leukemia: A Children's Oncology Group study. Pediatr Blood Cancer 65 (7): e27034, 2018.
- Yoshimi A, Bader P, Matthes-Martin S, et al.: Donor leukocyte infusion after hematopoietic stem cell transplantation in patients with juvenile myelomonocytic leukemia. Leukemia 19 (6): 971-7, 2005.
- Yoshimi A, Mohamed M, Bierings M, et al.: Second allogeneic hematopoietic stem cell transplantation (HSCT) results in outcome similar to that of first HSCT for patients with juvenile myelomonocytic leukemia. Leukemia 21 (3): 556-60, 2007.
Leucemia mielógena crónica
Incidencia
La leucemia mielógena crónica (LMC) representa menos del 5 % de todas las leucemias infantiles, y, en el intervalo de edad pediátrica, se presenta con mayor frecuencia en los adolescentes mayores.[
Anomalías moleculares
La anomalía citogenética más característica de la LMC es el cromosoma Filadelfia (Ph), una translocación de los cromosomas 9 y 22 (t(9;22)) que produce la proteína de fusión BCR::ABL1.[
Cuadro clínico inicial
La LMC se caracteriza por una leucocitosis pronunciada que a menudo se relaciona con trombocitosis y, a veces, con un funcionamiento plaquetario anormal. Una aspiración o biopsia de médula ósea revela hipercelularidad con maduración granulocítica relativamente normal y no se observa un aumento importante en los blastocitos leucémicos. A pesar de que se observa actividad reducida de la fosfatasa alcalina leucocitaria en la LMC, este no es un hallazgo específico.
La LMC tiene las siguientes 3 fases clínicas:
- Fase crónica. La fase crónica, que dura cerca de 3 años cuando no se trata, por lo general se manifiesta con síntomas secundarios a la hiperleucocitosis como debilidad, fiebre, sudores nocturnos, dolor óseo, disnea, priapismo, dolor en el cuadrante superior izquierdo (esplenomegalia), y, en pocas ocasiones, hipoacusia y trastornos visuales.
- Fase acelerada. La fase acelerada se caracteriza por esplenomegalia progresiva, trombocitopenia y un porcentaje alto de blastocitos periféricos y en la médula ósea, junto con acumulación de anomalías cariotípicas además del cromosoma Ph.
- Fase de crisis blástica. La crisis blástica es visible en la médula ósea, con más del 20 % de blastocitos o lesiones cloromatosas y una imagen clínica indistinguible de una leucemia aguda. Cerca de dos tercios de las crisis blásticas son mieloides, y el resto es linfoide, por lo usual de linaje B. Los pacientes en crisis blástica morirán al cabo de pocos meses.[
3 ]
Tratamiento de la leucemia mielógena crónica: Perspectiva histórica
Antes de la era de los inhibidores de tirosina–cinasas (ITC), el trasplante alogénico de células madre hematopoyéticas era el tratamiento primario para los niños con leucemia mielógena crónica (LMC). En informes publicados en este período, se describen tasas de supervivencia del 70 % al 80 % cuando se utilizó un donante emparentado con compatibilidad (DEC) de HLA para tratar a los niños durante la fase crónica temprana; las tasas de supervivencia fueron más bajas cuando se utilizaron donantes no emparentados con compatibilidad de HLA.[
Las tasas de recaída fueron bajas (menos del 20 %) cuando los trasplantes se hicieron durante la fase crónica.[
En comparación con el trasplante en la fase crónica, el trasplante durante la fase acelerada o la crisis blástica, así como y durante la segunda fase crónica, produce una disminución significativa de la supervivencia.[
La introducción del ITC imatinib como opción terapéutica dirigida a inhibir la cinasa de la fusión BCR::ABL1 revolucionó el tratamiento de los pacientes con LMC, tanto en niños como en adultos.[
Tratamiento de la leucemia mielógena crónica con inhibidores de tirosina–cinasas
El imatinib es un inhibidor potente de tirosina–cinasas ABL1 y también de los receptores (α y β) del factor de crecimiento derivado de las plaquetas (PDGF) y de KIT. El tratamiento con imatinib produce remisiones clínicas, citogenéticas y moleculares (definidas por la ausencia de transcritos de la fusión BCR::ABL1) en una proporción alta de pacientes de LMC tratados durante la fase crónica.[
Evidencia (imatinib para adultos):
- El imatinib reemplazó el uso del interferón α recombinante para el tratamiento inicial de la LMC a partir de los resultados de un gran ensayo numeroso de fase III en el que se comparó el imatinib con el interferón combinado con citarabina (IRIS).[
11 ,12 ]- Los pacientes que recibieron imatinib presentaron tasas de respuesta citogenética completa más altas (76 % vs. 14 % a los 18 meses).[
11 ] La tasa de fracaso del tratamiento disminuyó con el paso del tiempo, del 3,3 % y 7,5 % en el primer y segundo año de tratamiento con imatinib, respectivamente, a menos del 1 % al quinto año de tratamiento.[12 ] - Después de eliminar del análisis a los pacientes que murieron por causas no relacionadas con la LMC o el trasplante, la tasa de supervivencia general calculada para los pacientes asignados al azar a imatinib fue del 95 % a los 60 meses.[
12 ]
- Los pacientes que recibieron imatinib presentaron tasas de respuesta citogenética completa más altas (76 % vs. 14 % a los 18 meses).[
Se establecieron pautas de tratamiento con imatinib para los adultos con LMC de acuerdo con la respuesta del paciente al tratamiento, que incluye el tiempo para lograr una respuesta hematológica completa, una respuesta citogenética completa y una respuesta molecular mayor (definida como el logro de una disminución de 3 log en el cociente fusión génica BCR::ABL1/gen de control).[
El cumplimiento inadecuado del tratamiento es una causa importante de pérdida de la respuesta citogenética completa y de fracaso del imatinib para los adultos con LMC durante el tratamiento a largo plazo.[
Se demostró que otros 2 ITC, dasatinib y nilotinib, son eficaces en pacientes con reacción inadecuada al imatinib, pero no en los pacientes con mutación en T315I. Tanto dasatinib como nilotinib recibieron la aprobación reglamentaria para el tratamiento de adultos con LMC en fase crónica recién diagnosticada, a partir de los siguientes estudios:
- Dasatinib. El dasatinib se aprobó a partir de un ensayo de fase III en el que se comparó el dasatinib (100 mg al día) con el imatinib (400 mg al día).[
21 ] No hubo diferencias significativas en la supervivencia sin progresión (SSP) o la SG. Sin embargo, después de 12 meses de tratamiento, el dasatinib se relacionó con una tasa más alta de respuesta citogenética completa (83 % vs. 72 %, P = 0,001) y respuesta molecular mayor (46 % vs. 28 %, P < 0,0001). Las respuestas se lograron en menos tiempo con el dasatinib (P < 0,0001). - Nilotinib. El nilotinib (dosis de 300 o 400 mg 2 veces al día) se comparó con el imatinib (400 mg al día) en un ensayo de fase III.[
22 ] Al cabo de 12 meses, las tasas de respuesta citogenética completa fueron significativamente más altas con el nilotinib (80 % para la dosis de 300 mg y 78 % para la dosis de 400 mg) que con el imatinib (65 %) (P < 0,001 para ambas comparaciones). Además, el nilotinib se relacionó con tasas más altas de respuesta molecular mayor (44 % para la dosis de 300 mg y 43 % para la dosis de 400 mg en comparación con 22 % del imatinib, P < 0,001 para ambas comparaciones). El nilotinib en dosis de 300 mg 2 veces al día se relacionó con un perfil de inocuidad más favorable en comparación con la dosis de 400 mg.
Debido a la superioridad sobre el imatinib en términos de tasas de respuesta citogenética completa y respuesta molecular mayor, tanto el dasatinib como el nilotinib se usan mucho como tratamiento de primera línea en adultos con LMC. Sin embargo, la SSP y la SG son similares para los tres fármacos a pesar de que se observan respuestas más rápidas con dasatinib o nilotinib que con imatinib cuando se usan como tratamiento primario.[
El bosutinib es otro ITC que actúa sobre la fusión génica BCR::ABL1 y que la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) aprobó para el tratamiento de adultos con LMC en todas las fases, o para pacientes con intolerancia o enfermedad resistente a un tratamiento previo con otro ITC. El bosutinib no se ha estudiado en la población pediátrica.
El ponatinib es un inhibidor de la proteína de fusión BCR::ABL1 eficaz contra la mutación en T315I.[
En los adultos con LMC que se someten a TCMH alogénico no hay evidencia de que el imatinib pretrasplante afecte de manera adversa el desenlace.
Evidencia (imatinib seguido de trasplante de células madre hematopoyéticas en adultos):
- En un estudio retrospectivo en el que se comparó a 145 pacientes que recibieron imatinib antes del trasplante con una cohorte histórica de 231 pacientes, no se observaron diferencias en los efectos tóxicos hepáticos tempranos o en el retraso de la incorporación del injerto.[
28 ]- Además, la SG, la supervivencia sin enfermedad, la recaída y la mortalidad no relacionada con la recaída fueron similares en las dos cohortes.
- El único factor relacionado con un desenlace precario en la cohorte que recibió imatinib fue una respuesta inicial precaria al imatinib.
- En un informe del Center for International Blood and Marrow Transplant Research, se proporcionó evidencia adicional sobre la falta de efecto del imatinib pretrasplante en los desenlaces postrasplante; en este informe se compararon los desenlaces de 181 adultos y niños con LMC en la primera fase crónica tratados con imatinib antes del TCMH con los desenlaces de 657 personas que no recibieron imatinib antes del TCMH.[
29 ]- Entre los pacientes en la primera fase crónica, la terapia con imatinib antes del TCMH se relacionó con mejor SG.
- En un tercer informe de imatinib seguido por TCMH alogénico se respalda la eficacia de esta estrategia de trasplante para los pacientes con fracaso del imatinib en la primera fase crónica.[
13 ]- En este grupo (n = 37), la tasa de SG a 3 años fue del 94 %, y alrededor del 90 % de los pacientes logró una remisión molecular completa después del TCMH.
En los pacientes adultos tratados con un ITC solo (sin TCMH), se desconoce la duración óptima del tratamiento y a la mayoría de los pacientes se les sigue administrando tratamiento con un ITC por tiempo indefinido. Se están realizando estudios para determinar si es seguro suspender el tratamiento con ITC y los criterios pronósticos más importantes de remisión prolongada después de finalizar la terapia con ITC.
Evidencia (duración de la terapia con imatinib en adultos):
- en un esfuerzo por contestar la pregunta sobre la duración del tratamiento, en un estudio prospectivo se notificó que 69 adultos tratados con imatinib durante más de 2 años lograron una respuesta citogené[
30 ]- De este grupo, el 61 % presentó recaída de la enfermedad, pero alrededor del 38 % todavía presentaba una respuesta citogenética importante a los 24 meses.
- Cabe destacar que todos los pacientes con recidiva de la enfermedad volvieron a responder al reiniciarse el imatinib.
- En otro estudio se notificó sobre 40 pacientes con LMC en fase crónica que suspendieron el tratamiento con imatinib después de por lo menos 2 años de enfermedad residual mínima (ERM) indetectable mediante reacción en cadena de la polimerasa (PCR).[
31 ]- A los 24 meses, la probabilidad de remisión molecular prolongada en los pacientes que ya no recibían imatinib fue del 47,1 %.
- La mayoría de las recidivas se presentaron en el transcurso de 4 meses después de suspender el tratamiento con imatinib, y no se observaron recaídas después de los 27 meses.
- Todos los pacientes con recaída molecular presentaron una respuesta favorable cuando se reinició el imatinib; al cabo de una mediana de seguimiento de 42 meses ningún paciente presentó enfermedad progresiva ni la fusión génica BCR::ABL1.
Se necesita más investigación antes de que se pueda recomendar en la práctica clínica habitual suspender el imatinib u otras terapias dirigidas a BCR::ABL1 en determinados pacientes con LMC que se encuentra en remisión molecular.
Tratamiento de la leucemia mielógena crónica infantil
Las opciones de tratamiento para los niños con leucemia mielógena crónica (LMC) son las siguientes:
- Terapia con un inhibidor de tirosina–cinasas (ITC), como el imatinib.
Con el imatinib se observó un grado alto de actividad en los niños con LMC, que es comparable con la actividad observada en adultos.[
Evidencia (imatinib en niños):
- En un ensayo prospectivo, 44 pacientes pediátricos con diagnóstico nuevo de LMC se trataron con imatinib (260 mg/día).[
36 ]- La tasa de SSP a los 36 meses fue del 98 %.
- Se logró una respuesta hematológica completa en el 98 % de los pacientes.
- La tasa de respuesta citogenética completa fue del 61 % y la tasa de respuesta molecular mayor fue del 31 % a los 12 meses, similar a las tasas observadas en pacientes adultos con LMC en fase crónica tratados con imatinib.
Como resultado de este grado alto de actividad, es común iniciar el tratamiento con imatinib en niños con LMC en lugar de proceder de inmediato con el trasplante alogénico de células madre.[
Las dosis de imatinib usadas en los ensayos de fase II para niños con LMC han oscilado entre 260 mg/m2 y 340 mg/m2; esto provee exposiciones farmacológicas comparables a las dosis fijas en adultos de 400 a 600 mg.[
Evidencia (dosis de imatinib en niños):
- En un estudio italiano de 47 pacientes pediátricos con LMC en fase crónica tratados con 340 mg/m2 al día de imatinib, se logró una respuesta citogenética completa en el 91,5 % de los pacientes durante una mediana de tiempo de 6 meses; la tasa de respuesta molecular mayor fue del 66,6 % a los 12 meses.[
36 ]Por lo tanto, comenzar con la dosis más alta de 340 mg/m2 tiene una eficacia superior y por lo general se tolera bien; si es necesario, se ajusta la dosis de acuerdo a los efectos tóxicos.[
35 ,36 ] - Las respuestas moleculares tempranas en niños se han relacionado con mejora de la SSP, semejante a los datos de respuesta molecular temprana en adultos. La respuesta molecular temprana se definió como aquellos niños que presentaron un cociente BCR::ABL1/ABL ≤10 % (usando una medición de la ERM por PCR) 3 meses después de comenzar con el imatinib.[
39 ]
Es adecuado usar en los niños las directrices de seguimiento para los adultos con LMC descritas antes.
Los niños por lo general toleran bien el imatinib, los efectos adversos a menudo son leves a moderados y reversibles una vez se interrumpe el tratamiento o se reduce la dosis.[
Hay menos datos publicados sobre la eficacia y la toxicidad de los otros dos ITC (dasatinib y nilotinib) aprobados por la FDA para su uso en niños con LMC.
Evidencia (dasatinib en niños):
- En un ensayo de fase I de dasatinib en niños, se observó que la distribución farmacológica, la tolerabilidad y la eficacia de este fármaco son similares a las observadas en los adultos.[
42 ,43 ] - En un ensayo de fase II en el que participaron 84 niños con LMC en fase crónica recién diagnosticada, se administró una dosis diaria de 60 mg/m2 (comprimidos) o de 72 mg/m2 (solución oral).[
44 ]- Después de 12 meses de tratamiento se logró una respuesta citogenética completa y una respuesta molecular mayor (disminución ≥3 log o ≤0,1 % en la escala internacional) en el 92 % y el 52 % de los pacientes, respectivamente, y una tasa de SSP a 4 años del 93 %.
- El dasatinib se toleró bien, con muy pocos efectos adversos de grados 3 o 4. No se observaron derrames pleurales o pericárdicos, ni complicaciones pulmonares.
Evidencia (nilotinib en niños):
- A partir de los resultados de dos ensayos patrocinados, la FDA aprobó el nilotinib en marzo de 2018 para el tratamiento de los niños con LMC.[
45 ,46 ]En un estudio inicial (NCT01077544 [CAMN107A2120]) de 11 pacientes, se evaluaron los datos farmacocinéticos, la inocuidad y la eficacia preliminar; en un segundo estudio (NCT01844765 [CAMN107A2203; AAML1321]) de 58 pacientes, se evaluaron la eficacia y la inocuidad. Se combinaron los datos de ambos estudios para un análisis conjunto de 69 pacientes, que abarcó a 25 pacientes con LMC recién diagnosticada y 44 pacientes con LMC resistente o intolerante al tratamiento. En ambos estudios se administró una dosis de 230 mg/m2 2 veces al día (redondeada a la dosis de 50 mg más cercana, con una dosis máxima de 400 mg).[
46 ]- En el ensayo de fase II, el 64 % de los pacientes con LMC recién diagnosticada obtuvo una respuesta molecular mayor al cabo de 1 año.
- La tolerabilidad del nilotinib en niños fue similar a la observada en los adultos. Los efectos secundarios principales que afectaron a más del 30 % de los niños fueron cefaleas, fiebre e hiperbilirrubinemia.
- La prolongación del intervalo de QT corregido (QTc, que en este ensayo se definió como un aumento de >30 ms con respecto al valor de referencia), es un efecto secundario conocido del nilotinib, y se observó en el 25 % de los niños en estos ensayos. Los investigadores recomiendan obtener un electrocardiograma al inicio, otro al cabo de 1 semana, y luego en forma periódica y después de cada ajuste de la dosis.
No se ha establecido una dosis pediátrica inocua para los otros ITC (por ejemplo, bosutinib y ponatinib).
Suspensión de la terapia con inhibidores de tirosina–cinasas
La suspensión del tratamiento con inhibidores de tirosina–cinasas (ITC) es una estrategia aceptada para los adultos con LMC que cumplen criterios estrictos relacionados con la duración del tratamiento y la respuesta al tratamiento. La European LeukemiaNet (ELN) y la U.S. National Comprehensive Cancer Network (NCCN) elaboraron directrices para la interrupción de los ITC.[
- Terapia con ITC por una duración mínima de 4 a 5 años según la ELN y de 3 años según la NCCN.
- Una duración mínima de respuesta molecular intensa (RMI o RM4) (concentración del transcrito de la proteína BCR::ABL1 ≤0,01 % de la International Scale [IS]) de 2 años para la ELN y la NCCN.
Estas directrices especifican una vigilancia estrecha de las concentraciones de transcritos de BCR::ABL1 después de la suspensión de los ITC. La pérdida de la respuesta molecular principal (RMP o RM3) (concentración del transcrito BCR::ABL1 ≤0,1 % IS) por lo general se usa como desencadenante para el reinicio de la terapia con ITC.
La pérdida de RMP suele ocurrir dentro de los primeros 6 meses de la suspensión de los ITC. La pérdida de RMP es mucho menos frecuente después de 1 año de la suspensión de los ITC. En un metanálisis se incluyeron 3105 pacientes adultos que comenzaron con un primer intento de suspensión de los ITC. En el estudio se encontró que la probabilidad de recidiva molecular era del 35 % después de 0 a 6 meses, del 8 % después de 6 a 12 meses, del 3 % después de 12 a 18 meses y del 3 % después de 18 a 24 meses.[
Hay pocos datos sobre la suspensión de ITC en niños con LMC. La poca experiencia se explica, en parte, por la baja incidencia de LMC en niños. Además, solo una minoría de niños con LMC que se tratan con ITC cumplen los criterios para la suspensión de los ITC. Por ejemplo, entre los pacientes que se incluyen en el International Chronic Myeloid Leukemia Pediatric Study (I-CML-Ped [NCT01281735]), solo el 9 % de los niños con LMC que se trataron con ITC cumplieron los criterios para su suspensión.[
- El Japan Pediatric Leukemia and Lymphoma Study Group (JPSLG) informó sobre 22 niños con LMC que cumplieron con sus criterios para la suspensión de los ITC, los cuales eran similares a los criterios de suspensión de los ITC de la NCCN.[
52 ] La mediana de edad en el momento del diagnóstico de la LMC fue de 9 años y la mediana de edad en el momento de la suspensión de los ITC fue de 16 años. La mediana de duración de la terapia con ITC superó los 8 años, y la mediana de duración de RM4 antes de la suspensión de ITC superó los 4 años. De los 22 niños, 11 experimentaron pérdida de la RMP en una mediana de 90 días después de la suspensión de ITC. Todos estos niños recuperaron más tarde la RM4, después de que reiniciaran la terapia con ITC. La tasa de remisión sin tratamiento a 12 meses fue del 50 % y no se observaron recaídas más allá de los 4 meses de la suspensión de los ITC.El síndrome de abstinencia de los ITC se observa en alrededor del 20 % al 30 % de los adultos cuando se suspende el tratamiento con ITC.[
53 ] El síndrome incluye dolor musculoesquelético que por lo general se presenta en los primeros 2 meses desde la suspensión de los ITC y continúa durante varios meses. En el estudio del JPLSG no se observó dolor musculoesquelético en los niños después de la suspensión de los ITC. - El International Registry of Childhood Chronic Myeloid Leukemia informó sobre 18 pacientes con LMC que, en el momento del diagnóstico, tenían menos de 18 años. Estos pacientes suspendieron el imatinib después de cumplir los criterios para la suspensión de ITC (es decir, en fase crónica con una RMI al imatinib prolongada [MR4; concentración de transcritos BCR::ABL1 ≤0,01 % IS]) durante al menos 2 años.[
50 ]De los 18 niños que dejaron de tomar imatinib, 9 (50 %), pasado cierto tiempo, reanudaron el tratamiento.[
50 ] De estos 9 pacientes, 7 presentaron pérdida de RMP (concentración de transcritos BCR::ABL1 ≤0,1 % IS). De los 7 pacientes, 6 recuperaron MR4 dentro de una mediana de cerca de 5 meses después reiniciar el tratamiento con ITC. El otro paciente logró una RMP después de reiniciar el tratamiento con ITC. En el caso de 2 pacientes adicionales que tenían un aumento de un logaritmo en las concentraciones del transcrito BCR::ABL1, pero que no cumplían con los criterios para la pérdida de MMR, fueron sus médicos quienes les indicaron reiniciar el tratamiento con imatinib. En los otros 9 pacientes que permanecieron en remisión sin tratamiento, la mediana del período de seguimiento después de la suspensión del imatinib fue de 50 meses. No se notificó síndrome de abstinencia de los ITC en ninguno de los pacientes que suspendió el imatinib.
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de ensayo clínico nacional o institucional en curso:
- AAML18P1 (NCT03817398) (Stopping TKIs in Affecting Treatment-Free Remission in Patients With Chronic-Phase CML): Este estudio del COG es un estudio longitudinal, prospectivo, no aleatorizado en el que se incluyeron niños que alcanzaron remisión profunda (BCR-ABL1 <0,01 % mediante reacción en cadena de la polimerasa cuantitativa en tiempo real de sangre periférica [>MR4, es decir, disminución >4-log]) por al menos 2 años después de recibir el mismo ITC durante al menos 3 años. En este ensayo se examina la incidencia de recidiva molecular después de suspender la terapia con ITC y la capacidad de los pacientes para volver a alcanzar la remisión si se produce una recidiva.
Tratamiento de la leucemia mielógena crónica recidivante o resistente al tratamiento
Las opciones de tratamiento para los niños con leucemia mielógena crónica (LMC) recidivante o resistente al tratamiento son las siguientes:
- Otros inhibidores de tirosina–cinasas como el dasatinib o el nilotinib.
- Trasplante alogénico de células madre hematopoyéticas.
En los niños que presentan una recaída hematológica o citogenética durante el tratamiento con imatinib o que tienen una respuesta inicial inadecuada al imatinib, se debería considerar analizar el estado de la mutación en el dominio de la cinasa BCR::ABL1 para ayudar a guiar el tratamiento posterior. De acuerdo con el estado de la mutación en el paciente, se pueden considerar otros inhibidores de tirosina–cinasas como el dasatinib o el nilotinib, a partir de la experiencia con estos fármacos en adultos y niños.[
Evidencia (dasatinib en niños con LMC resistente o intolerante al tratamiento):
- En un estudio de 14 niños con LMC resistente o intolerante al tratamiento, se observaron los siguientes resultados:[
44 ]- De los pacientes, el 76 % tuvo remisión citogenética completa, y el 41 % tuvo respuesta molecular mayor después de 12 meses de tratamiento con dasatinib.
- La tasa de SSP fue del 78 % a los 48 meses.
Evidencia (nilotinib en niños con LMC resistente o intolerante al tratamiento):
- En un estudio de 44 niños con LMC resistente o intolerante al imatinib o al dasatinib, se observaron los siguientes resultados:[
45 ]- De los pacientes, el 40,7 % alcanzaron una respuesta molecular mayor después de 12 meses de terapia con nilotinib.
- Después de una mediana de 11,3 meses, ningún paciente tuvo progresión de la enfermedad.
El dasatinib y el nilotinib son activos contra muchas mutaciones de fusión génica BCR::ABL1 que confieren resistencia al imatinib, aunque son ineficaces para los pacientes con la mutación T315I. Cuando hay una mutación T315I, resistente a todos los inhibidores de tirosina–cinasas aprobados por la FDA, se debe considerar un trasplante alogénico. El ponatinib, un inhibidor de la proteína de fusión BCR::ABL1 que es eficaz contra la mutación T315I en adultos, no se ha estudiado de forma prospectiva en la población pediátrica.
La pregunta sobre si un paciente pediátrico de LMC debe recibir un trasplante alogénico cuando se dispone de varios ITC sigue sin respuesta; sin embargo, en informes recientes se indica que la SSP no mejora cuando se utiliza un TCMH en comparación con la continuación del imatinib.[
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
- Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649. Also available online. Last accessed August 8, 2022.
- Quintás-Cardama A, Cortes J: Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 113 (8): 1619-30, 2009.
- O'Dwyer ME, Mauro MJ, Kurilik G, et al.: The impact of clonal evolution on response to imatinib mesylate (STI571) in accelerated phase CML. Blood 100 (5): 1628-33, 2002.
- Millot F, Esperou H, Bordigoni P, et al.: Allogeneic bone marrow transplantation for chronic myeloid leukemia in childhood: a report from the Société Française de Greffe de Moelle et de Thérapie Cellulaire (SFGM-TC). Bone Marrow Transplant 32 (10): 993-9, 2003.
- Cwynarski K, Roberts IA, Iacobelli S, et al.: Stem cell transplantation for chronic myeloid leukemia in children. Blood 102 (4): 1224-31, 2003.
- Weisdorf DJ, Anasetti C, Antin JH, et al.: Allogeneic bone marrow transplantation for chronic myelogenous leukemia: comparative analysis of unrelated versus matched sibling donor transplantation. Blood 99 (6): 1971-7, 2002.
- Lee SJ, Klein J, Haagenson M, et al.: High-resolution donor-recipient HLA matching contributes to the success of unrelated donor marrow transplantation. Blood 110 (13): 4576-83, 2007.
- Horowitz MM, Gale RP, Sondel PM, et al.: Graft-versus-leukemia reactions after bone marrow transplantation. Blood 75 (3): 555-62, 1990.
- Druker BJ: Translation of the Philadelphia chromosome into therapy for CML. Blood 112 (13): 4808-17, 2008.
- Kantarjian H, Sawyers C, Hochhaus A, et al.: Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med 346 (9): 645-52, 2002.
- O'Brien SG, Guilhot F, Larson RA, et al.: Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 348 (11): 994-1004, 2003.
- Druker BJ, Guilhot F, O'Brien SG, et al.: Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 355 (23): 2408-17, 2006.
- Saussele S, Lauseker M, Gratwohl A, et al.: Allogeneic hematopoietic stem cell transplantation (allo SCT) for chronic myeloid leukemia in the imatinib era: evaluation of its impact within a subgroup of the randomized German CML Study IV. Blood 115 (10): 1880-5, 2010.
- Hughes TP, Hochhaus A, Branford S, et al.: Long-term prognostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: an analysis from the International Randomized Study of Interferon and STI571 (IRIS). Blood 116 (19): 3758-65, 2010.
- Kantarjian H, Cortes J: Considerations in the management of patients with Philadelphia chromosome-positive chronic myeloid leukemia receiving tyrosine kinase inhibitor therapy. J Clin Oncol 29 (12): 1512-6, 2011.
- Bisen A, Claxton DF: Tyrosine kinase targeted treatment of chronic myelogenous leukemia and other myeloproliferative neoplasms. Adv Exp Med Biol 779: 179-96, 2013.
- Ibrahim AR, Eliasson L, Apperley JF, et al.: Poor adherence is the main reason for loss of CCyR and imatinib failure for chronic myeloid leukemia patients on long-term therapy. Blood 117 (14): 3733-6, 2011.
- Soverini S, Hochhaus A, Nicolini FE, et al.: BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood 118 (5): 1208-15, 2011.
- Hazarika M, Jiang X, Liu Q, et al.: Tasigna for chronic and accelerated phase Philadelphia chromosome--positive chronic myelogenous leukemia resistant to or intolerant of imatinib. Clin Cancer Res 14 (17): 5325-31, 2008.
- Brave M, Goodman V, Kaminskas E, et al.: Sprycel for chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate. Clin Cancer Res 14 (2): 352-9, 2008.
- Kantarjian H, Shah NP, Hochhaus A, et al.: Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 362 (24): 2260-70, 2010.
- Saglio G, Kim DW, Issaragrisil S, et al.: Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med 362 (24): 2251-9, 2010.
- Jabbour E, Kantarjian HM, Saglio G, et al.: Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION). Blood 123 (4): 494-500, 2014.
- Hochhaus A, Saglio G, Hughes TP, et al.: Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia 30 (5): 1044-54, 2016.
- O'Hare T, Shakespeare WC, Zhu X, et al.: AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 16 (5): 401-12, 2009.
- Cortes JE, Kim DW, Pinilla-Ibarz J, et al.: A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med 369 (19): 1783-96, 2013.
- Prasad V, Mailankody S: The accelerated approval of oncologic drugs: lessons from ponatinib. JAMA 311 (4): 353-4, 2014 Jan 22-29.
- Oehler VG, Gooley T, Snyder DS, et al.: The effects of imatinib mesylate treatment before allogeneic transplantation for chronic myeloid leukemia. Blood 109 (4): 1782-9, 2007.
- Lee SJ, Kukreja M, Wang T, et al.: Impact of prior imatinib mesylate on the outcome of hematopoietic cell transplantation for chronic myeloid leukemia. Blood 112 (8): 3500-7, 2008.
- Mahon FX, Réa D, Guilhot J, et al.: Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 11 (11): 1029-35, 2010.
- Ross DM, Branford S, Seymour JF, et al.: Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood 122 (4): 515-22, 2013.
- Champagne MA, Capdeville R, Krailo M, et al.: Imatinib mesylate (STI571) for treatment of children with Philadelphia chromosome-positive leukemia: results from a Children's Oncology Group phase 1 study. Blood 104 (9): 2655-60, 2004.
- Millot F, Guilhot J, Nelken B, et al.: Imatinib mesylate is effective in children with chronic myelogenous leukemia in late chronic and advanced phase and in relapse after stem cell transplantation. Leukemia 20 (2): 187-92, 2006.
- Millot F, Baruchel A, Guilhot J, et al.: Imatinib is effective in children with previously untreated chronic myelogenous leukemia in early chronic phase: results of the French national phase IV trial. J Clin Oncol 29 (20): 2827-32, 2011.
- Champagne MA, Fu CH, Chang M, et al.: Higher dose imatinib for children with de novo chronic phase chronic myelogenous leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 57 (1): 56-62, 2011.
- Giona F, Putti MC, Micalizzi C, et al.: Long-term results of high-dose imatinib in children and adolescents with chronic myeloid leukaemia in chronic phase: the Italian experience. Br J Haematol 170 (3): 398-407, 2015.
- Andolina JR, Neudorf SM, Corey SJ: How I treat childhood CML. Blood 119 (8): 1821-30, 2012.
- Menon-Andersen D, Mondick JT, Jayaraman B, et al.: Population pharmacokinetics of imatinib mesylate and its metabolite in children and young adults. Cancer Chemother Pharmacol 63 (2): 229-38, 2009.
- Millot F, Guilhot J, Baruchel A, et al.: Impact of early molecular response in children with chronic myeloid leukemia treated in the French Glivec phase 4 study. Blood 124 (15): 2408-10, 2014.
- Shima H, Tokuyama M, Tanizawa A, et al.: Distinct impact of imatinib on growth at prepubertal and pubertal ages of children with chronic myeloid leukemia. J Pediatr 159 (4): 676-81, 2011.
- Millot F, Guilhot J, Baruchel A, et al.: Growth deceleration in children treated with imatinib for chronic myeloid leukaemia. Eur J Cancer 50 (18): 3206-11, 2014.
- Aplenc R, Blaney SM, Strauss LC, et al.: Pediatric phase I trial and pharmacokinetic study of dasatinib: a report from the children's oncology group phase I consortium. J Clin Oncol 29 (7): 839-44, 2011.
- Zwaan CM, Rizzari C, Mechinaud F, et al.: Dasatinib in children and adolescents with relapsed or refractory leukemia: results of the CA180-018 phase I dose-escalation study of the Innovative Therapies for Children with Cancer Consortium. J Clin Oncol 31 (19): 2460-8, 2013.
- Gore L, Kearns PR, de Martino ML, et al.: Dasatinib in Pediatric Patients With Chronic Myeloid Leukemia in Chronic Phase: Results From a Phase II Trial. J Clin Oncol 36 (13): 1330-1338, 2018.
- Novartis Pharmaceuticals Corporation: TASIGNA (nilotinib): Prescribing Information. East Hanover, NJ: Novartis, 2018. Available online. Last accessed April 7, 2022.
- Hijiya N, Maschan A, Rizzari C, et al.: Phase 2 study of nilotinib in pediatric patients with Philadelphia chromosome-positive chronic myeloid leukemia. Blood 134 (23): 2036-2045, 2019.
- Hochhaus A, Baccarani M, Silver RT, et al.: European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 34 (4): 966-984, 2020.
- National Comprehensive Cancer Network: NCCN Guidelines for Patients: Chronic Myeloid Leukemia, 2021. Plymouth Meeting, PA: National Comprehensive Cancer Network, 2021. Available online with free subscription. Last accessed August 29, 2022.
- Dulucq S, Astrugue C, Etienne G, et al.: Risk of molecular recurrence after tyrosine kinase inhibitor discontinuation in chronic myeloid leukaemia patients: a systematic review of literature with a meta-analysis of studies over the last ten years. Br J Haematol 189 (3): 452-468, 2020.
- Millot F, Suttorp M, Ragot S, et al.: Discontinuation of Imatinib in Children with Chronic Myeloid Leukemia: A Study from the International Registry of Childhood CML. Cancers (Basel) 13 (16): , 2021.
- de Bruijn CMA, Millot F, Suttorp M, et al.: Discontinuation of imatinib in children with chronic myeloid leukaemia in sustained deep molecular remission: results of the STOP IMAPED study. Br J Haematol 185 (4): 718-724, 2019.
- Shima H, Kada A, Tanizawa A, et al.: Discontinuation of tyrosine kinase inhibitors in pediatric chronic myeloid leukemia. Pediatr Blood Cancer 69 (8): e29699, 2022.
- Berger MG, Pereira B, Rousselot P, et al.: Longer treatment duration and history of osteoarticular symptoms predispose to tyrosine kinase inhibitor withdrawal syndrome. Br J Haematol 187 (3): 337-346, 2019.
- Hochhaus A, Baccarani M, Deininger M, et al.: Dasatinib induces durable cytogenetic responses in patients with chronic myelogenous leukemia in chronic phase with resistance or intolerance to imatinib. Leukemia 22 (6): 1200-6, 2008.
- le Coutre P, Ottmann OG, Giles F, et al.: Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood 111 (4): 1834-9, 2008.
- Kantarjian H, O'Brien S, Talpaz M, et al.: Outcome of patients with Philadelphia chromosome-positive chronic myelogenous leukemia post-imatinib mesylate failure. Cancer 109 (8): 1556-60, 2007.
Supervivencia y secuelas adversas tardías
Aunque el tema de las complicaciones a largo plazo del cáncer y su tratamiento abarca muchas categorías de enfermedad, hay varios aspectos importantes que se relacionan con el tratamiento de las neoplasias mieloides malignas que vale la pena destacar. Para obtener más información consultar Efectos tardíos del tratamiento anticanceroso en la niñez.
A continuación se presentan estudios seleccionados sobre los efectos tardíos del tratamiento de la LMA en los adultos sobrevivientes que no recibieron trasplante de células madre hematopoyéticas (TCMH):
- Cardíacos.
- En el Childhood Cancer Survivor Study (CCSS) se examinaron 272 sobrevivientes de leucemia mieloide aguda (LMA) que no se habían sometido a TCMH.[
1 ]- En este estudio se identificaron segundas neoplasias malignas (incidencia acumulada, 1,7 %) y cardiotoxicidad (incidencia acumulada, 4,7 %) como riesgos a largo plazo significativos.
- Se notificó cardiomiopatía en el 4,3 % de los sobrevivientes de LMA de acuerdo con los estudios del grupo Berlin-Frankfurt-Münster. De estos, el 2,5 % presentaron síntomas clínicos.[
2 ]
- En un estudio retrospectivo del funcionamiento cardíaco de niños tratados con regímenes del United Kingdom Medical Research Council, al cabo de una mediana de 13 meses del tratamiento se notificó una media de cambio nocivo en el volumen sistólico ventricular izquierdo del 8,4 % en comparación con los valores iniciales.[
3 ] - Para los pacientes pediátricos, el riesgo de presentar toxicidad temprana fue del 13,7 %, y el riesgo de padecer cardiotoxicidad tardía (definida al año de completar el tratamiento de primera línea) fue del 17,4 %. La cardiotoxicidad temprana fue un factor pronóstico importante de cardiotoxicidad tardía y de la aparición de cardiomiopatía clínica con necesidad de tratamiento a largo plazo.[
4 ] - En un análisis retrospectivo de un solo estudio se indicó que el riesgo cardíaco quizá sea elevado para los niños con síndrome de Down,[
5 ] pero se necesitan estudios prospectivos para corroborar estos resultados.
- En el Childhood Cancer Survivor Study (CCSS) se examinaron 272 sobrevivientes de leucemia mieloide aguda (LMA) que no se habían sometido a TCMH.[
- Psicosociales.
- En un ensayo retrospectivo del Nordic Society for Pediatric Hematology and Oncology de niños con LMA tratados con quimioterapia sola se encontró que, al cabo de una mediana de 11 años de seguimiento, el uso de servicios de salud y el estado civil fueron semejantes al de sus hermanos de acuerdo con la notificación de los pacientes.[
6 ] - En un estudio poblacional de sobrevivientes de LMA infantil que no se habían sometido a un TCMH, se notificaron tasas de logros educativos, empleo y estado civil equivalentes a las de sus hermanos. Sin embargo, los sobrevivientes de LMA fueron más propensos a recibir fármacos recetados; en especial para el asma, en comparación con sus hermanos (23 % vs. 9 %; P = 0,03). También se demostró que la fatiga crónica es un efecto tardío adverso significativamente más probable en los sobrevivientes de LMA infantil que en los sobrevivientes de otras neoplasias malignas.[
7 ] - En un informe del CCSS se evaluaron los sobrevivientes de LMA infantil tratados entre 1970 y 1999 (mediana de edad en el momento de la evaluación, 30 a 32 años) y se compararon sus desenlaces con los datos de sus hermanos. Los sobrevivientes que recibieron quimioterapia de consolidación intensiva (n = 299) o TCMH (n = 183) tuvieron desenlaces significativamente peores que sus hermanos en las medidas de síntomas somáticos (prevalencia, 8,4–12 %), funcionamiento neurocognitivo (prevalencia, 17,7–25,7 %), medidas de calidad de vida relacionada con la salud (prevalencia, 8,2–24,6 %) y medidas de logros a nivel social. En ninguna de las medidas se observó diferencia estadísticamente significativa entre las dos cohortes de consolidación en la prevalencia de los problemas identificados.[
8 ]
- En un ensayo retrospectivo del Nordic Society for Pediatric Hematology and Oncology de niños con LMA tratados con quimioterapia sola se encontró que, al cabo de una mediana de 11 años de seguimiento, el uso de servicios de salud y el estado civil fueron semejantes al de sus hermanos de acuerdo con la notificación de los pacientes.[
Se ha notificado que son muy poco frecuentes los efectos adversos tardíos renales, gastrointestinales y hepáticos en los niños que solo reciben quimioterapia para el tratamiento de la LMA.[
A continuación se describen los efectos tardíos notificados en estudios seleccionados sobre el tratamiento de la LMA en adultos sobrevivientes tratados con TCMH:
- En una revisión de una institución, la frecuencia más alta de secuelas adversas a largo plazo en los niños tratados por LMA incluyó las siguientes tasas de incidencia: anomalías del crecimiento (51 %), anomalías neurocognitivas (30 %), hepatitis adquirida por transfusión (28 %), esterilidad (25 %), endocrinopatías (16 %), enfermedad pulmonar restrictiva (20 %), enfermedad de injerto contra huésped crónica (20 %), neoplasias malignas secundarias (14 %) y cataratas (12 %).[
10 ]- La mayoría de estas secuelas adversas se deben al TCMH alogénico mielosupresor. A pesar de que se observaron anomalías cardíacas en solo el 8 % de los pacientes, este es un tema que quizá sea muy relevante dado el aumento actual del uso de antraciclinas en los ensayos clínicos para niños con LMA recién diagnosticada.
- En otro estudio se examinaron los desenlaces de niños menores de 3 años con LMA o leucemia linfoblástica aguda (LLA) sometidos a TCMH.[
11 ]- Los efectos tóxicos notificados fueron deficiencia de la hormona del crecimiento (59 %), dislipidemias (59 %), hipotiroidismo (35 %), osteocondromas (24 %) y disminución de la densidad mineral ósea (24 %).
- Se presentaron neoplasias malignas secundarias en 2 de los 33 pacientes.
- Los sobrevivientes tenían inteligencia promedio, pero presentaban problemas frecuentes de déficit de atención y anomalías en la motricidad fina en comparación con los controles de la población.
- Por el contrario, en el The Bone Marrow Transplant Survivor Study se compararon sobrevivientes con LMA o LLA infantil con sus hermanos y se usó un cuestionario de autonotificación.[
12 ] La mediana de seguimiento fue de 8,4 años, y el 86 % de los pacientes recibieron irradiación corporal total (ICT) como parte de su régimen de preparación para el trasplante.- Los sobrevivientes de leucemia que recibieron un TCMH presentaron frecuencias significativamente más altas de varios efectos adversos, como diabetes, hipotiroidismo, osteoporosis, cataratas, osteonecrosis, disnea inducida por el ejercicio, dificultades neurosensoriales y problemas de equilibrio, temblores y debilidad en comparación con sus hermanos.
- La evaluación general de la salud disminuyó significativamente en los sobrevivientes en comparación con sus hermanos (oportunidad relativa = 2,2; P = 0,03).
- No se observaron diferencias significativas entre los regímenes de ICT en comparación con quimioterapia sola, que la mayoría de las veces incluyó busulfano.
- Los desenlaces fueron similares para los pacientes con LMA y LLA; ello indica que la causa principal subyacente de los efectos adversos tardíos fue el TCMH.
- En un estudio del Children's Oncology Group (COG) en el que se usó una comparación de calidad de vida relacionada con la salud, se notificó que el 21 % de los sobrevivientes a 5 años presentaban una afección crónica grave o que ponía en peligro la vida; cuando se hizo la comparación según el tipo de tratamiento, se encontró que este porcentaje fue del 16 % en el grupo tratado con quimioterapia sola, el 21 % en el grupo tratado con TCMH autógeno y el 33 % en los que recibieron TCMH alogénico.[
13 ] - En un análisis de cohortes del CCSS se examinó la mortalidad a largo plazo y el estado de salud de 856 niños (sobrevivientes a 5 años) que habían recibido tratamiento para la LMA, con TCMH o sin este, entre 1970 y 1999.[
14 ]- Las tasas acumulativas de afecciones de salud crónicas de grados 3 a 5 disminuyeron significativamente entre los receptores de TCMH entre las décadas de 1970 y 1990 (de 76,1 % a 43,5 %; P = 0,04) pero permanecieron estables en los pacientes que solo recibieron quimioterapia (de 12,2 % a 27,6 %; P = 0,06).
- Hubo una reducción significativa en la mortalidad acumulada tardía por todas las causas de los receptores de TCMH en el mismo período de tiempo (de 38,9 % a 8,5 %; P < 0,0001). Esto se debió sobre todo a una reducción en la recaída, mientras que no se observó una disminución significativa en la mortalidad tardía de los sobrevivientes que solo recibieron quimioterapia (de 6,8 % a 2,6 %; P = 0,35).
- En los informes autonotificados, los estados de salud entre todos los sobrevivientes fueron excelentes, muy buenos o buenos en el 85 % de los receptores de TCMH y en el 90 % de los receptores de quimioterapia sola. Sin embargo, el estado de salud de los sobrevivientes en ambos grupos de tratamiento fue significativamente más precario que el de sus hermanos o hermanas (CRI, 3,8; 95 % IC, 2,7–5,4 vs. CRI, 2,6; 95 % IC, 1,8–3,6, respectivamente).
Se necesitan nuevos enfoques terapéuticos para reducir las secuelas adversas a largo plazo, en especial, para disminuir las secuelas tardías relacionadas con el TCMH mielosupresor.
Se crearon recursos importantes sobre los pormenores del seguimiento y los riesgos de los sobrevivientes de cáncer, entre ellos el documento del COG Long-Term Follow-Up Guidelines for Survivors of Childhood, Adolescent, and Young Adult Cancers y el documento de la National Comprehensive Cancer Network Guidelines for Acute Myeloid Leukemia. Además, cada vez se reconoce más la importancia que tiene para los sobrevivientes de cáncer contar con el acceso a los antecedentes médicos y que estos se puedan compartir con otros proveedores de salud.
Referencias:
- Mulrooney DA, Dover DC, Li S, et al.: Twenty years of follow-up among survivors of childhood and young adult acute myeloid leukemia: a report from the Childhood Cancer Survivor Study. Cancer 112 (9): 2071-9, 2008.
- Creutzig U, Diekamp S, Zimmermann M, et al.: Longitudinal evaluation of early and late anthracycline cardiotoxicity in children with AML. Pediatr Blood Cancer 48 (7): 651-62, 2007.
- Orgel E, Zung L, Ji L, et al.: Early cardiac outcomes following contemporary treatment for childhood acute myeloid leukemia: a North American perspective. Pediatr Blood Cancer 60 (9): 1528-33, 2013.
- Temming P, Qureshi A, Hardt J, et al.: Prevalence and predictors of anthracycline cardiotoxicity in children treated for acute myeloid leukaemia: retrospective cohort study in a single centre in the United Kingdom. Pediatr Blood Cancer 56 (4): 625-30, 2011.
- O'Brien MM, Taub JW, Chang MN, et al.: Cardiomyopathy in children with Down syndrome treated for acute myeloid leukemia: a report from the Children's Oncology Group Study POG 9421. J Clin Oncol 26 (3): 414-20, 2008.
- Molgaard-Hansen L, Glosli H, Jahnukainen K, et al.: Quality of health in survivors of childhood acute myeloid leukemia treated with chemotherapy only: a NOPHO-AML study. Pediatr Blood Cancer 57 (7): 1222-9, 2011.
- Jóhannsdóttir IM, Hjermstad MJ, Moum T, et al.: Increased prevalence of chronic fatigue among survivors of childhood cancers: a population-based study. Pediatr Blood Cancer 58 (3): 415-20, 2012.
- Stefanski KJ, Anixt JS, Goodman P, et al.: Long-Term Neurocognitive and Psychosocial Outcomes After Acute Myeloid Leukemia: A Childhood Cancer Survivor Study Report. J Natl Cancer Inst 113 (4): 481-495, 2021.
- Skou AS, Glosli H, Jahnukainen K, et al.: Renal, gastrointestinal, and hepatic late effects in survivors of childhood acute myeloid leukemia treated with chemotherapy only--a NOPHO-AML study. Pediatr Blood Cancer 61 (9): 1638-43, 2014.
- Leung W, Hudson MM, Strickland DK, et al.: Late effects of treatment in survivors of childhood acute myeloid leukemia. J Clin Oncol 18 (18): 3273-9, 2000.
- Perkins JL, Kunin-Batson AS, Youngren NM, et al.: Long-term follow-up of children who underwent hematopoeitic cell transplant (HCT) for AML or ALL at less than 3 years of age. Pediatr Blood Cancer 49 (7): 958-63, 2007.
- Baker KS, Ness KK, Weisdorf D, et al.: Late effects in survivors of acute leukemia treated with hematopoietic cell transplantation: a report from the Bone Marrow Transplant Survivor Study. Leukemia 24 (12): 2039-47, 2010.
- Schultz KA, Chen L, Chen Z, et al.: Health conditions and quality of life in survivors of childhood acute myeloid leukemia comparing post remission chemotherapy to BMT: a report from the children's oncology group. Pediatr Blood Cancer 61 (4): 729-36, 2014.
- Turcotte LM, Whitton JA, Leisenring WM, et al.: Chronic conditions, late mortality, and health status after childhood AML: a Childhood Cancer Survivor Study report. Blood 141 (1): 90-101, 2023.
Modificaciones a este resumen (08 / 17 / 2023)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene información nueva. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha antes mencionada.
Información general sobre la leucemia mieloide aguda infantil
Se añadió hipodiploidía como una anormalidad genética vinculada con pronóstico desfavorable. También se añadió texto para indicar que la hipodiploidía se define como un número modal de cromosomas igual o inferior a 45. En un análisis de una cohorte retrospectiva, el Berlin-Frankfurt-Münster AML Study Group se enfocó en caracterizar la hipodiploidía en pacientes pediátricos con leucemia mieloide aguda (LMA) (se citó a Hammer et al. como referencia 111).
Tratamiento de la leucemia mieloide aguda
Se añadió texto sobre los resultados del ensayo AML1031 del Children's Oncology Group (COG), en el que se demostró que el sorafenib mejoró la supervivencia sin complicaciones de pacientes pediátricos con LMA de novo y proporción alélica alta de mutaciones por duplicaciones internas en tándem de FLT3 (se citó a Pollard et al. como referencia 36).
Leucemia promielocítica aguda
Se añadió texto para indicar que en el ensayo COG AAML1331, los pacientes con leucemia promielocítica aguda (LPA) de riesgo estándar no recibieron idarrubicina en el ciclo de inducción. En los ciclos de consolidación se eliminaron la mitoxantrona, las dosis altas de citarabina y la idarrubicina. Luego, se quitaron la mercaptopurina y el metotrexato de los ciclos de mantenimiento. Las dosis intratecales de citarabina también se eliminaron.
Síndromes mielodisplásicos
Se añadió texto para indicar que en un análisis retrospectivo, se observó que solo el subgrupo de riesgo muy alto del International Prognostic Scoring System (R-IPSS) corregido, definido por la presencia de características citogenéticas complejas, tenía un efecto pronóstico adverso significativo sobre la supervivencia general y el riesgo de recaída después del trasplante (se citó a Yamamoto et al. como referencia 34).
Se añadió texto sobre los grupos pronóstico del R-IPSS y las anomalías citogenéticas relacionadas.
Leucemia mielógena crónica
Se añadió Suspensión de la terapia con inhibidores de tirosina–cinasas como una subsección nueva.
Supervivencia y secuelas adversas tardías
Se añadió texto sobre los resultados de un análisis de cohortes del Childhood Cancer Survivor Study en el que se examinó la mortalidad a largo plazo y el estado de salud de 856 niños que habían recibido tratamiento para la LMA, con trasplante de células madre hematopoyéticas o sin este, entre 1970 y 1999 (se citó a Turcotte et al. como referencia 14).
El Consejo editorial del PDQ sobre el tratamiento pediátrico es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este sumario del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este sumario del PDQ
Propósito de este sumario
Este sumario del PDQ con información sobre el cáncer para profesionales de la salud proporciona información integral revisada por expertos y fundamentada en evidencia científica sobre el tratamiento de la leucemia mieloide aguda infantil y otras neoplasias malignas mieloides. El propósito es servir como fuente de información y ayuda para los médicos que atienden a pacientes de cáncer. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El Consejo editorial del PDQ sobre el tratamiento pediátrico, cuya función editorial es independiente del Instituto Nacional del Cáncer (NCI), revisa con regularidad este sumario y, en caso necesario, lo actualiza. Este sumario refleja una revisión bibliográfica independiente y no constituye una declaración de la política del Instituto Nacional del Cáncer ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los miembros de este Consejo examinan artículos publicados recientemente para determinar si se deben:
- tratar en una reunión,
- citar textualmente, o
- sustituir o actualizar, si ya se citaron con anterioridad.
Los cambios en los sumarios se deciden mediante consenso, una vez que los integrantes del Consejo evalúan la solidez de la evidencia científica de los artículos publicados y determinan la forma en que se incorporarán al sumario.
Los revisores principales del sumario sobre Tratamiento de la leucemia mieloide aguda y otras neoplasias mieloides malignas infantiles son:
- William L. Carroll, MD (Laura and Isaac Perlmutter Cancer Center at NYU Langone)
- Alan Scott Gamis, MD, MPH (Children's Mercy Hospital)
- Karen J. Marcus, MD, FACR (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Jessica Pollard, MD (Dana-Farber/Boston Children's Cancer and Blood Disorders Center)
- Michael A. Pulsipher, MD (Children's Hospital Los Angeles)
- Rachel E. Rau, MD (Texas Medical Center)
- Lewis B. Silverman, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Malcolm A. Smith, MD, PhD (National Cancer Institute)
- Sarah K. Tasian, MD (Children's Hospital of Philadelphia)
Cualquier comentario o pregunta sobre el contenido de este sumario se debe enviar mediante el formulario de comunicación en Cancer.gov/espanol del NCI. No se comunique con los miembros del Consejo para enviar preguntas o comentarios sobre los sumarios. Los miembros del Consejo no responderán a preguntas del público.
Niveles de evidencia científica
En algunas referencias bibliográficas de este sumario se indica el nivel de evidencia científica. El propósito de estas designaciones es ayudar al lector a evaluar la solidez de la evidencia científica que respalda el uso de ciertas intervenciones o abordajes. El Consejo editorial del PDQ sobre el tratamiento pediátrico emplea un sistema de jerarquización formal para establecer las designaciones del nivel de evidencia científica.
Permisos para el uso de este sumario
PDQ (Physician Data Query) es una marca registrada. Se autoriza el libre uso del texto de los documentos del PDQ. Sin embargo, no se podrá identificar como un sumario de información sobre cáncer del PDQ del NCI, salvo que se reproduzca en su totalidad y se actualice con regularidad. Por otra parte, se permitirá que un autor escriba una oración como "En el sumario del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del sumario]".
Se sugiere citar la referencia bibliográfica de este sumario del PDQ de la siguiente forma:
PDQ® sobre el tratamiento pediátrico. PDQ Tratamiento de la leucemia mieloide aguda y otras neoplasias mieloides malignas infantiles. Bethesda, MD: National Cancer Institute. Actualización: <MM/DD/YYYY>. Disponible en: https://www.cancer.gov/espanol/tipos/leucemia/pro/tratamiento-lma-infantil-pdq. Fecha de acceso: <MM/DD/YYYY>.
Las imágenes en este sumario se reproducen con el permiso del autor, el artista o la editorial para uso exclusivo en los sumarios del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este sumario o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia científica, las opciones de tratamiento se clasifican como "estándar" o "en evaluación clínica". Estas clasificaciones no deben fundamentar ninguna decisión sobre reintegros de seguros. Para obtener más información sobre cobertura de seguros, consultar la página Manejo de la atención del cáncer disponible en Cancer.gov/espanol.
Para obtener más información
En Cancer.gov/espanol, se ofrece más información sobre cómo comunicarse o recibir ayuda en ¿En qué podemos ayudarle?. También se puede enviar un mensaje de correo electrónico mediante este formulario.
Última revisión: 2023-08-17
Esta información no reemplaza el consejo de un médico. Healthwise, Incorporated, niega toda garantía y responsabilidad por el uso de esta información. El uso que usted haga de esta información implica que usted acepta los
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Healthwise, Incorporated.
Page Footer
Quiero...
Audiencia
Sitios seguros para miembros
Información sobre The Cigna Group
Aviso legal
Los planes individuales y familiares de seguro médico y dental están asegurados por Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc. y Cigna HealthCare of Texas, Inc. Los planes de beneficios de salud y de seguro de salud de grupo están asegurados o administrados por CHLIC, Connecticut General Life Insurance Company (CGLIC) o sus afiliadas (puedes ver
Todas las pólizas de seguros y los planes de beneficios de grupo contienen exclusiones y limitaciones. Para conocer la disponibilidad, los costos y detalles completos de la cobertura, comunícate con un agente autorizado o con un representante de ventas de Cigna. Este sitio web no está dirigido a los residentes de New Mexico.