Tratamiento del carcinoma de células de Merkel (PDQ®) : Tratamiento - información para profesionales de salud [NCI]
Información general sobre el carcinoma de células de Merkel
El carcinoma de células de Merkel (CCM) fue descrito por primera vez por Toker en 1972 como carcinoma trabecular de la piel.[
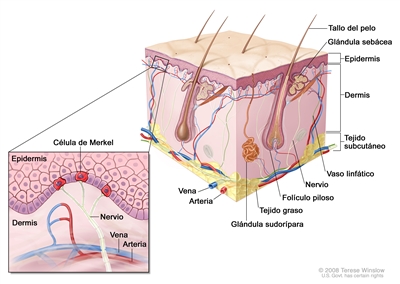
El CCM es un carcinoma neuroendocrino de crecimiento rápido que aparece en la unión dermoepidérmica (consultar la
Históricamente, las opciones de tratamiento para los pacientes con enfermedad en estadio avanzado han sido limitadas; no obstante, los nuevos abordajes de inmunoterapia producen respuestas duraderas.[
Características anatómicas
Incidencia y mortalidad
La incidencia del CCM aumenta de forma progresiva con la edad. Se han notificado pocos casos en pacientes menores de 50 años y la mediana de edad en el momento del diagnóstico es de alrededor de 65 años (consultar la 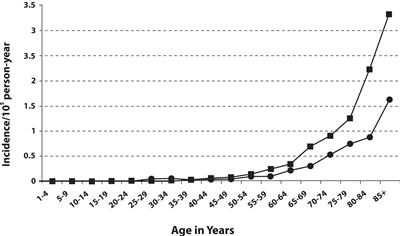
El CCM se presenta con mayor frecuencia en áreas de la piel expuestas al sol; en particular, la cabeza y el cuello, seguido por las extremidades y luego, el tronco.[
En 2013, se notificó que el CCM tuvo una incidencia anual de 0,7 casos por 100 000 personas en los Estados Unidos.[
| Localización anatómica | Casos (%) |
|---|---|
| SAI = sin otra indicación; SEER = Surveillance, Epidemiology, and End Results Program. | |
| a Albores-Saavedra J et al: Merkel cell carcinoma demographics, morphology, and survival based on 3,870 cases: A population-based study. J Cutan Pathol. Reproducción autorizada. © 2009 Publicado por Wiley-Blackwell. Todos los derechos reservados.[ |
|
| Piel, cara | 1041 (26,9) |
| Piel de miembro superior y hombro | 853 (22,0) |
| Piel de miembro inferior y cadera | 578 (14,9) |
| Piel del tronco | 410 (10,6) |
| Piel de cuero cabelludo y cuello | 348 (9,0) |
| Piel, SAI | 234 (6,0) |
| Oído externo | 120 (3,1) |
| Párpado | 98 (2,5) |
| Piel del labio | 91 (2,4) |
| Sitio primario desconocido | 31 (0,8) |
| Total | 3804 (98,3) |
En varias series de casos, hasta el 97 % de los CCM aparecen en la piel. Los CCM primarios en otras localizaciones fueron muy poco frecuentes, así como los CCM de sitio primario desconocido.[
En los datos del registro SEER se observó un exceso en el riesgo de CCM como primero o segundo cáncer en pacientes con varios cánceres primarios.[
Patogenia
Se ha observado un aumento de la incidencia de CCM en personas tratadas por psoriasis con dosis altas de metoxaleno (psoraleno) y radiación ultravioleta A (PUVA) (3 de 1380 pacientes, un 0,2 %). Esto también se ha observado en personas con depresión inmunitaria crónica, en especial, por leucemia linfocítica crónica, infección por el VIH y trasplante previo de un órgano sólido.[
En 2008, se notificó por primera vez un poliomavirus nuevo (poliomavirus de células de Merkel, [MCPyV]) en muestras tumorales de CCM [
Se detectaron índices muy bajos de MCPyV en la piel normal distante del tumor primario de un CCM en un porcentaje significativo de pacientes con trastornos cutáneos no relacionados con el CCM, en la piel con apariencia normal de personas sanas y en cánceres de piel no melanoma en personas con depresión inmunitaria.[
La importancia de los nuevos hallazgos sobre el MCPyV permanece incierta. El significado pronóstico de la carga viral, las concentraciones de los títulos de anticuerpos y la función de inmunodepresión subyacente en las personas portadoras de este virus (debido a enfermedades y medicamentos) siguen en investigación.
La prevalencia del MCPyV difiere entre los pacientes con CCM de los Estados Unidos y Europa en oposición con Australia. Es posible que hayan dos vías independientes para el surgimiento del CCM: una que es impulsada por la presencia del MCPyV y otra impulsada principalmente por el daño solar, en especial, según se observó en los pacientes de series australianas.[
Aunque no se ha identificado un marcador único para el CCM, se informó de una variedad de marcadores moleculares y citogenéticos del CCM.[
Presentación clínica
El CCM se presenta por lo general como un nódulo dérmico solitario, indoloro, endurecido, de color ligeramente eritematoso a violeta intenso y, con poca frecuencia, ulcerado. El CCM puede infiltrar localmente a través de los vasos linfáticos dérmicos, lo que da lugar a múltiples lesiones satélite. Debido a su apariencia clínica inespecífica, pocas veces se presume la presencia de CCM antes de realizar una biopsia.[
Se propuso la siguiente fórmula nemotécnica en inglés para resumir las características clínicas del CCM:[
AEIOU
- A = asymptomatic (asintomático).
- E = expanding rapidly (expansión rápida).
- I = immune suppressed (inmunodepresión).
- O = older than 50 years (mayor de 50 años).
- U = UV-exposed skin (piel expuesta a rayos ultravioleta).
No todos los pacientes cuentan con todos los elementos de esta fórmula mnemónica; sin embargo, el 89 % de los pacientes cumplieron con 3 o más criterios, el 52 % cumplieron con 4 o más criterios y 7 % cumplieron con los 5 criterios.[
Evaluación clínica inicial
Debido a que la diseminación locorregional es común, los pacientes con diagnóstico reciente de CCM necesitan un examen físico cuidadoso que incluya la búsqueda de lesiones satélite y compromiso ganglionar regional.
Las pruebas con imágenes se adaptan de acuerdo con el cuadro clínico y se deben considerar todos los signos y síntomas relevantes. No se ha realizado un estudio sistemático de las pruebas con imágenes óptimas para los pacientes con diagnóstico reciente ni está claro si todos estos pacientes, en especial, aquellos con tumores primarios más pequeños, se benefician de imágenes detalladas.
Si se realizan pruebas con imágenes, estas tal vez incluyan una tomografía computarizada (TC) del tórax y el abdomen para descartar un cáncer primario de células pequeñas de pulmón, así como metástasis a distancia y regionales. A veces también se recomiendan estudios con imágenes diseñados para evaluar los signos y síntomas sospechosos. En una serie, las TC rindieron una tasa de falsos negativos del 80 % para las metástasis regionales.[
Resultados de la estadificación inicial
De acuerdo con las series de casos retrospectivos notificados en el transcurso de varias décadas, los resultados de la estadificación clínica inicial de un CCM varían mucho en la literatura. De los cánceres invasivos, el 48,6 % tenían invasión local, el 31,1 % tenían invasión regional y el 8,2 % tenían invasión a distancia.[
El CCM que se manifiesta en ganglios regionales sin una lesión primaria identificable se encuentra en una minoría de pacientes y el porcentaje de estos casos varía en las series notificadas. Los tumores sin una lesión primaria identificable se atribuyeron a una regresión espontánea del tumor primario o a un carcinoma neuroendocrino metastásico de sitio oculto en la exploración clínica.[
Progresión clínica
En un análisis de pacientes de 18 series de casos, 279 de 926 pacientes (30,1 %) presentaron recidiva local durante el seguimiento, excepto aquellos que presentaron al inicio una enfermedad metastásica a distancia. Estos casos se atribuyeron normalmente a márgenes quirúrgicos inadecuados o a la falta de radioterapia adyuvante. Además, 545 de 982 pacientes (55,5 %) tenían metástasis en los ganglios linfáticos en el momento del diagnóstico o durante el seguimiento.[
En el mismo análisis de 18 series de casos, los sitios más comunes de metástasis a distancia fueron los ganglios linfáticos distantes (60,1 %), una parte distante de la piel (30,3 %), el pulmón (23,4 %), el sistema nervioso central (18,4 %) y el hueso (15,2 %).[
En una serie de 237 pacientes que se presentaron al inicio con enfermedad local o regional, la mediana de tiempo hasta la recidiva fue de 9 meses (intervalo, 2–70 meses). Noventa y uno por ciento de las recidivas se presentaron dentro de los 2 años a partir del diagnóstico.[
Posibles factores pronósticos
El grado de diseminación de la enfermedad en el momento de la presentación inicial quizás proporcione el cálculo más útil para pronóstico.[
Los procedimientos diagnósticos, como la biopsia del ganglio linfático centinela, quizás ayuden a diferenciar la enfermedad local y regional en el momento de la presentación inicial. Un tercio de los pacientes que carecen de ganglios clínicamente palpables o radiológicamente visibles tendrán enfermedad regional evidente en el microscopio.[
En muchos estudios retrospectivos se evaluó la relación entre una variedad amplia de factores biológicos e histológicos con la supervivencia y el control locorregional.[
En un estudio retrospectivo amplio de una sola institución se evaluaron los factores histológicos potencialmente relacionados con el pronóstico de 156 pacientes con CCM durante una mediana de seguimiento de 51 meses (intervalo, 2–224 meses).[
En un estudio realizado en 2009 se investigó si la presencia del MCPyV recién identificado en muestras tumorales de CCM influyó en los desenlaces clínicos de 114 pacientes con CCM en Finlandia. En este pequeño estudio, los pacientes cuyos tumores exhibieron positividad para el MCPyV presentaron una mejor supervivencia que los pacientes cuyos tumores exhibieron negatividad para el MCPyV.[
Pronóstico
Los parámetros más importantes para el pronóstico del CCM son el tamaño del tumor y la presencia de metástasis locorregionales o a distancia. Estos factores constituyen la base del sistema de estadificación del American Joint Committee on Cancer para el CCM.[
El grueso de la bibliografía sobre el CCM corresponde a series de casos pequeñas sujetas a muchos factores de confusión. Por esta razón, las tasas de recaída y supervivencia por estadio de las que se tienen informes varían mucho en la bibliografía. En general, la enfermedad en los estadios más bajos se relaciona con una mejor supervivencia general.[
En un informe se notificó que los desenlaces clínicos de pacientes que presentan al inicio una enfermedad local de volumen pequeño y ganglios linfáticos sin compromiso tumoral confirmados mediante estudio patológico tienen una tasa de supervivencia por causa específica a 5 años superior al 90 %.[
En un resumen tabulado de los resultados de 12 series de tratamiento del CCM se ilustra la dificultad de comparar los datos de los resultados entre las series.[
En la 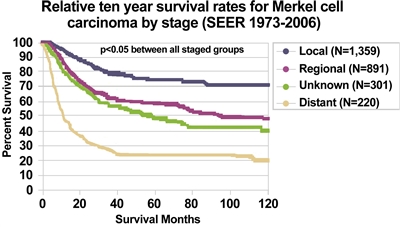
Referencias:
- Toker C: Trabecular carcinoma of the skin. Arch Dermatol 105 (1): 107-10, 1972.
- Schwartz RA, Lambert WC: The Merkel cell carcinoma: a 50-year retrospect. J Surg Oncol 89 (1): 5, 2005.
- Agelli M, Clegg LX, Becker JC, et al.: The etiology and epidemiology of merkel cell carcinoma. Curr Probl Cancer 34 (1): 14-37, 2010 Jan-Feb.
- Harms PW: Update on Merkel Cell Carcinoma. Clin Lab Med 37 (3): 485-501, 2017.
- Nghiem P, McKee PH, Haynes HA: Merkel cell (cutaneous neuroendocrine) carcinoma. In: Sober AJ, Haluska FG, eds.: Skin Cancer. BC Decker Inc., 2001, pp 127-141.
- Nghiem P, James N: Merkel cell carcinoma. In: Wolff K, Goldsmith LA, Katz SI, et al., eds.: Fitzpatrick's Dermatology in General Medicine. 7th ed. McGraw-Hill , 2008, pp 1087-94.
- Eng TY, Boersma MG, Fuller CD, et al.: A comprehensive review of the treatment of Merkel cell carcinoma. Am J Clin Oncol 30 (6): 624-36, 2007.
- Medina-Franco H, Urist MM, Fiveash J, et al.: Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol 8 (3): 204-8, 2001.
- Busse PM, Clark JR, Muse VV, et al.: Case records of the Massachusetts General Hospital. Case 19-2008. A 63-year-old HIV-positive man with cutaneous Merkel-cell carcinoma. N Engl J Med 358 (25): 2717-23, 2008.
- Rockville Merkel Cell Carcinoma Group: Merkel cell carcinoma: recent progress and current priorities on etiology, pathogenesis, and clinical management. J Clin Oncol 27 (24): 4021-6, 2009.
- Calder KB, Smoller BR: New insights into merkel cell carcinoma. Adv Anat Pathol 17 (3): 155-61, 2010.
- Cassler NM, Merrill D, Bichakjian CK, et al.: Merkel Cell Carcinoma Therapeutic Update. Curr Treat Options Oncol 17 (7): 36, 2016.
- Agelli M, Clegg LX: Epidemiology of primary Merkel cell carcinoma in the United States. J Am Acad Dermatol 49 (5): 832-41, 2003.
- Hodgson NC: Merkel cell carcinoma: changing incidence trends. J Surg Oncol 89 (1): 1-4, 2005.
- Young JL, Ward KC, Ries LAG: Cancer of rare sites. In: Ries LAG, Young JL, Keel GE, et al., eds.: SEER Survival Monograph: Cancer Survival Among Adults: U. S. SEER Program, 1988-2001, Patient and Tumor Characteristics. National Cancer Institute, 2007. NIH Pub. No. 07-6215, pp 251-61.
- Miller RW, Rabkin CS: Merkel cell carcinoma and melanoma: etiological similarities and differences. Cancer Epidemiol Biomarkers Prev 8 (2): 153-8, 1999.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al.: Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol 37 (1): 20-7, 2010.
- Heath M, Jaimes N, Lemos B, et al.: Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol 58 (3): 375-81, 2008.
- Paulson KG, Park SY, Vandeven NA, et al.: Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J Am Acad Dermatol 78 (3): 457-463.e2, 2018.
- Ma JE, Brewer JD: Merkel cell carcinoma in immunosuppressed patients. Cancers (Basel) 6 (3): 1328-50, 2014.
- Howard RA, Dores GM, Curtis RE, et al.: Merkel cell carcinoma and multiple primary cancers. Cancer Epidemiol Biomarkers Prev 15 (8): 1545-9, 2006.
- Bzhalava D, Bray F, Storm H, et al.: Risk of second cancers after the diagnosis of Merkel cell carcinoma in Scandinavia. Br J Cancer 104 (1): 178-80, 2011.
- Lunder EJ, Stern RS: Merkel-cell carcinomas in patients treated with methoxsalen and ultraviolet A radiation. N Engl J Med 339 (17): 1247-8, 1998.
- Feng H, Shuda M, Chang Y, et al.: Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319 (5866): 1096-100, 2008.
- Garneski KM, Warcola AH, Feng Q, et al.: Merkel cell polyomavirus is more frequently present in North American than Australian Merkel cell carcinoma tumors. J Invest Dermatol 129 (1): 246-8, 2009.
- Becker JC, Houben R, Ugurel S, et al.: MC polyomavirus is frequently present in Merkel cell carcinoma of European patients. J Invest Dermatol 129 (1): 248-50, 2009.
- Kassem A, Schöpflin A, Diaz C, et al.: Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of a unique deletion in the VP1 gene. Cancer Res 68 (13): 5009-13, 2008.
- Houben R, Schrama D, Becker JC: Molecular pathogenesis of Merkel cell carcinoma. Exp Dermatol 18 (3): 193-8, 2009.
- Paik JY, Hall G, Clarkson A, et al.: Immunohistochemistry for Merkel cell polyomavirus is highly specific but not sensitive for the diagnosis of Merkel cell carcinoma in the Australian population. Hum Pathol 42 (10): 1385-90, 2011.
- Andres C, Belloni B, Puchta U, et al.: Prevalence of MCPyV in Merkel cell carcinoma and non-MCC tumors. J Cutan Pathol 37 (1): 28-34, 2010.
- Kassem A, Technau K, Kurz AK, et al.: Merkel cell polyomavirus sequences are frequently detected in nonmelanoma skin cancer of immunosuppressed patients. Int J Cancer 125 (2): 356-61, 2009.
- Foulongne V, Dereure O, Kluger N, et al.: Merkel cell polyomavirus DNA detection in lesional and nonlesional skin from patients with Merkel cell carcinoma or other skin diseases. Br J Dermatol 162 (1): 59-63, 2010.
- DeCaprio JA: Does detection of Merkel cell polyomavirus in Merkel cell carcinoma provide prognostic information? J Natl Cancer Inst 101 (13): 905-7, 2009.
- Laude HC, Jonchère B, Maubec E, et al.: Distinct merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with merkel cell carcinoma. PLoS Pathog 6 (8): , 2010.
- Buck CB, Lowy DR: Immune readouts may have prognostic value for the course of merkel cell carcinoma, a virally associated disease. J Clin Oncol 29 (12): 1506-8, 2011.
- Lemos B, Nghiem P: Merkel cell carcinoma: more deaths but still no pathway to blame. J Invest Dermatol 127 (9): 2100-3, 2007.
- Seattle Cancer Care Alliance: Merkel Cell Carcinoma Information for Patients and Their Physicians: Clinical Photos/Images. Seattle, Wa: Seattle Cancer Care Alliance Skin Oncology Clinic, 2009.
Available online. Last accessed April 19, 2024. - Gupta SG, Wang LC, Peñas PF, et al.: Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: The Dana-Farber experience and meta-analysis of the literature. Arch Dermatol 142 (6): 685-90, 2006.
- Anderson SE, Beer KT, Banic A, et al.: MRI of merkel cell carcinoma: histologic correlation and review of the literature. AJR Am J Roentgenol 185 (6): 1441-8, 2005.
- Iagaru A, Quon A, McDougall IR, et al.: Merkel cell carcinoma: Is there a role for 2-deoxy-2-[f-18]fluoro-D-glucose-positron emission tomography/computed tomography? Mol Imaging Biol 8 (4): 212-7, 2006 Jul-Aug.
- Belhocine T, Pierard GE, Frühling J, et al.: Clinical added-value of 18FDG PET in neuroendocrine-merkel cell carcinoma. Oncol Rep 16 (2): 347-52, 2006.
- Missotten GS, de Wolff-Rouendaal D, de Keizer RJ: Merkel cell carcinoma of the eyelid review of the literature and report of patients with Merkel cell carcinoma showing spontaneous regression. Ophthalmology 115 (1): 195-201, 2008.
- Richetta AG, Mancini M, Torroni A, et al.: Total spontaneous regression of advanced merkel cell carcinoma after biopsy: review and a new case. Dermatol Surg 34 (6): 815-22, 2008.
- Allen PJ, Bowne WB, Jaques DP, et al.: Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol 23 (10): 2300-9, 2005.
- Stokes JB, Graw KS, Dengel LT, et al.: Patients with Merkel cell carcinoma tumors < or = 1.0 cm in diameter are unlikely to harbor regional lymph node metastasis. J Clin Oncol 27 (23): 3772-7, 2009.
- Jabbour J, Cumming R, Scolyer RA, et al.: Merkel cell carcinoma: assessing the effect of wide local excision, lymph node dissection, and radiotherapy on recurrence and survival in early-stage disease--results from a review of 82 consecutive cases diagnosed between 1992 and 2004. Ann Surg Oncol 14 (6): 1943-52, 2007.
- Henness S, Vereecken P: Management of Merkel tumours: an evidence-based review. Curr Opin Oncol 20 (3): 280-6, 2008.
- Skelton HG, Smith KJ, Hitchcock CL, et al.: Merkel cell carcinoma: analysis of clinical, histologic, and immunohistologic features of 132 cases with relation to survival. J Am Acad Dermatol 37 (5 Pt 1): 734-9, 1997.
- Sandel HD, Day T, Richardson MS, et al.: Merkel cell carcinoma: does tumor size or depth of invasion correlate with recurrence, metastasis, or patient survival? Laryngoscope 116 (5): 791-5, 2006.
- Llombart B, Monteagudo C, López-Guerrero JA, et al.: Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology 46 (6): 622-34, 2005.
- Senchenkov A, Barnes SA, Moran SL: Predictors of survival and recurrence in the surgical treatment of merkel cell carcinoma of the extremities. J Surg Oncol 95 (3): 229-34, 2007.
- Goldberg SR, Neifeld JP, Frable WJ: Prognostic value of tumor thickness in patients with Merkel cell carcinoma. J Surg Oncol 95 (8): 618-22, 2007.
- Heath ML, Nghiem P: Merkel cell carcinoma: if no breslow, then what? J Surg Oncol 95 (8): 614-5, 2007.
- Tai P: Merkel cell cancer: update on biology and treatment. Curr Opin Oncol 20 (2): 196-200, 2008.
- Andea AA, Coit DG, Amin B, et al.: Merkel cell carcinoma: histologic features and prognosis. Cancer 113 (9): 2549-58, 2008.
- Paulson KG, Iyer JG, Tegeder AR, et al.: Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral CD8+ lymphocyte invasion as an independent predictor of survival. J Clin Oncol 29 (12): 1539-46, 2011.
- Fields RC, Busam KJ, Chou JF, et al.: Recurrence and survival in patients undergoing sentinel lymph node biopsy for merkel cell carcinoma: analysis of 153 patients from a single institution. Ann Surg Oncol 18 (9): 2529-37, 2011.
- Sihto H, Kukko H, Koljonen V, et al.: Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J Natl Cancer Inst 101 (13): 938-45, 2009.
- Harms KL, Healy MA, Nghiem P, et al.: Analysis of Prognostic Factors from 9387 Merkel Cell Carcinoma Cases Forms the Basis for the New 8th Edition AJCC Staging System. Ann Surg Oncol 23 (11): 3564-3571, 2016.
- Iyer JG, Storer BE, Paulson KG, et al.: Relationships among primary tumor size, number of involved nodes, and survival for 8044 cases of Merkel cell carcinoma. J Am Acad Dermatol 70 (4): 637-643, 2014.
- Schwartz JL, Griffith KA, Lowe L, et al.: Features predicting sentinel lymph node positivity in Merkel cell carcinoma. J Clin Oncol 29 (8): 1036-41, 2011.
- Ko JS, Prieto VG, Elson PJ, et al.: Histological pattern of Merkel cell carcinoma sentinel lymph node metastasis improves stratification of Stage III patients. Mod Pathol 29 (2): 122-30, 2016.
- Eng TY, Boersma MG, Fuller CD, et al.: Treatment of merkel cell carcinoma. Am J Clin Oncol 27 (5): 510-5, 2004.
Clasificación celular del carcinoma de células de Merkel
Aunque el origen y la función exactas de las células de Merkel permanecen bajo investigación, se piensa que tienen características de origen tanto epitelial como neuroendocrino y que surgen de células con función de sensibilidad al tacto (mecanorreceptores).[
Las características histopatológicas incluyen gránulos neurosecretores en el citoplasma con centros densos detectados mediante microscopia electrónica y análisis inmunohistoquímico con citoqueratina 20 (consultar la
Un panel de reactivos inmunitarios (consultar la 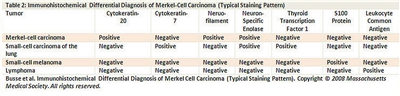
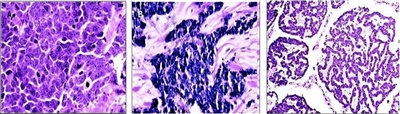
Desde el punto de vista histológico, el CCM se clasificó en tres subtipos diferentes:[
- Trabecular: estructura clásica, tipo de célula grande, densidad alta o gránulos detectados mediante examen ecográfico.
- Intermedio: estructura sólida (más común).
- Células pequeñas: difuso, pocos gránulos de densidad alta detectados mediante examen ecográfico (segundo tipo más común).
Son comunes las mezclas de variantes.[
Un grupo indicó una lista de 12 elementos que se deberían describir en los informes patológicos de las lesiones primarias resecadas y 9 elementos que se describen en los informes de patología de los ganglios linfáticos centinela. La importancia pronóstica de estos elementos no ha sido validada de forma prospectiva.[
Si los siguientes datos quedan registrados para cada paciente con CCM, se puede estadificar a cualquier paciente con el sistema de estadificación disponible o con un nuevo sistema de estadificación:
- Tamaño del tumor primario (dimensión patológica o clínica máxima en centímetros).
- Presencia o ausencia de invasión tumoral primaria en el hueso, el músculo, la fascia o el cartílago.
- Presencia o ausencia de metástasis ganglionares.
- Método usado en la evaluación del estado del compromiso ganglionar (examen clínico o patológico).
- Presencia o ausencia de metástasis a distancia.
El College of American Pathologists publicó un protocolo para el examen de muestras de pacientes con CCM de la piel.[
Para obtener más información, consultar la sección
Las variantes histológicas del CCM se presentan en la
Referencias:
- Nghiem P, McKee PH, Haynes HA: Merkel cell (cutaneous neuroendocrine) carcinoma. In: Sober AJ, Haluska FG, eds.: Skin Cancer. BC Decker Inc., 2001, pp 127-141.
- Bichakjian CK, Nghiem P, Johnson T, et al.: Merkel Cell Carcinoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp 549-62.
- Eng TY, Boersma MG, Fuller CD, et al.: A comprehensive review of the treatment of Merkel cell carcinoma. Am J Clin Oncol 30 (6): 624-36, 2007.
- Medina-Franco H, Urist MM, Fiveash J, et al.: Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol 8 (3): 204-8, 2001.
- Busse PM, Clark JR, Muse VV, et al.: Case records of the Massachusetts General Hospital. Case 19-2008. A 63-year-old HIV-positive man with cutaneous Merkel-cell carcinoma. N Engl J Med 358 (25): 2717-23, 2008.
- Haag ML, Glass LF, Fenske NA: Merkel cell carcinoma. Diagnosis and treatment. Dermatol Surg 21 (8): 669-83, 1995.
- Ratner D, Nelson BR, Brown MD, et al.: Merkel cell carcinoma. J Am Acad Dermatol 29 (2 Pt 1): 143-56, 1993.
- Gould VE, Moll R, Moll I, et al.: Neuroendocrine (Merkel) cells of the skin: hyperplasias, dysplasias, and neoplasms. Lab Invest 52 (4): 334-53, 1985.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al.: Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol 37 (1): 20-7, 2010.
- Alam M: Management of Merkel cell carcinoma: What we know. Arch Dermatol 142 (6): 771-4, 2006.
- Heath ML, Nghiem P: Merkel cell carcinoma: if no breslow, then what? J Surg Oncol 95 (8): 614-5, 2007.
- Andea AA, Coit DG, Amin B, et al.: Merkel cell carcinoma: histologic features and prognosis. Cancer 113 (9): 2549-58, 2008.
- Bichakjian CK, Lowe L, Lao CD, et al.: Merkel cell carcinoma: critical review with guidelines for multidisciplinary management. Cancer 110 (1): 1-12, 2007.
- Rao P, Balzer BL, Lemos BD, et al.: Protocol for the examination of specimens from patients with merkel cell carcinoma of the skin. Arch Pathol Lab Med 134 (3): 341-4, 2010.
- Goessling W, McKee PH, Mayer RJ: Merkel cell carcinoma. J Clin Oncol 20 (2): 588-98, 2002.
Esta información no reemplaza el consejo de un médico. Ignite Healthwise, LLC, niega toda garantía y responsabilidad por el uso de esta información. El uso que usted haga de esta información implica que usted acepta los
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
Page Footer
Quiero...
Audiencia
Sitios seguros para miembros
Información sobre The Cigna Group
Aviso legal
Los planes individuales y familiares de seguro médico y dental están asegurados por Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc. y Cigna HealthCare of Texas, Inc. Los planes de beneficios de salud y de seguro de salud de grupo están asegurados o administrados por CHLIC, Connecticut General Life Insurance Company (CGLIC) o sus afiliadas (puedes ver
Todas las pólizas de seguros y los planes de beneficios de grupo contienen exclusiones y limitaciones. Para conocer la disponibilidad, los costos y detalles completos de la cobertura, comunícate con un agente autorizado o con un representante de ventas de Cigna. Este sitio web no está dirigido a los residentes de New Mexico.