Tratamiento del cáncer de hígado infantil (PDQ®) : Tratamiento - información para profesionales de salud [NCI]
Estratificación tumoral por imágenes
Un objetivo principal del tratamiento para niños y adolescentes con cáncer de hígado es la extirpación quirúrgica del tumor primario. La agrupación por riesgo depende en gran medida de factores determinados mediante imágenes que se relacionan con una resección quirúrgica segura del tumor, así como de la agrupación PRETEXT. Estos hallazgos de las imágenes incluyen la sección o secciones del hígado afectadas por el tumor y otros hallazgos, denominados factores de anotación, que inciden en la toma de decisiones quirúrgicas y en el pronóstico.
La estratificación del riesgo del cáncer de hígado durante la niñez y la adolescencia conlleva el uso de imágenes transversales de alta calidad. Se usan la tomografía trifásica computarizada (sin contraste, arterial y venosa) o las imágenes por resonancia magnética (IRM) con contraste. La IRM con gadoxetato disódico, un fármaco a base de gadolinio que los hepatocitos absorben y excretan preferentemente, se usa cada vez más y puede mejorar la detección de la enfermedad multifocal.[
Definiciones de los grupos PRETEXT y POSTTEXT
Los sistemas de agrupación por imágenes que se usan para definir radiológicamente la extensión del compromiso tumoral en el hígado son los siguientes:
- PRETEXT (PRE-Treatment EXTent of disease). La extensión del compromiso hepático se define antes del tratamiento.
- POSTTEXT (POST-Treatment EXTent of disease). La extensión del compromiso hepático se define en respuesta al tratamiento.
PRETEXT
Los principales grupos de ensayos multicéntricos utilizan PRETEXT como un componente central de los esquemas de estratificación del riesgo que guían el tratamiento del hepatoblastoma. PRETEXT se basa en la estructura anatómica de Couinaud de 8 segmentos del hígado evaluados en imágenes transversales.
El sistema PRETEXT divide el hígado en 4 partes llamadas secciones. El lóbulo izquierdo del hígado se compone de una sección lateral (segmentos de Couinaud I, II y III) y una sección medial (segmento IV), mientras que el lóbulo derecho se compone de una sección anterior (segmentos V y VIII) y una sección posterior (segmentos VI y VII) (consultar la
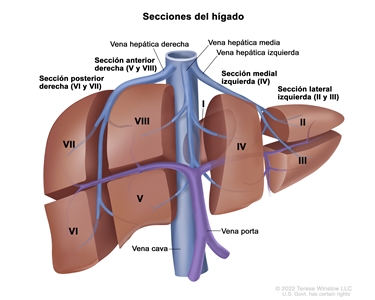
La asignación al grupo PRETEXT I, II, III, o IV se determina por el número de secciones del hígado sin compromiso. La clasificación PRETEXT se complementa con los factores de anotación. Los factores de anotación incluyen hallazgos que son importantes para el tratamiento quirúrgico e indicios de extensión tumoral fuera del parénquima de las secciones principales del hígado, incluso la enfermedad metastásica. Para obtener una descripción detallada de los grupos PRETEXT, consultar el
Los factores de anotación identifican la extensión del compromiso tumoral en los vasos principales y su efecto en el flujo venoso de entrada y salida. Estos factores proporcionan información esencial para el cirujano y pueden afectar los desenlaces quirúrgicos. En un momento dado, las definiciones de compromiso vascular macroscópico utilizadas por el Children's Oncology Group (COG) y los principales centros de cirugía hepática en los Estados Unidos difirieron de las utilizadas por SIOPEL y en Europa. Estas diferencias se han resuelto y las nuevas definiciones se están utilizando en un ensayo internacional.[
Si bien PRETEXT se usa para predecir la resecabilidad del tumor, tiene limitaciones. Puede ser complicado distinguir la invasión real más allá del borde anatómico de una sección hepática determinada de la compresión y el desplazamiento causados por el tumor, en especial en el momento del diagnóstico. Además, en ocasiones es difícil distinguir entre la coartación vascular y el compromiso vascular, en especial si no se usa la imagen adecuada. La asignación al grupo PRETEXT tiene un grado moderado de variabilidad entre observadores. En un informe en el que se usaron datos del estudio SIOPEL-1, el grupo preoperatorio PRETEXT se alineó con los hallazgos patológicos posoperatorios solo en el 51 % de los casos, se presentó sobreestadificación en el 37 % de los pacientes y subestadificación en el 12 % de los pacientes.[
Debido a que es difícil distinguir la asignación al grupo PRETEXT, la revisión central de las imágenes es fundamental y se suele hacer en todos los ensayos clínicos importantes. Cuando los pacientes no participan en ensayos clínicos, se debe considerar la revisión radiológica por expertos para los casos cuestionables cuya asignación al grupo PRETEXT afecta la elección del tratamiento.
| Grupos PRETEXT y POSTTEXT | Definición | Imagen | |
|---|---|---|---|
| a Adaptación de Roebuck et al.[ |
|||
| I | Compromiso de 1 sola sección; 3 secciones adyacentes sin tumor. | 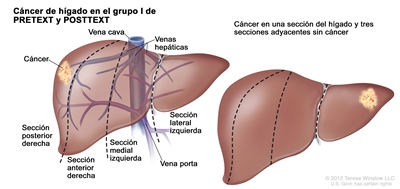 |
|
| II | Compromiso de 1 o 2 secciones; 2 secciones adyacentes sin tumor. | 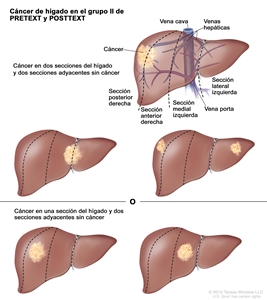 |
|
| III | Compromiso de 2 o 3 secciones; 1 sección adyacente sin tumor. | 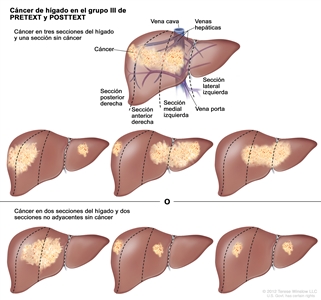 |
|
| IV | Compromiso de 4 secciones. | 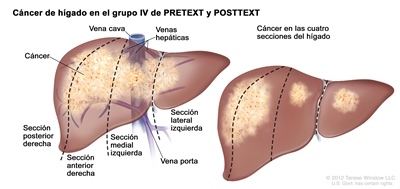 |
|
| Factores de anotación | Definición | ||
|---|---|---|---|
| TC = tomografía computarizada; IRM = imágenes por resonancia magnética; UH = unidad de Hounsfield. | |||
| a Adaptación de Roebuck et al.[ |
|||
| b Se publicaron detalles adicionales que describen los factores de anotación.[ |
|||
| Vb | Compromiso venoso: Compromiso vascular de la vena cava retrohepática o compromiso de las3 venas hepáticas principales (derecha, media e izquierda). | ||
| V0 | Tumor a menos de 1 cm del vaso. | ||
| V1 | Tumor adyacente al vaso. | ||
| V2 | Tumor que comprime o distorsiona el vaso. | ||
| V3 | Crecimiento tumoral infiltrante, atrapamiento vascular o trombo. | ||
| Pb | Compromiso portal: Compromiso vascular de la vena porta principal o deambas venas portas, derecha e izquierda. | ||
| P0 | Tumor a menos de 1 cm del vaso. | ||
| P1 | Tumor adyacente a la vena porta principal, las venas portas derecha e izquierda o la bifurcación de la vena porta. | ||
| P2 | Tumor que comprime la vena porta principal, las venas portas derecha e izquierda o la bifurcación de la vena porta. | ||
| P3 | Crecimiento tumoral infiltrante, atrapamiento vascular (>50 % o >180 grados) o trombo intravascular en la vena porta principal, las venas portas derecha e izquierda o la bifurcación de la vena porta. | ||
| Eb | Diseminación extrahepática de la enfermedad. Se cumple cualquiera de los siguientes criterios: | ||
| E1 | Tumor que cruza los límites o los planos tisulares. | ||
| E2 | Tumor rodeado de tejido normal en más de 180 grados. | ||
| E3 | Presencia de nódulos peritoneales (que no son ganglios linfáticos); al menos 1 nódulo ≥10 mm o 2 nódulos ≥5 mm. | ||
| Mb | Metástasis a distancia. Se cumple cualquiera de los siguientes criterios: | ||
| M1 | 1 nódulo pulmonar no calcificado de diámetro ≥5 mm. | ||
| M2 | 2 o más nódulos no calcificados, cada uno de diámetro ≥3 mm. | ||
| M3 | Enfermedad metastásica confirmada mediante estudio patológico. | ||
| C | Tumor que compromete el lóbulo caudado. | ||
| F | Multifocalidad. 2 o más tumores hepáticos aislados con tejido hepático intermedio normal. | ||
| Nb | Metástasis ganglionares. Se cumple cualquiera de los siguientes criterios: | ||
| N1 | Ganglio linfático con un diámetro de eje corto >1 cm. | ||
| N2 | Ganglio linfático portocavo con un diámetro de eje corto >1,5 cm. | ||
| N3 | Ganglio linfático esférico con pérdida del hilio graso. | ||
| Rb | Ruptura del tumor. Líquido libre en el abdomen o la pelvis con uno o más de los siguientes signos de hemorragia: | ||
| R1 | Complejidad interna o tabicaciones que separan el líquido. | ||
| R2 | Se observa líquido de alta densidad en la TC (>25 UH). | ||
| R3 | Se observan imágenes características de sangre o de productos de degradación de la sangre en la IRM. | ||
| R4 | Se observa líquido heterogéneo con partículas ecogénicas en la ecografía. | ||
| R5 | Cápsula tumoral con anomalía visibleo células tumorales en el líquido peritonealo ruptura diagnosticada mediante estudio patológico en pacientes que se sometieron a una resección inicial. | ||
POSTTEXT
El grupo POSTTEXT se determina después de que los pacientes reciben quimioterapia. La respuesta más alta a la quimioterapia, medida como disminución del tamaño del tumor y la concentración de alfafetoproteína (AFP), se produce después de los 2 primeros ciclos de quimioterapia.[
Estadificación quirúrgica de Evans para el cáncer de hígado infantil
El sistema de estadificación de Evans y del COG, basado en los hallazgos operatorios y la resecabilidad quirúrgica, se utilizó durante muchos años en los Estados Unidos para agrupar a los niños con cáncer de hígado y determinar su tratamiento (consultar el
| Estadio quirúrgico de Evans | Definición |
|---|---|
| Estadio I | El tumor se resecó por completo. |
| Estadio II | Queda tumor residual microscópico después de la resección. |
| Estadio III | No hay metástasis a distancia y se presenta al menos una de las siguientes situaciones: 1) El tumor es irresecable, o se extirpó pero quedan residuos tumorales macroscópicos; 2) hay compromiso ganglionar extrahepático. |
| Estadio IV | Hay metástasis a distancia, con independencia de la extensión del compromiso hepático. |
Referencias:
- Meyers AB, Towbin AJ, Geller JI, et al.: Hepatoblastoma imaging with gadoxetate disodium-enhanced MRI--typical, atypical, pre- and post-treatment evaluation. Pediatr Radiol 42 (7): 859-66, 2012.
- Brown J, Perilongo G, Shafford E, et al.: Pretreatment prognostic factors for children with hepatoblastoma-- results from the International Society of Paediatric Oncology (SIOP) study SIOPEL 1. Eur J Cancer 36 (11): 1418-25, 2000.
- Roebuck DJ, Aronson D, Clapuyt P, et al.: 2005 PRETEXT: a revised staging system for primary malignant liver tumours of childhood developed by the SIOPEL group. Pediatr Radiol 37 (2): 123-32; quiz 249-50, 2007.
- Towbin AJ, Meyers RL, Woodley H, et al.: 2017 PRETEXT: radiologic staging system for primary hepatic malignancies of childhood revised for the Paediatric Hepatic International Tumour Trial (PHITT). Pediatr Radiol 48 (4): 536-554, 2018.
- Aronson DC, Schnater JM, Staalman CR, et al.: Predictive value of the pretreatment extent of disease system in hepatoblastoma: results from the International Society of Pediatric Oncology Liver Tumor Study Group SIOPEL-1 study. J Clin Oncol 23 (6): 1245-52, 2005.
- Lovvorn HN, Ayers D, Zhao Z, et al.: Defining hepatoblastoma responsiveness to induction therapy as measured by tumor volume and serum alpha-fetoprotein kinetics. J Pediatr Surg 45 (1): 121-8; discussion 129, 2010.
- Venkatramani R, Stein JE, Sapra A, et al.: Effect of neoadjuvant chemotherapy on resectability of stage III and IV hepatoblastoma. Br J Surg 102 (1): 108-13, 2015.
- Ortega JA, Krailo MD, Haas JE, et al.: Effective treatment of unresectable or metastatic hepatoblastoma with cisplatin and continuous infusion doxorubicin chemotherapy: a report from the Childrens Cancer Study Group. J Clin Oncol 9 (12): 2167-76, 1991.
- Douglass EC, Reynolds M, Finegold M, et al.: Cisplatin, vincristine, and fluorouracil therapy for hepatoblastoma: a Pediatric Oncology Group study. J Clin Oncol 11 (1): 96-9, 1993.
- Ortega JA, Douglass EC, Feusner JH, et al.: Randomized comparison of cisplatin/vincristine/fluorouracil and cisplatin/continuous infusion doxorubicin for treatment of pediatric hepatoblastoma: A report from the Children's Cancer Group and the Pediatric Oncology Group. J Clin Oncol 18 (14): 2665-75, 2000.
Hepatoblastoma
Incidencia
La incidencia anual de hepatoblastoma en los Estados Unidos aumentó (más del doble), de 0,8 (1975–1983) a 2,3 (2020) casos por millón personas de 19 años o menos.[
La edad de presentación del cáncer de hígado en la niñez se relaciona con el subtipo histológico del tumor. Por lo general, los hepatoblastomas se presentan antes de los 3 años de edad, y cerca del 90 % de los tumores malignos de hígado en niños de 4 años o menos son hepatoblastomas.[
Factores de riesgo
Las afecciones relacionadas con un aumento del riesgo de hepatoblastoma se describen en el
| Síndromes, enfermedades y trastornos relacionados | Hallazgos clínicos |
|---|---|
| Síndrome de Aicardi[ |
Para obtener más información, consultar la sección |
| Síndrome de Beckwith-Wiedemann[ |
Para obtener más información, consultar la sección |
| Poliposis adenomatosa familiar[ |
Para obtener más información, consultar la sección |
| Glucogenosis I–IV (enfermedades por almacenamiento de glucógeno)[ |
Los síntomas varían según la enfermedad específica. |
| Lactantes con bajo peso al nacer[ |
Neonatos prematuros y neonatos pequeños para la edad gestacional. |
| Síndrome de Simpson-Golabi-Behmel[ |
Macroglosia, macrosomía, anomalías renales y esqueléticas, y aumento de riesgo de tumor de Wilms. |
| Trisomía 18 y otras trisomías[ |
Trisomía 18: microcefalia y micrognatia, puños cerrados con dedos superpuestos y retraso del desarrollo. La mayoría de los pacientes (>90 %) mueren durante el primer año de vida. |
Síndrome de Aicardi
Se presume que el síndrome de Aicardi es una afección ligada al cromosoma X que se presenta exclusivamente en mujeres, lo que lleva a la hipótesis de que un gen alterado en el cromosoma X es mortal en varones. El síndrome se define, de manera clásica, como la presencia de agenesia del cuerpo calloso, laguna coriorretiniana y espasmos infantiles, con una facies característica. A menudo se encuentran otras alteraciones encefálicas, oculares y costovertebrales.[
Síndrome de Beckwith-Wiedemann y hemihiperplasia
La incidencia de hepatoblastoma aumenta de 1000 a 10 000 veces en lactantes y niños con síndrome de Beckwith-Wiedemann.[
La causa más común del síndrome de Beckwith-Wiedemann son cambios epigenéticos y en este caso el síndrome es esporádico. En ocasiones, también obedece a variantes genéticas y en este caso el síndrome es familiar. Cualquiera de estos mecanismos se relaciona con un aumento de la incidencia de tumores embrionarios, como el tumor de Wilms y el hepatoblastoma.[
Para identificar neoplasias malignas abdominales en estadio temprano, todos los niños con síndrome de Beckwith-Wiedemann o hemihiperplasia aislada se someten a exámenes de detección regulares para muchos tipos de tumores mediante ecografía abdominal.[
Poliposis adenomatosa familiar
El hepatoblastoma se relaciona con la poliposis adenomatosa familiar (PAF). El riesgo de hepatoblastoma es 800 veces mayor en los niños de familias portadoras del gen APC. La detección del hepatoblastoma en familias con PAF mediante ecografías y análisis de las concentraciones de AFP es polémica porque se notificó que el hepatoblastoma se presenta en menos de un 1 % de los miembros de este grupo.[
La evidencia actual no permite descartar la posibilidad de que la predisposición al hepatoblastoma se limite a un subconjunto específico de variantes de APC. En otro estudio de niños con hepatoblastoma, se observó un predominio de la variante en la región 5' del gen, pero algunos pacientes presentaron variantes más cercanas a la región 3'.[
En ausencia de variantes germinales de APC, los hepatoblastomas infantiles no tienen variantes somáticas en el gen APC. Sin embargo, los hepatoblastomas con frecuencia tienen variantes del gen CTNNB1, cuya función está muy relacionada con APC.[
Exámenes de detección para niños con predisposición al hepatoblastoma
En una publicación de la American Association for Cancer Research, se recomendó que todos los niños con síndromes genéticos que otorgan un riesgo de hepatoblastoma del 1 % o más se sometan a exámenes de detección. Este grupo incluye a pacientes con síndrome de Beckwith-Wiedemann, hemihiperplasia, síndrome de Simpson-Golabi-Behmel y trisomía 18. Los exámenes de detección con ecografía abdominal y análisis de AFP se hacen cada 3 meses desde el nacimiento (o diagnóstico) hasta los 4 años, lo que identificará entre el 90 % al 95 % de los hepatoblastomas que se presentan en estos niños.[
Características genómicas del hepatoblastoma
Características moleculares del hepatoblastoma
Los hallazgos genómicos relacionados con el hepatoblastoma son los siguientes:
- La frecuencia de variantes del hepatoblastoma, según lo determinaron 3 grupos mediante secuenciación del exoma completo, fue muy baja (cerca de 3 variantes por tumor) en niños menores de 5 años.[
31 ,32 ,33 ,34 ] En un estudio genómico de todos los tipos de cáncer en pediatría se encontró que el hepatoblastoma tuvo la tasa de variantes génicas más baja entre todos los tipos de cáncer infantil analizados.[35 ] - El hepatoblastoma es primariamente una enfermedad de la activación de la vía WNT. El principal mecanismo de activación de la vía WNT son las variantes activadoras o deleciones que comprometen el exón 3 de CTNNB1. Se notificaron variantes de CTNNB1 en más del 80 % de los casos.[
31 ,33 ,34 ,36 ,37 ] Una causa menos común de activación de la vía WNT en el hepatoblastoma son las variantes de APC, que se asocian con la poliposis adenomatosa familiar.[36 ] - En tres estudios se identificaron variantes de NFE2L2 en 10 de 174 (6 %), 4 de 88 (5 %) y 5 de 112 (4 %) casos de hepatoblastoma.[
33 ,34 ,37 ] La presencia de variantes de NFE2L2 se asoció con una tasa más baja de supervivencia.[37 ] - De manera comparable, las variantes de NFE2L2 se han encontrado en muchos tipos de cáncer, como el carcinoma hepatocelular. Estas variantes hacen que NFE2L2 sea insensible a la degradación mediada por KEAP1, lo que lleva a la activación de la vía NFE2L2-KEAP1, que a su vez activa la resistencia al estrés oxidativo y se cree que confiere resistencia ante la quimioterapia.
- Las variantes de TERT y TP53, que son frecuentes en el carcinoma hepatocelular de adultos,[
38 ] son infrecuentes en niños con hepatoblastoma.[31 ,33 ,34 ,36 ] Los pacientes pediátricos con variantes de TERT presentan hepatoblastoma a una edad significativamente mayor, en comparación con los pacientes sin variantes de TERT (mediana de edad en el momento del diagnóstico, alrededor de 10 años vs. 1,4 años).[37 ] - La disomía uniparental en 11p15.5 con pérdida del alelo materno se notificó en 6 de 15 casos de hepatoblastoma.[
39 ] Este hallazgo se confirmó en los estudios de caracterización genómica, donde se observó un desequilibrio alélico en el locus 11p15 en un 30 % a un 40 % de los casos.[34 ,36 ,37 ]
La expresión génica y el perfil epigenético se han usado para identificar los subtipos biológicos de hepatoblastoma y para evaluar la importancia pronóstica de cada uno.[
- Una firma de expresión de 16 genes dividió los casos de hepatoblastoma en 2 subtipos: C1 y C2.[
37 ,40 ] El subtipo C1 abarcó la mayoría de los casos de tipo histológico fetal bien diferenciado (fetal puro). El subtipo C2 exhibió una configuración más inmadura y se asoció con tasas más altas de enfermedad metastásica en el momento del diagnóstico. En un estudio de 174 pacientes con hepatoblastoma, el subtipo C2 fue un factor de predicción significativo de desenlace precario en un análisis multivariante.[37 ] - Otro grupo de investigación también encontró que el perfil de expresión génica se puede usar para identificar subtipos de hepatoblastoma con pronóstico favorable versus pronóstico desfavorable.[
33 ] El grupo de pacientes con pronóstico desfavorable mostró expresión elevada de genes asociados con las células madre embrionarias y células progenitoras (por ejemplo, LIN28B, SALL4 y HMGA2). El grupo de pacientes con pronóstico favorable mostró expresión elevada de genes asociados con la diferenciación hepática (por ejemplo, HNF1A). - Se identificó una firma de expresión génica del cromosoma 14q32 (por ejemplo, DLK1), con una señal de expresión más fuerte vinculada con un riesgo más alto de fracaso del tratamiento.[
34 ] Una firma de expresión 14q32 fuerte también se observó en tejido hepático fetal, lo que apoya más el concepto de que los pacientes con hepatoblastoma que tienen tumores con características biológicas similares a las de las células precursoras hepáticas exhiben un pronóstico más precario. - El perfil epigenético del hepatoblastoma se ha usado para identificar subtipos de hepatoblastoma definidos por características moleculares. Se evaluaron los tumores de 113 pacientes con hepatoblastoma usando matrices de metilación del DNA. Se identificaron dos subtipos diferenciados, los grupos epigenéticos A y B (Epi-CA y Epi-CB).[
34 ] El perfil de metilación del grupo Epi-CB se parece al perfil del tejido hepático en fases iniciales de desarrollo embrionario o fetal. El perfil de metilación del grupo Epi-CA fue similar al del tejido hepático en fases fetales tardías o después del nacimiento. La supervivencia sin complicaciones fue significativamente más baja en los pacientes con el subtipo Epi-CB que en los pacientes con el subtipo Epi-CA.[34 ]
La delimitación de las aplicaciones clínicas de los métodos para obtener el perfil genómico, transcriptómico y epigenómico con el fin de clasificar el riesgo en pacientes con hepatoblastoma exige una validación independiente, que es uno de los objetivos del Paediatric Hepatic International Tumour Trial (PHITT [NCT03017326]).
Diagnóstico
Biopsia
Siempre se indica una biopsia para confirmar el diagnóstico de un tumor hepático en pediatría, excepto por alguna de las siguientes circunstancias:
- Hemangioma hepático infantil (lactantes). La biopsia no se indica en los lactantes con hemangioma hepático infantil cuando se observan hallazgos clásicos en las imágenes por resonancia magnética (IRM). Si el diagnóstico resulta dudoso luego de imágenes de alta calidad, solo entonces se confirma mediante biopsia.
- Hiperplasia nodular focal. Es posible que no se indique o que se pueda retrasar una biopsia en los pacientes con hiperplasia nodular focal que muestran características clásicas en las IRM cuando se usa un contraste específico para hepatocitos. Si el diagnóstico resulta dudoso, se confirma mediante biopsia.
- En las directrices quirúrgicas del Children's Oncology Group (COG) (apéndice AHEP0731 [NCT00980460]), se recomienda la resección del tumor en el momento del diagnóstico sin quimioterapia preoperatoria para los niños con tumores de los grupos I y II de PRE-Treatment EXTent of disease (PRETEXT) con más de 1 cm de márgenes radiográficos en la vena cava y las venas hepáticas media y porta. En consecuencia, no se suele recomendar una biopsia en estas circunstancias.
- Coriocarcinoma hepático infantil (lactantes). A menudo se indica quimioterapia sin biopsia en lactantes con coriocarcinoma hepático infantil, que se puede diagnosticar mediante imágenes y concentraciones muy elevadas de gonadotropina coriónica humana (GCH-ß).[
41 ]
Marcadores tumorales
Los marcadores tumorales AFP y GCH-β son útiles para el diagnóstico y tratamiento de los tumores hepáticos. Si bien la AFP está elevada en la mayoría de los niños con neoplasias hepáticas malignas, no es patognomónica de un tumor hepático maligno.[
Pronóstico y factores pronósticos
Pronóstico
La tasa de supervivencia general (SG) a 5 años para los niños con hepatoblastoma es del 70 %.[
Las tasas de supervivencia a 5 años, con independencia de los factores de anotación, fueron las siguientes:
- 90 % en pacientes con tumores del grupo PRETEXT I.
- 83 % en pacientes con tumores del grupo PRETEXT II.
- 73 % en pacientes con tumores del grupo PRETEXT III.
- 52 % en pacientes con tumores del grupo PRETEXT IV.
Cuando se examinó cada factor de anotación por separado, sin tener en cuenta el grupo PRETEXT u otros factores de anotación, las tasas de SG a 5 años fueron las siguientes:
- 51 % en pacientes con factor de anotación V (compromiso de las 3 venas hepáticas o de la vena cava inferior).
- 49 % en pacientes con factor de anotación P (compromiso de las venas portas derecha e izquierda).
- 53 % en pacientes con factor de anotación E (tumor extrahepático adyacente).
- 52 % en pacientes con factor de anotación F (compromiso multifocal).
- 51 % en pacientes con factor de anotación R (ruptura del tumor).
- 41 % en pacientes con factor de anotación M (metástasis a distancia).
Para obtener más información sobre la agrupación de PRETEXT y los factores de anotación, consultar la sección
Pronóstico del hepatoblastoma según el estadio quirúrgico de Evans. Los protocolos de estudio actuales utilizan la estadificación PRETEXT para determinar el pronóstico. El pronóstico, de acuerdo al estadio de Evans, se indica a continuación. Para obtener más información, consultar la sección
- Estadios I y II.
Cerca del 20 % al 30 % de los niños con hepatoblastoma tienen una enfermedad en estadio l o II. El pronóstico varía según el subtipo de hepatoblastoma:
- Los pacientes con tumores de subtipo histológico fetal bien diferenciado (antes conocidos como fetales puros), que corresponden al 4 % de los hepatoblastomas, tienen una tasa de SG a 3 y 5 años del 100 % con quimioterapia mínima o sin quimioterapia, ya sean de los grupos PRETEXT I, II o III.[
50 ,51 ,52 ] - Los pacientes con hepatoblastomas de otro subtipo histológico diferente al fetal bien diferenciado o al indiferenciado de células pequeñas tienen una tasa de SG a 3 y 4 años del 90 % al 100 % con quimioterapia adyuvante.[
50 ,51 ] - Si hay algún elemento indiferenciado de células pequeñas en pacientes con hepatoblastoma en estadio I o II, la tasa de supervivencia a 3 años es del 40 % al 70 %.[
50 ,53 ]
- Los pacientes con tumores de subtipo histológico fetal bien diferenciado (antes conocidos como fetales puros), que corresponden al 4 % de los hepatoblastomas, tienen una tasa de SG a 3 y 5 años del 100 % con quimioterapia mínima o sin quimioterapia, ya sean de los grupos PRETEXT I, II o III.[
- Estadio III.
Cerca del 50 % al 70 % de los niños con hepatoblastoma tienen una enfermedad en estadio III. La tasa de SG a 3 y 5 años para estos niños es inferior al 70 %.[
50 ,51 ] - Estadio IV.
Cerca del 10 % al 20 % de los niños con hepatoblastoma tienen una enfermedad en estadio IV. La tasa de SG a 3 y 5 años para estos niños varía bastante, desde un 20 % hasta cerca de un 60 %, según los informes publicados.[
50 ,51 ,54 ,55 ,56 ,57 ] El estadio IV posquirúrgico es equivalente a cualquier grupo PRETEXT con un factor de anotación M.[58 ,59 ,60 ]
Factores pronósticos
Los grupo de estudio de cáncer infantil, de manera individual, han intentado definir la importancia relativa de varios factores pronósticos presentes en el momento del diagnóstico y en respuesta al tratamiento.[
- Grupo PRETEXT más alto.[
58 ] - Presencia de los siguientes factores de anotación PRETEXT:[
58 ]- V: compromiso de las 3 venas hepáticas o la vena cava intrahepática inferior.
- P: compromiso de las ramas portales izquierda y derecha.
- E: extensiones tumorales extrahepáticas adyacentes (por ejemplo, al diafragma u otros órganos adyacentes).
- F: tumores multifocales.
- R: ruptura del tumor.
- M: metástasis a distancia; con frecuencia en el pulmón.
- Concentración baja de AFP (<100 ng/ml o 100–1000 ng/ml para incluir a los lactantes con concentraciones elevadas de AFP).[
63 ] - Edad más avanzada. Los pacientes de 3 a 7 años tienen un desenlace más precario en el grupo PRETEXT IV.[
58 ] Los pacientes de 8 años o más tienen un desenlace más precario que los pacientes más jóvenes de todos los grupos PRETEXT. En un informe posterior del grupo CHIC, el riesgo de un episodio aumentó con la edad en todas las cohortes de edad.[64 ][Nivel de evidencia C1] El aumento de la edad atenuó el efecto de otros factores de riesgo, como las metástasis, la concentración de AFP inferior a 100 ng/ml, la ruptura tumoral y la presencia de un factor de anotación.Por el contrario, en los estudios SIOPEL 2 y 3, los lactantes menores de 6 meses presentaron grupos PRETEXT, factores de anotación y desenlaces similares a los de los niños mayores sometidos al mismo tratamiento.[
65 ][Nivel de evidencia C1]
En el estudio CHIC, el sexo, la prematuridad, el peso al nacer y el síndrome de Beckwith-Wiedemann no tuvieron efecto en la SSC.[
Se publicó un análisis multivariante de estos factores pronósticos para ayudar a formular una nueva clasificación de grupos de riesgo para el hepatoblastoma.[
En otros estudios se observaron los siguientes factores que afectan el pronóstico:
- Grupo PRETEXT. En los estudios de SIOPEL, presentar tumores de un grupo PRETEXT bajo en el momento del diagnóstico (tumores PRETEXT I, II y III) es un factor de pronóstico favorable, mientras que tener un grupo PRETEXT IV es un factor de pronóstico precario.[
58 ] Para obtener más información, consultar la secciónEstratificación tumoral por imágenes . - Estadio tumoral. En los estudios del COG, los pacientes con características histológicas de hepatoblastoma clásico y tumores en estadio I que se resecaron en el momento del diagnóstico tienen un desenlace favorable cuando se tratan con quimioterapia limitada. Los pacientes con tumores del subtipo histológico fetal bien diferenciado presentan un pronóstico excelente. Estos tumores por lo general no se tratan con quimioterapia. Los pacientes con tumores de otros estadios y subtipos histológicos reciben un tratamiento más intensivo.[
58 ] - Factores relacionados con el tratamiento:
Quimioterapia. La quimioterapia suele reducir el tamaño y la extensión del hepatoblastoma, lo que permite una resección completa.[
51 ,54 ,66 ,67 ,68 ] La respuesta favorable del tumor primario a la quimioterapia predice su resecabilidad; la respuesta favorable se define como una disminución del tamaño del tumor del 30 % según los Response Evaluation Criteria In Solid Tumors (RECIST) o una disminución del 90 % o más en las concentraciones de AFP. A su vez, esta respuesta favorable predijo la SG en todos los grupos de riesgo CHIC tratados con quimioterapia neoadyuvante en el ensayo clínico nacional japonés JPLT-2.[69 ][Nivel de evidencia B4]Cirugía. La cura del hepatoblastoma exige la resección macroscópica del tumor. Los hepatoblastomas a menudo son unifocales, por lo tanto, la resección suele ser posible. La mayoría de los pacientes sobreviven si se extirpa por completo el hepatoblastoma. Sin embargo, debido a compromiso vascular o de otro tipo, menos de un tercio de los pacientes tienen lesiones susceptibles de resección completa en el momento del diagnóstico.[
58 ] Es de vital importancia que un niño con probable hepatoblastoma sea evaluado por un cirujano pediátrico con experiencia en los métodos de resección hepática extrema con reconstrucción vascular. El niño también debe tener acceso a un programa de trasplante de hígado. Para los tumores en estadio avanzado, el tratamiento quirúrgico del hepatoblastoma es un procedimiento exigente. Las complicaciones posoperatorias en los pacientes de riesgo alto disminuyen la tasa de SG.[70 ]Trasplante ortotópico de hígado. Este tipo de trasplante es otra opción de tratamiento para los pacientes cuyo tumor permanece irresecable después de la quimioterapia preoperatoria.[
71 ,72 ] Sin embargo, la presencia de tumor residual microscópico en el margen quirúrgico no impide un desenlace favorable.[73 ,74 ] Esto puede ser el resultado de los otros ciclos de quimioterapia administrados antes o después de la resección.[66 ,67 ,73 ]Para obtener más información sobre los desenlaces relacionados con regímenes quimioterapéuticos específicos, consultar el
Cuadro 6 . - Factores relacionados con los marcadores tumorales:
El 90 % de los niños con hepatoblastoma y dos tercios de los niños con carcinoma hepatocelular exhiben concentraciones elevadas del marcador tumoral sérico AFP, que aumenta en forma paralela al grado de actividad de la enfermedad. Las concentraciones de AFP en el momento del diagnóstico y la tasa de disminución de estas durante el tratamiento se comparan con un intervalo de referencia ajustado por edad. La ausencia de una disminución significativa en las concentraciones de AFP durante el tratamiento en ocasiones predice una respuesta precaria al tratamiento.[
75 ] En un estudio exploratorio de 34 niños con hepatoblastoma, la tasa de disminución de la AFP y el volumen tumoral, pero no las mediciones RECIST I, después de 2 ciclos de tratamiento tras el diagnóstico fue predictiva de la SSC y la SG.[76 ]La ausencia de concentraciones elevadas de AFP en el momento del diagnóstico (AFP <100 ng/ml) se presenta en un porcentaje pequeño de niños con hepatoblastoma y parece vincularse con un pronóstico muy precario, del mismo modo que la variante de hepatoblastoma indiferenciado de células pequeñas.[
58 ] Algunas de estas variantes no expresan SMARCB1 y se pueden considerar tumores rabdoides de hígado, que requieren otro tipo de tratamiento. Todos los hepatoblastomas indiferenciados de células pequeñas se someten a pruebas inmunohistoquímicas para determinar la pérdida de expresión de SMARCB1 con el fin de identificar aquellos casos que se deben tratar como hepatoblastoma versus aquellos que se deben tratar como tumores rabdoides de hígado.[50 ,53 ,56 ,57 ,77 ,78 ]Las concentraciones de GCH-ß a veces también están elevadas en el hepatoblastoma o carcinoma hepatocelular durante la niñez, lo que lleva a precocidad isosexual en los niños varones.[
45 ,46 ] - Subtipo histológico del tumor:
Para obtener más información, consultar la sección
Características histológicas en Hepatoblastoma.
Se han indicado otras variables como factores de pronóstico precario, pero ha sido difícil definir su importancia. En el estudio SIOPEL-1, un análisis multivariante de factores pronósticos luego de una respuesta favorable a la quimioterapia, indicó que solo una variable, el grupo PRETEXT, predijo la SG, mientras que la presencia de metástasis y el grupo PRETEXT predijeron la SSC.[
Características histológicas
El hepatoblastoma surge de precursores de hepatocitos que pueden presentar diferente aspecto morfológico, como sigue:[
- Células pequeñas sin diferenciación epitelial ni estromal. Es imprescindible diferenciar entre un hepatoblastoma indiferenciado de células pequeñas que expresa SMARCB1 y un tumor rabdoide de hígado, que no tiene el gen SMARCB1 ni expresa SMARCB1. Es posible que ambas enfermedades compartan características histológicas similares. En ocasiones se necesitan abordajes y quimioterapias diferentes para el tratamiento óptimo de un tumor rabdoide de hígado y de un hepatoblastoma indiferenciado de células pequeñas. Para obtener más información sobre las diferencias entre estas dos enfermedades, consultar la sección
Hepatoblastoma de subtipo histológico indiferenciado de células pequeñas y tumores rabdoides de hígado . - Células epiteliales embrionarias que se asemejan al epitelio hepático entre las 6 y 8 semanas de gestación.
- Hepatocitos fetales bien diferenciados que, desde el punto de vista morfológico son indistinguibles de las células hepáticas fetales normales.
La mayoría de las veces, el tumor se compone de una mezcla de precursores de hepatocitos epiteliales. Cerca del 20 % de los tumores contienen derivados estromales como elementos osteoides, condroides y rabdoides. En ocasiones, se encuentran elementos neuronales, melanocíticos, escamosos y enteroendocrinos. Los siguientes subtipos histológicos tienen importancia clínica:
-
Hepatoblastoma de subtipo histológico fetal bien diferenciado (fetal puro) . - Hepatoblastoma de subtipo mixto con células epiteliales fetales y embrionarias.
-
Hepatoblastoma de subtipo histológico indiferenciado de células pequeñas y tumores rabdoides de hígado .- Hepatoblastoma indiferenciado de células pequeñas (positivo para SMARCB1).
- Tumor rabdoide de hígado (negativo para SMARCB1).
Hepatoblastoma de subtipo histológico fetal bien diferenciado (fetal puro)
En un análisis de pacientes con hepatoblastomas resecados al inicio (antes de recibir quimioterapia), se indicó que los pacientes con tumores del subtipo histológico fetal bien diferenciados (antes llamados fetales puros) tienen un pronóstico mejor que los pacientes con una mezcla de componentes embrionarios más primitivos y de división celular rápida, u otros tejidos indiferenciados. En los estudios se notificó lo que se indica a continuación.
- En un estudio de pacientes con hepatoblastoma y tumores de subtipo histológico fetal bien diferenciado, se observó lo siguiente:[
51 ]- La tasa de supervivencia en pacientes que recibieron 4 dosis de doxorrubicina en monoterapia fue del 100 %. Esto indica que los pacientes con tumores de subtipo histológico fetal bien diferenciado tal vez no necesiten quimioterapia después de la resección completa.[
81 ,82 ]
- La tasa de supervivencia en pacientes que recibieron 4 dosis de doxorrubicina en monoterapia fue del 100 %. Esto indica que los pacientes con tumores de subtipo histológico fetal bien diferenciado tal vez no necesiten quimioterapia después de la resección completa.[
- En un estudio del COG (COG-P9645), 16 pacientes con hepatoblastoma de subtipo histológico fetal bien diferenciado con 2 o menos mitosis por 10 campos de gran aumento no se trataron con quimioterapia. En retrospectiva, los grupos PRETEXT fueron el grupo I (n = 4), el grupo II (n = 6) y el grupo III (n = 2).[
52 ]- La tasa de SG fue del 100 %.
- Los 16 pacientes que participaron estaban vivos sin indicios de enfermedad al cabo de una mediana de seguimiento de 4,9 años (intervalo, 9 meses a 9,2 años).
Por tanto, la resección completa de un hepatoblastoma de subtipo histológico fetal bien diferenciado quizás indique que no se necesita quimioterapia.
Hepatoblastoma de subtipo histológico indiferenciado de células pequeñas y tumores rabdoides de hígado
El hepatoblastoma indiferenciado de células pequeñas (que conserva SMARCB1) es una variante poco común del hepatoblastoma. Desde el punto de vista histológico, el hepatoblastoma indiferenciado de células pequeñas se tipifica por una población difusa de células pequeñas con poco citoplasma que parecen neuroblastos.[
El hepatoblastoma de subtipo histológico indiferenciado de células pequeñas y los tumores rabdoides de hígado se distinguen por las siguientes anomalías características:
- Anomalías cromosómicas. Estas anomalías en los tumores rabdoides incluyen translocaciones que afectan un punto de ruptura en el cromosoma 22q11 y la deleción homocigótica en la región del cromosoma 22q12 que alberga el gen SMARCB1.[
84 ,85 ] - Ausencia de expresión de SMARCB1. La falta de detección de SMARCB1 en el análisis inmunohistoquímico es característica de los tumores rabdoides malignos.[
84 ]
Históricamente, se notificó que el hepatoblastoma indiferenciado de células pequeñas se presentaba a una edad más temprana (6–10 meses) que otros casos de hepatoblastoma [
En el ensayo Paediatric Hepatic International Tumour Trial (PHITT), se designa cualquier tumor hepático infantil como un tumor rabdoide de hígado si contiene células que carecen de la expresión de SMARCB1. Los pacientes con tumores negativos para SMARCB1, que se presume están relacionados con los tumores rabdoides, tal vez no se incluyan en el ensayo internacional en el que se aborda el tratamiento del hepatoblastoma que incluye el subtipo histológico indiferenciado de células pequeñas, el carcinoma hepatocelular y las neoplasias malignas hepáticas infantiles sin otra indicación (SAI), pero no el tumor rabdoide de hígado. En este ensayo, se requiere que todos los pacientes con características histológicas compatibles con hepatoblastoma indiferenciado de células pequeñas puro, según la evaluación del patólogo institucional, se sometan a pruebas del gen SMARCB1 por análisis inmunohistoquímico de acuerdo con las prácticas de la institución. Además, la presencia de un componente blastémico indica que es un hepatoblastoma convencional.[
Una característica compartida por el hepatoblastoma indiferenciado de células pequeñas y el tumor rabdoide maligno es el pronóstico precario.[
- En 2009, se notificaron los resultados de un estudio de 11 niños con concentraciones bajas de AFP y características morfológicas de células pequeñas. Entre estos niños, 10 murieron por progresión de la enfermedad y 1 murió por complicaciones. De los 6 niños sometidos a pruebas génicas, todos eran negativos para SMARCB1, pero solo 1 exhibía características morfológicas rabdoides. Estos hallazgos indican que muchos tumores de hígado, o quizás todos los tumores de hígado con características morfológicas de células pequeñas y concentraciones de AFP muy bajas en niños pequeños pueden ser tumores rabdoides hepáticos. Estos tumores tienen un pronóstico precario que se relaciona con la variante oncoiniciadora.[
84 ] - En un estudio de una sola institución de 7 niños con tumores de hígado con características morfológicas de células pequeñas, se encontró que todos conservaban la expresión de SMARCB1. Sobrevivieron 6 niños, 1 niño murió por complicaciones del trasplante de hígado.[
88 ] - En un estudio de 23 tumores de hígado del banco de tumores de Kiel, se encontraron 12 tumores con características morfológicas de células pequeñas. Nueve tumores exhibían características histológicas rabdoides malignas clásicas y 2 tumores tenían características histológicas mixtas de células pequeñas y rabdoides. No se proporcionaron desenlaces, pero se observó que los tumores rabdoides en el encéfalo tenían características de células pequeñas, en lugar de rabdoides clásicas.[
89 ] - En un estudio de una sola institución de 6 niños con tumores de hígado negativos para SMARCB1, murieron 2 niños con características morfológicas de células pequeñas. Los 4 niños restantes con características histológicas rabdoides clásicas no recibieron tratamiento a base de cisplatino; 3 niños sobrevivieron y 1 niño murió por complicaciones del trasplante.[
90 ] - En un informe del ensayo del COG AHEP0731 (NCT00980460), se identificó a 35 de 177 pacientes evaluables (19 %) con hepatoblastoma indiferenciado de células pequeñas confirmado por la revisión central.[
86 ] Se conservó la expresión nuclear de SMARCB1 en 33 de 35 pacientes. A diferencia de los informes anteriores, la presencia de características histológicas indiferenciadas de células pequeñas no se correlacionó con la edad, el sexo o las concentraciones de AFP en el momento del diagnóstico. Las tasas de SSC a 5 años en los pacientes con hepatoblastoma indiferenciado de células pequeñas de riesgo bajo, intermedio y alto fueron del 86 % (intervalo de confianza [IC] 95 %, 33–98 %), 81 % (IC 95 %, 51–92 %) y 29 % (IC 95 %, 4–81 %), respectivamente. Las tasas de SSC a 5 años en los pacientes con hepatoblastoma de riesgo bajo, intermedio y alto sin características histológicas indiferenciadas de células pequeñas fueron del 87 % (IC 95 %, 72–95 %), del 88 % (IC 95 %, 79–95 %) y del 55 % (IC 95 %, 33–74 %); P = 0,17), respectivamente. En este ensayo, la concordancia entre la revisión local y la revisión central fue precaria y solo fue concordante en 9 de 35 casos (26 %). Se hizo análisis inmunohistoquímico de expresión de SMARCB1 en todos los tumores. En este estudio, el hepatoblastoma, que de otro modo se consideraría de riesgo muy bajo o riesgo bajo, se actualizó a riesgo intermedio si se encontraba cualquier elemento indiferenciado de células pequeñas. Para obtener más información, consultar elCuadro 5 .
Es posible que los resultados del ensayo CHIC sobre tumores hepáticos infantiles aclaren algunos de los aspectos relacionados con estos hallazgos histológicos y genéticos.
Estratificación del riesgo
Hay diferencias importantes en la estratificación del riesgo que los grupos de estudio de cáncer infantil usan para determinar el tratamiento, esto dificulta la comparación de los resultados de los distintos tratamientos. En el
| | COG (AHEP-0731) | SIOPEL (SIOPEL-3, 3HR, 4, 6) | GPOH | JPLT (JPLT-2 y 3) |
|---|---|---|---|---|
| AFP = alfafetoproteína; COG = Children's Oncology Group; GPOH = Gesellschaft für Pädiatrische Onkologie und Hämatologie (Society for Paediatric Oncology and Haematology); JPLT = Japanese Study Group for Pediatric Liver Tumor; PRETEXT = PRE-Treatment EXTent of disease; SIOPEL = International Childhood Liver Tumors Strategy Group. | ||||
| a Adaptación de Czauderna et al.[ |
||||
| b Para obtener más información sobre las anotaciones utilizadas en PRETEXT, consultar el |
||||
| c Las definiciones del COG y PRETEXT sobre el compromiso vascular difieren. | ||||
| Riesgo muy bajo | PRETEXT I o II; subtipo histológico fetal bien diferenciado; resección primaria en el momento del diagnóstico | |||
| Riesgo bajo o riesgo estándar | PRETEXT I o II de cualquier subtipo histológico con resección primaria en el momento del diagnóstico | PRETEXT I, II o III | PRETEXT I, II, o III | PRETEXT I, II, o III |
| Riesgo intermediob | PRETEXT II, III o IV con tumor irresecable en el momento del diagnóstico; o V+c, P+, E+ | PRETEXT IV o cualquier PRETEXT con ruptura del tumor; o N1, P2, P2a, V3, V3a; o multifocal | ||
| Riesgo altob | Cualquier PRETEXT con M+; concentración de AFP <100 ng/ml | Cualquier PRETEXT; V+, P+, E+, M+; concentración de AFP <100 ng/ml; ruptura del tumor | Cualquier PRETEXT con V+, E+, P+, M+ o multifocal | Cualquier PRETEXT con M1 o N2; o concentración de AFP <100 ng/ml |
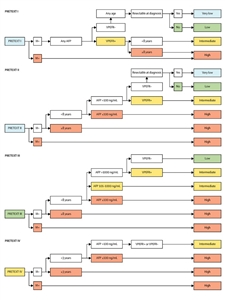
Modelo internacional de clasificación del riesgo
El grupo CHIC formuló un sistema novedoso de estratificación del riesgo para su uso en ensayos clínicos internacionales de acuerdo con las características pronósticas presentes en el momento del diagnóstico. El CHIC uniformó las diferentes definiciones y los sistemas de estadificación utilizados por los grupos de ensayos multicéntricos de cooperación del ámbito pediátrico, a fin de permitir la comparación de estudios dirigidos por grupos heterogéneos en distintos países.[
Según el análisis univariante inicial de los datos combinados con los patrones tradicionales de tratamiento clínico y los datos de ensayos clínicos grandes previos, se seleccionaron 5 grupos básicos, lo que permitió una estratificación del riesgo aún mayor. En el análisis multivariante posterior que se hizo de acuerdo a estos grupos básicos se definieron los siguientes factores clínicos pronósticos: grupo PRETEXT (I, II, III o IV), presencia de metástasis (sí o no) y AFP (≤100 ng/ml). Los grupos principales son los siguientes:[
- Grupo básico 1: PRETEXT I/II, no metastásico, AFP mayor de 100 ng/ml.
- Grupo básico 2: PRETEXT III, no metastásico, AFP mayor de 100 ng/ml.
- Grupo básico 3: PRETEXT IV, no metastásico, AFP mayor de 100 ng/ml.
- Grupo básico 4: cualquier grupo PRETEXT, enfermedad metastásica en el momento del diagnóstico, AFP mayor de 100 ng/ml.
- Grupo básico 5: cualquier grupo PRETEXT, metastásico o no metastásico, AFP de 100 ng/ml o menos en el momento del diagnóstico.
Se consultaron otros factores diagnósticos (por ejemplo, la edad) para cada una de las categorías principales, como la presencia de al menos una de las siguientes anotaciones PRETEXT (definidas como VPEFR+, ver
- V: compromiso de la vena cava, compromiso de las 3 venas hepáticas, o ambos.
- P: compromiso de la bifurcación portal, compromiso de las venas portales derecha e izquierda, o ambos.
- E: extensión tumoral extrahepática adyacente.
- F: tumor hepático multifocal.
- R: ruptura del tumor en el momento del diagnóstico.
Para los pacientes de los grupos PRETEXT I y II, se añadió una evaluación de la resecabilidad quirúrgica en el momento del diagnóstico. Los pacientes de cada una de las 5 categorías de los grupos básicos se estratificaron en subcategorías a partir de un análisis multivariante de eliminación gradual retrógrada usando otras características del paciente, como la edad y la presencia o ausencia de factores de anotación de PRETEXT (V, P, E, F y R). Cada una de estas subcategorías recibió 1 de 4 designaciones de riesgo (muy bajo, bajo, intermedio o alto). El resultado del análisis multivariante se utilizó para asignar a los pacientes a categorías de riesgo muy bajo, bajo, intermedio y alto, como se observa en la
Tratamiento del hepatoblastoma
Las opciones de tratamiento del hepatoblastoma recién diagnosticado dependen de los siguientes aspectos:
- La resecabilidad del cáncer en el momento del diagnóstico.
- Subtipo histológico del tumor.
- La respuesta del cáncer a la quimioterapia.
- La presencia de metástasis del cáncer.
La quimioterapia a base de cisplatino produjo una tasa de supervivencia de más del 90 % en los niños con enfermedad resecable PRETEXT y POST-Treatment EXTent (POSTTEXT) I y II antes o después de la quimioterapia.[
Los regímenes quimioterapéuticos que se usan para el tratamiento del hepatoblastoma y sus respectivos desenlaces se describen en el
| Estudio | Régimen quimioterapéutico | Número de pacientes | Resultados |
|---|---|---|---|
| AFP = alfafetoproteína; C5V = cisplatino, fluorouracilo (5-FU) y vincristina; CARBO = carboplatino; CCG = Children's Cancer Group; CDDP = cisplatino; CITA = pirarrubicina y cisplatino; COG = Children's Oncology Group; DOXO = doxorrubicina; SSC = supervivencia sin complicaciones, GPOH = Gesellschaft für Pädiatrische Onkologie und Hämatologie (Society for Paediatric Oncology and Haematology); H+ = ruptura o hemorragia intraperitoneal; RA = riesgo alto; IFOS = ifosfamida; IPA = ifosfamida, cisplatino y doxorrubicina; ITEC = ifosfamida, pirarrubicina, etopósido y carboplatino; JPLT = Japanese Study Group for Pediatric Liver Tumor; RB = riesgo bajo; SN = sin notificación; SG = supervivencia general; PLADO = cisplatino y doxorrubicina; POG = Pediatric Oncology Group; PRETEXT = PRE-Treatment EXTent of disease; SIOPEL = International Childhood Liver Tumors Strategy Group; RE = riesgo estándar; SUPERPLADO = cisplatino, doxorrubicina y carboplatino; THP = adriamicina tetrahidropiranilo (pirarrubicina); VP = vinorelbina y cisplatino; VPE+ = compromiso venoso, portal y extrahepático; VP16 = etopósido. | |||
| a Adaptación de Czauderna et al.,[ |
|||
| b El estudio se cerró antes de tiempo debido a los desenlaces inferiores en el grupo de CDDP/CARBO. | |||
| INT0098 (CCG/POG) 1989–1992 | C5V vs. CDDP/DOXO | Estadios I/II: 50 | SSC/SG a 4 años: |
| I/II = 88 %/100 % vs. 96 %/96 % | |||
| Estadio III: 83 | III = 60 %/68 % vs. 68 %/71 % | ||
| Estadio IV: 40 | IV = 14 %/33 % vs. 37 %/42 % | ||
| P9645 (COG) b 1999–2002 | C5V vs. CDDP/CARBO | Estadio III: 38 | SSC/SG a 3 años: |
| III/IV: C5V = 60 %/74 %; CDDP/CARBO = 38 %/54 % | |||
| Estadio IV: 50 | |||
| AHEP0731 (COG) 2010–2014[ |
RB: C5V (2 ciclos) | RB (estadios I/II): 49 | SSC a 5 años: 88 %;SG a 5 años: 91 % |
| HB 94 (GPOH) 1994–1997 | I/II: IFOS/CDDP/DOXO | Estadio I: 27 | SSC/SG a 4 años: |
| I = 89 %/96 % | |||
| Estadio II: 3 | II = 100 %/100 % | ||
| III/IV: IFOS/CDDP/DOXO + VP/CARBO | Estadio III: 25 | III = 68 %/76 % | |
| Estadio IV: 14 | IV = 21 %/36 % | ||
| HB 99 (GPOH) 1999–2004 | RE: IPA | RE: 58 | SSC/SG a 3 años: |
| RE: 90 %/88 % | |||
| RA: CARBO/VP16 | RA: 42 | CRI: 52 %/55 % | |
| SIOPEL-2 1994–1998 | RE: PLADO | PRETEXT I: 6 | SSC/SG a 3años: |
| RE: 73 %/–91 % | |||
| PRETEXT II: 36 | |||
| PRETEXT III: 25 | |||
| RA: CDDP/CARBO/DOXO | PRETEXT IV: 21 | RA: IV = 48 %/61 % | |
| Metástasis: 25 | RA: metástasis = 36 %/44 % | ||
| SIOPEL-3 1998–2006 | RE: CDDP vs. PLADO | RE: PRETEXT I: 18 | SSC/SG a 3 años: |
| RE: CDDP = 83 %/95 %; PLADO = 85 %/93 % | |||
| PRETEXT II: 133 | |||
| PRETEXT III: 104 | |||
| RA: SUPERPLADO | RA: PRETEXT IV: 74 | RA: General = 65 %/69 % | |
| VPE+: 70 | |||
| Metástasis: 70 | Metástasis = 57 %/63 % | ||
| AFP <100 ng/ml: 12 | |||
| SIOPEL-4 2005–2009 | RA: Bloque A: Semanal; CDDP/3 semanal DOXO; Bloque B: CARBO/DOXO | PRETEXT I: 2 | SSC/SG a 3 años: |
| Todos RA = 76 %/83 % | |||
| PRETEXT II: 17 | |||
| PRETEXT III: 27 | |||
| PRETEXT IV: 16 | RA: IV = 75 %/88 % | ||
| Metástasis: 39 | RA: Metástasis = 77 %/79 % | ||
| JPLT-1 1991–1999 | I/II: CDDP(30)/THP-DOXO | Estadio I: 9 | SSC/SG a 5 años: |
| I = SN/100 % | |||
| Estadio II: 32 | II = SN/76 % | ||
| III/IV: CDDP(60)/THP-DOXO | Estadio IIIa: 48 | IIIa = SN/50 % | |
| Estadio IIIb: 25 | IIIb = SN/64 % | ||
| Estadio IV: 20 | IV = SN/77 % | ||
| JPLT-2 1999–2010[ |
Cirugía inicial y 2 ciclos de CITA | Estrato 1: PRETEXT I/II, sin factores de anotación excepto por H+ (n = 40) | SSC/SG a 5 años: |
| 74,2 %/89,9 % | |||
| 2 ciclos de CITA seguidos de cirugía y 2–4 ciclos de CITA | Estrato 2: PRETEXT II con multifocalidad (n = 80) | 84,8 %/90,8 % | |
| 2 ciclos de CITA seguidos de 2 ciclos de CITA (pacientes que responden al tratamiento); intento de cirugía que incluye trasplante | Estrato 3: PRETEXT I/II (con factores de anotación) y III/IV (n = 176) que responden al tratamiento | 71,6%/85,9% | |
| 2 ciclos de CITA seguidos de 2 ciclos de ITEC (que no responden al tratamiento); intento de cirugía que incluye trasplante | Estrato 4: PRETEXT I/II (con factores de anotación) y III/IV (n = 59) que no responden al tratamiento | 59,1 %/67,3 % | |
Opciones de tratamiento del hepatoblastoma resecable en el momento del diagnóstico
Cerca del 20 % al 30 % de los niños con hepatoblastoma presentan enfermedad resecable en el momento del diagnóstico. En las directrices quirúrgicas del Children's Oncology (COG) (apéndice AHEP0731 [NCT00980460]), se recomienda la resección del tumor en el momento del diagnóstico sin quimioterapia preoperatoria para los niños con tumores de los grupos PRETEXT I y PRETEXT II con más de 1 cm de márgenes radiográficos en la vena cava y las venas hepáticas media y porta. Los desenlaces para los pacientes después de someterse a una resección completa en el momento del diagnóstico en comparación con los pacientes con compromiso microscópico de los márgenes en el momento de la resección, son similares después de recibir quimioterapia.[
Según el subtipo histológico, el pronóstico varía de la siguiente manera:
- Los pacientes con el subtipo histológico fetal bien diferenciado (4 % de los hepatoblastomas) tienen una tasa de SG a 3 y 5 años del 100 % con quimioterapia adyuvante mínima o sin esta.[
50 ,51 ,52 ,96 ] - Los pacientes con hepatoblastomas de otro subtipo histológico diferente al fetal bien diferenciado o al indiferenciado de células pequeñas tienen una tasa de SG a 3 y 4 años del 90 % al 100 % con quimioterapia adyuvante.[
50 ,51 ,54 ,56 ,97 ] - Si hay algún elemento indiferenciado de células pequeñas, la tasa de supervivencia a 3 años es del 40 % al 70 %.[
50 ,53 ]
Las opciones de tratamiento del hepatoblastoma resecable en el momento del diagnóstico y de subtipo diferente al fetal bien diferenciado son las siguientes:
- Resección seguida de 2 a 4 ciclos de quimioterapia.[
58 ]
Es posible que no sea necesaria una nueva resección de márgenes con compromiso microscópico. No se dispone de evidencia concluyente sobre los tumores resecados en el momento del diagnóstico en comparación con aquellos que presentan márgenes con compromiso microscópico resecados después de la quimioterapia preoperatoria.
Evidencia (resección quirúrgica macroscópica, con márgenes con compromiso microscópico o sin este, y quimioterapia posoperatoria):
- En el ensayo del COG AHEP0731 (NCT00980460), 49 de 51 pacientes con hepatoblastoma en estadio I o estadio II (sin características histológicas fetales puras) recibieron 2 ciclos de quimioterapia adyuvante con cisplatino, fluorouracilo y vincristina.[
93 ][Nivel de evidencia C1]- La tasa de SSC a 5 años fue del 88 % y la tasa de SG a 5 años fue del 91 %.
- Este desenlace es comparable a los desenlaces de niños tratados con 4 ciclos después de la resección inicial, así como a los desenlaces de los niños tratados con 2 ciclos de quimioterapia neoadyuvante antes de la resección seguida de 2 ciclos de quimioterapia después de la resección.
- No se dispone de datos confiables sobre el riesgo de recidiva local en pacientes con márgenes con compromiso microscópico que se resecaron en el momento del diagnóstico.[
68 ] Los estudios de SIOPEL indican que en los pacientes que recibieron quimioterapia preoperatoria, la presencia de algún margen con compromiso microscópico no aumentó el riesgo de recidiva local.[56 ,57 ,73 ]; [95 ][Nivel de evidencia C1]- En un estudio europeo realizado entre 1990 y 1994, 11 pacientes presentaron tejido tumoral en los márgenes quirúrgicos luego de una resección hepática y 2 pacientes murieron; ninguno presentó recidiva local. De los 11 pacientes, ninguno se sometió a una segunda resección y solo 1 paciente recibió radioterapia posoperatoria. Todos los pacientes recibieron 4 ciclos de cisplatino y doxorrubicina antes de la cirugía y 2 cursos de quimioterapia posoperatoria.[
73 ] - En otro estudio europeo de hepatoblastoma de riesgo alto, 11 pacientes presentaron tumor residual microscópico después de la cirugía inicial y recibieron de 2 a 4 ciclos de quimioterapia posoperatoria sin cirugía adicional. De estos 11 pacientes, 9 sobrevivieron.[
57 ] - En el estudio SIOPEL-2, sobrevivieron 13 de 13 pacientes con compromiso microscópico en los márgenes de resección.[
56 ] - En un estudio retrospectivo no planificado de los ensayos SIOPEL-2 y SIOPEL-3, se encontró que después de 4 cursos de cisplatino administrados a pacientes de riesgo estándar y 7 cursos de cisplatino alternado con doxorrubicina y carboplatino para los pacientes de riesgo alto, se realizó una resección cuando, a partir de las imágenes, se determinó que no sería peligrosa. De los 431 niños tratados en estos ensayos, 58 exhibieron márgenes tumorales con compromiso microscópico y 371 presentaron remisión completa. No hubo diferencias estadísticamente significativas en las tasas de recidiva local, SSC o SG entre los dos grupos.[
95 ][Nivel de evidencia C1]
- En un estudio europeo realizado entre 1990 y 1994, 11 pacientes presentaron tejido tumoral en los márgenes quirúrgicos luego de una resección hepática y 2 pacientes murieron; ninguno presentó recidiva local. De los 11 pacientes, ninguno se sometió a una segunda resección y solo 1 paciente recibió radioterapia posoperatoria. Todos los pacientes recibieron 4 ciclos de cisplatino y doxorrubicina antes de la cirugía y 2 cursos de quimioterapia posoperatoria.[
- En un ensayo clínico aleatorizado se demostró una eficacia comparable de la administración de cisplatino, vincristina y fluorouracilo posoperatorios y la administración de cisplatino y doxorrubicina para el tratamiento de pacientes con hepatoblastoma.[
51 ]- Si bien los desenlaces de supervivencia fueron nominalmente más altos en los niños que recibieron cisplatino y doxorrubicina, esta diferencia no fue estadísticamente significativa.
- La administración de una combinación de cisplatino, vincristina y fluorouracilo presentó una toxicidad significativamente inferior a la administración de dosis de cisplatino y doxorrubicina.
Los resultados de los ensayos clínicos de quimioterapia se describen en el
Las opciones de tratamiento del hepatoblastoma de subtipo histológico fetal bien diferenciado (fetal puro) resecable en el momento del diagnóstico son las siguientes:
- Resección quirúrgica completa seguida de conducta expectante o quimioterapia.[
52 ]
Evidencia (resección quirúrgica completa seguida de conducta expectante o quimioterapia):
- En un ensayo clínico prospectivo del COG (INT0098), 9 niños con tumor en estadio I de subtipo histológico fetal bien diferenciado (completamente resecado) y menos de 2 mitosis por campo de gran aumento recibieron 4 ciclos de doxorrubicina adyuvante.[
51 ]- Al cabo de una mediana de seguimiento de 5,1 años, las tasas de SSC y SG fueron del 100 % en los 9 niños.
- En el estudio del COG P9645 (NCT00003994), 16 pacientes con tumores en estadio I (completamente resecados) de tipo histológico fetal bien diferenciado no recibieron quimioterapia adyuvante. En 21 de estos 25 pacientes se estableció la clasificación PRETEXT (de manera retrospectiva y usando datos adecuados) y se encontraron tumores PRETEXT I, II y III en 7, 10 y 4 pacientes, respectivamente.[
52 ]- Las tasas de SSC y SG fueron del 100 % en los pacientes con tumores del subtipo histológico fetal bien diferenciado en estadio I, incluso 1 paciente que se sometió a una segunda cirugía para tratar un margen tumoral comprometido.
Opciones de tratamiento del hepatoblastoma irresecable o que no se resecó en el momento del diagnóstico
Cerca del 70 % al 80 % de los niños con hepatoblastoma tienen tumores que no se resecaron en el momento del diagnóstico. En las directrices quirúrgicas del COG (apéndice AHEP0731 [NCT00980460]), se recomienda una biopsia diagnóstica sin intentar resecar el tumor en niños con tumores del grupo PRETEXT II con menos de 1 cm de margen radiográfico en la vena cava y la vena hepática media, así como en todos los niños con tumores de los grupos PRETEXT III y IV.
Las opciones de tratamiento del hepatoblastoma irresecable o que no se resecó en el momento del diagnóstico son las siguientes:
- Quimioterapia seguida de una nueva evaluación de la resecabilidad quirúrgica y resección quirúrgica completa.
- Quimioterapia seguida de una nueva evaluación de la resecabilidad quirúrgica y trasplante ortotópico de hígado.[
54 ,71 ,98 ,99 ,100 ,101 ,102 ,103 ] - Quimioembolización transarterial (QETA) y radioembolización transarterial (RETA). La QETA y la RETA se pueden usar para mejorar la resecabilidad antes de la cirugía definitiva.[
104 ,105 ,106 ]
La ruptura tumoral en el momento de la presentación inicial, que produce una hemorragia importante que se puede controlar mediante embolización arterial transcatéter o resección parcial para estabilizar al paciente, no impide un desenlace favorable cuando se sigue de quimioterapia y cirugía definitiva.[
En años recientes, la mayoría de los niños con hepatoblastoma se han tratado con quimioterapia. En los centros oncológicos europeos, los niños con hepatoblastoma resecable en el momento del diagnóstico reciben quimioterapia preoperatoria, lo que en ocasiones reduce la incidencia de complicaciones quirúrgicas en el momento de la resección.[
Los pacientes cuyos tumores permanecen irresecables después de la quimioterapia se deben considerar candidatos para un trasplante de hígado.[
Evidencia (quimioterapia seguida de una nueva evaluación de la resecabilidad quirúrgica y resección quirúrgica completa o trasplante de hígado):
- En el estudio SIOPEL-1, la quimioterapia preoperatoria (doxorrubicina y cisplatino) se administró a todos los niños con hepatoblastoma con metástasis o sin esta. Después de la quimioterapia, y excluyendo a quienes se sometieron a un trasplante de hígado (<5 % de los pacientes), se realizó una resección completa.[
54 ]- La quimioterapia se toleró bien.
- La resección completa se obtuvo en el 87 % de los niños.
- Esta estrategia produjo una SG del 75 % a los 5 años del diagnóstico.
- Se observaron resultados idénticos en un estudio internacional de seguimiento (SIOPEL-2).[
56 ] - En el estudio SIOPEL-3, se comparó la administración de cisplatino solo con la administración de cisplatino y doxorrubicina en pacientes con hepatoblastoma de riesgo preoperatorio estándar. El riesgo estándar se definió como un tumor confinado al hígado y que afecta hasta 3 sectores.[
97 ][Nivel de evidencia A1]- Las tasas de resección y SG fueron similares en los grupos de cisplatino (95 %) y cisplatino con doxorrubicina (93 %).
- En un estudio piloto, SIOPEL-3HR, se administró cisplatino alternado con carboplatino y doxorrubicina en dosis intensivas a pacientes de riesgo alto con hepatoblastoma.[
57 ]- De 74 pacientes con tumores PRETEXT IV (22 de los cuales también tenían metástasis), 31 pacientes tenían tumores que se volvieron resecables y 26 pacientes se sometieron a trasplante. La tasa de SG a 3 años fue del 69 % (± 11 %).
- En los 70 pacientes con metástasis inscritos en este ensayo, la tasa de SSC a 3 años fue del 56 % y la tasa de SG fue del 62 %. De los pacientes con metástasis pulmonares, el 50 % logró la remisión completa de las metástasis con quimioterapia sola (sin cirugía pulmonar).
- El estudio SIOPEL-4 (NCT00077389) fue un ensayo multinacional de viabilidad de la administración de quimioterapia con dosis densas de cisplatino y doxorrubicina, y cirugía radical para un grupo de niños con hepatoblastoma de riesgo alto. En la medida de lo posible, se realizó la resección quirúrgica de todas las lesiones tumorales restantes después de la quimioterapia (incluso trasplante de hígado y metastasectomía, cuando fue necesario). Los pacientes que se sometieron a resección hepática o trasplante de hígado después de 3 ciclos de quimioterapia recibieron más tarde 2 ciclos posoperatorios de carboplatino y doxorrubicina. Los pacientes cuyos tumores permanecieron irresecables después de 3 ciclos de quimioterapia recibieron 2 ciclos muy intensivos de carboplatino y doxorrubicina antes de la cirugía. Las masas tumorales primarias se clasificaron como PRETEXT II (27 %), III (44 %) y IV (26 %).[
74 ][Nivel de evidencia B4]- El 97 % (60 de 61) de los pacientes presentaron respuesta parcial a la quimioterapia.
- El 85 % (53) de los pacientes se sometieron a resección macroscópica completa; se encontró tumor microscópico en 5 pacientes, todos sobrevivientes sin enfermedad.
- De los pacientes, 2 murieron después de la cirugía.
- Se realizaron 37 hepatectomías parciales y 16 trasplantes de hígado.
- En el estudio participaron 62 pacientes de riesgo alto; el 74 % de los pacientes (62–84 %) se sometió a resección.
- La tasa de supervivencia sin enfermedad (SSE) a 3 años fue del 76 % (IC 95 %, 65–87 %).
- La tasa de SG a 3 años fue del 83 % (IC 95 %, 73–93 %).
- De los 16 pacientes con tumores PRETEXT IV, 11 se reclasificaron a un grupo más bajo después de la quimioterapia (6 pacientes al grupo PRETEXT III, 4 pacientes al grupo PRETEXT II y 1 paciente al grupo PRETEXT I). De los tumores, 12 se volvieron resecables; y de estos, 4 pacientes, se sometieron a hepatectomía parcial mientras que 8 pacientes se sometieron a trasplante de hígado. En los pacientes que presentaron enfermedad PRETEXT IV:
- La tasa de SG a 3 años fue del 73 % (IC 95 %, 51–96 %).
- La tasa de SG a 3 años fue del 80 % (IC 95 %, 60–100 %).
- En cerca del 75 % de los niños y adolescentes con hepatoblastoma irresecable al inicio, los tumores se vuelven resecables gracias a la administración de quimioterapia preoperatoria a base de cisplatino, y entre un 60 % y un 65 % de los pacientes sobrevivirán sin enfermedad.[
108 ]
En los Estados Unidos, los tumores irresecables se han tratado con quimioterapia antes de la resección o el trasplante.[
El COG llevó a cabo un ensayo de fase III de un solo grupo (AHEP0731 [NCT00980460]) para pacientes con hepatoblastoma de riesgo intermedio. En el estudio se incluyeron 93 pacientes con enfermedad no metastásica irresecable y 9 pacientes con resección completa en el momento del diagnóstico. Todos los tumores eran de subtipo histológico indiferenciado de células pequeñas. Se evaluó la viabilidad y eficacia de la adición de doxorrubicina al tratamiento estándar (cisplatino, fluorouracilo y vincristina). En los 93 pacientes con enfermedad irresecable al inicio, la tasa de SSC a 5 años fue del 85 % (IC 95 %, 79–93 %) y la tasa de SG fue del 95 % (IC 95 %, 87–98 %).[
La quimioterapia seguida de QETA y ultrasonidos focalizados de alta intensidad mostró resultados prometedores en China para pacientes con tumores PRETEXT lll y lV, algunos de los cuales eran resecables. Los pacientes no se sometieron a resección quirúrgica debido a la negativa de los padres.[
Opciones de tratamiento del hepatoblastoma con metástasis en el momento del diagnóstico
Los desenlaces de los pacientes con hepatoblastoma metastásico en el momento del diagnóstico son precarios, pero es posible la supervivencia a largo plazo y la curación.[
Las opciones de tratamiento del hepatoblastoma metastásico en el momento del diagnóstico son las siguientes:
- Quimioterapia seguida de una nueva evaluación de la resecabilidad quirúrgica.
- Si el tumor primario y la enfermedad extrahepática (por lo común, nódulos pulmonares) son resecables luego de la quimioterapia, se hace una resección quirúrgica seguida de más quimioterapia.
- Si la enfermedad metastásica extrahepática está en remisión completa luego de la quimioterapia o de la resección quirúrgica de un nódulo pulmonar, pero el tumor primario permanece irresecable, se hace un trasplante ortotópico de hígado.
- Si la enfermedad metastásica extrahepática no es resecable o el paciente no es candidato para un trasplante, es posible que se indique más quimioterapia adicional, QETA, RETA o radioterapia.[
106 ]
El régimen de quimioterapia combinada estándar en América del Norte es de 4 ciclos de cisplatino, vincristina y fluorouracilo [
La quimioterapia de dosis altas con rescate de células madre no es más eficaz que la quimioterapia multifarmacológica estándar.[
Evidencia (quimioterapia seguida de cirugía para tratar la enfermedad metastásica en el momento del diagnóstico):
- Un subconjunto de 39 pacientes que presentaban metástasis desde el inicio participaron en el ensayo SIOPEL-4 (NCT00077389), un ensayo multinacional de viabilidad de quimioterapia con dosis densas de cisplatino y doxorrubicina y cirugía radical para un grupo de niños con hepatoblastoma de riesgo alto. Los pacientes sometidos a resección o trasplante hepático después de 3 ciclos de quimioterapia recibieron más tarde 2 ciclos posoperatorios de carboplatino y doxorrubicina. Los pacientes cuyos tumores eran irresecables después de 3 ciclos de quimioterapia recibieron 2 ciclos adicionales de carboplatino y doxorrubicina muy intensivos antes de la cirugía.[
74 ][Nivel de evidencia B4]- Después de 3 ciclos de quimioterapia, se obtuvo respuesta completa (solo de las metástasis) en 20 de 39 pacientes y respuesta parcial en 18 de 39 pacientes. De los pacientes que lograron una respuesta completa, 19 estaban vivos sin enfermedad 3 años después del diagnóstico.
- De los pacientes que lograron una respuesta parcial, 7 se sometieron a metastasectomía cerca del momento de la resección o el trasplante de hígado, con una tasa de SG del 100 %. Además, otros 7 pacientes que tenían nódulos pulmonares residuales pequeños se sometieron a resección sin metastasectomía; de ellos, 6 pacientes evolucionaron bien y 1 paciente presentó una recidiva.
- Con el tiempo, 2 pacientes que tenían metástasis iniciales, recidivaron.
- Se necesitó trasplante de hígado, en lugar de resección sola, para el tratamiento de 7 de los 39 pacientes que presentaban metástasis.
- Del subgrupo de 39 pacientes que presentaban metástasis desde el inicio, la tasa de SSE a 3 años fue del 77 % (IC 95 %, 63–90 %), y la tasa de SG fue del 79 % (IC 95 %, 66–92 %).
En los pacientes con tumores primarios resecados, cualquier remanente de metástasis pulmonar se debe extirpar mediante cirugía, de ser posible.[
Si la enfermedad extrahepática está en remisión completa después de la quimioterapia y el tumor primario sigue siendo irresecable, es posible realizar un trasplante ortotópico de hígado.[
Se observan discrepancias en los desenlaces de pacientes con metástasis pulmonares en el momento del diagnóstico que se someten a trasplante ortotópico de hígado luego de la resolución completa de la enfermedad pulmonar como reacción a la quimioterapia pretrasplante. En algunos estudios se notificaron desenlaces favorables para estos pacientes,[
Si la enfermedad extrahepática no es resecable después de la quimioterapia o el paciente no es apto para un trasplante, los abordajes alternativos de tratamiento son los siguientes:
- Otros fármacos quimioterapéuticos. Se han utilizado fármacos quimioterapéuticos como irinotecán, dosis altas de cisplatino y etopósido o infusión continua de doxorrubicina.[
120 ,121 ,122 ]; [123 ][Nivel de evidencia C1] - QETA.[
105 ,124 ] - Radioterapia.[
125 ]
Tratamiento del hepatoblastoma progresivo o recidivante
El pronóstico de un paciente con hepatoblastoma progresivo o recidivante depende de varios factores, como los siguientes:[
- Sitio de la recidiva.
- Tratamiento previo.
- Consideraciones individuales del paciente.
Las opciones de tratamiento del hepatoblastoma recidivante o progresivo son las siguientes:
- Resección quirúrgica. En los pacientes con hepatoblastoma que se resecó por completo en el momento del diagnóstico inicial, es posible que el tratamiento quirúrgico radical de las metástasis pulmonares aisladas que se presentan durante el curso de la enfermedad, junto con una estrategia general que incluya quimioterapia, haga posible una SSE prolongada.[
117 ,126 ,127 ]De ser posible, las metástasis aisladas se deben resecar en su totalidad en aquellos pacientes cuyo tumor primario está controlado.[
128 ] En un análisis retrospectivo de pacientes de los estudios SIOPEL 1, 2 y 3, se observó una incidencia de recidiva del 12 % después de la remisión completa mediante imágenes y concentraciones de AFP. Los desenlaces después de la recidiva fueron mejores si el tumor era susceptible de cirugía. De los pacientes sometidos a quimioterapia y cirugía, la tasa de SSC a 3 años fue del 34 % y la tasa de SG fue del 43 %.[126 ][Nivel de evidencia C1]Si no se puede extirpar mediante cirugía toda la enfermedad recidivante, los pacientes deben considerar la posibilidad de inscribirse en un ensayo clínico. Es posible que los ensayos clínicos de fase l y fase II sean apropiados.
- Quimioterapia. En los análisis de supervivencia después de la recidiva, se demostró que algunos pacientes tratados con cisplatino, vincristina y fluorouracilo podían recuperarse con regímenes que contenían doxorrubicina, pero los pacientes tratados con doxorrubicina y cisplatino no se podían recuperar con vincristina y fluorouracilo.[
129 ] La adición de doxorrubicina a la combinación de vincristina, fluorouracilo y cisplatino se evaluó clínicamente en el estudio del COG AHEP0731 (NCT00980460).La combinación de vincristina e irinotecán y la administración de irinotecán en monoterapia se han utilizado con cierto éxito.[
123 ]; [122 ][Nivel de evidencia C1]En una revisión de los estudios de fase I y II del COG, no se encontraron fármacos prometedores para el hepatoblastoma en recaída.[
130 ] - Trasplante de hígado. Se debe considerar el trasplante de hígado para los pacientes con recidiva de enfermedad no metastásica en el hígado que no es susceptible de resección.[
71 ,98 ,101 ] - Ablación percutánea. La ablación percutánea por radiofrecuencia se ha utilizado como alternativa a la resección quirúrgica del hepatoblastoma oligometastático.[
131 ][Nivel de evidencia C1] También se pueden considerar técnicas de ablación percutánea como terapia paliativa.[132 ]
Opciones de tratamiento en evaluación clínica para el hepatoblastoma
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el
Referencias:
- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations). National Cancer Institute, 2012, Section 29.
Also available online . Last accessed August 11, 2022. - Bulterys M, Goodman MT, Smith MA, et al.: Hepatic tumors. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 91-98.
Also available online . Last accessed August 11, 2022. - National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute.
Available online . Last accessed February 25, 2025. - Turcotte LM, Georgieff MK, Ross JA, et al.: Neonatal medical exposures and characteristics of low birth weight hepatoblastoma cases: a report from the Children's Oncology Group. Pediatr Blood Cancer 61 (11): 2018-23, 2014.
- Tanimura M, Matsui I, Abe J, et al.: Increased risk of hepatoblastoma among immature children with a lower birth weight. Cancer Res 58 (14): 3032-5, 1998.
- McLaughlin CC, Baptiste MS, Schymura MJ, et al.: Maternal and infant birth characteristics and hepatoblastoma. Am J Epidemiol 163 (9): 818-28, 2006.
- Darbari A, Sabin KM, Shapiro CN, et al.: Epidemiology of primary hepatic malignancies in U.S. children. Hepatology 38 (3): 560-6, 2003.
- Kamien BA, Gabbett MT: Aicardi syndrome associated with hepatoblastoma and pulmonary sequestration. Am J Med Genet A 149A (8): 1850-2, 2009.
- DeBaun MR, Tucker MA: Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr 132 (3 Pt 1): 398-400, 1998.
- Weksberg R, Shuman C, Smith AC: Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 137C (1): 12-23, 2005.
- Iwama T, Mishima Y: Mortality in young first-degree relatives of patients with familial adenomatous polyposis. Cancer 73 (8): 2065-8, 1994.
- Garber JE, Li FP, Kingston JE, et al.: Hepatoblastoma and familial adenomatous polyposis. J Natl Cancer Inst 80 (20): 1626-8, 1988.
- Giardiello FM, Petersen GM, Brensinger JD, et al.: Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut 39 (96): 867-9, 1996.
- Ito E, Sato Y, Kawauchi K, et al.: Type 1a glycogen storage disease with hepatoblastoma in siblings. Cancer 59 (10): 1776-80, 1987.
- Ikeda H, Hachitanda Y, Tanimura M, et al.: Development of unfavorable hepatoblastoma in children of very low birth weight: results of a surgical and pathologic review. Cancer 82 (9): 1789-96, 1998.
- Spector LG, Birch J: The epidemiology of hepatoblastoma. Pediatr Blood Cancer 59 (5): 776-9, 2012.
- Buonuomo PS, Ruggiero A, Vasta I, et al.: Second case of hepatoblastoma in a young patient with Simpson-Golabi-Behmel syndrome. Pediatr Hematol Oncol 22 (7): 623-8, 2005 Oct-Nov.
- Tan ZH, Lai A, Chen CK, et al.: Association of trisomy 18 with hepatoblastoma and its implications. Eur J Pediatr 173 (12): 1595-8, 2014.
- Trobaugh-Lotrario AD, Venkatramani R, Feusner JH: Hepatoblastoma in children with Beckwith-Wiedemann syndrome: does it warrant different treatment? J Pediatr Hematol Oncol 36 (5): 369-73, 2014.
- Hoyme HE, Seaver LH, Jones KL, et al.: Isolated hemihyperplasia (hemihypertrophy): report of a prospective multicenter study of the incidence of neoplasia and review. Am J Med Genet 79 (4): 274-8, 1998.
- Clericuzio CL, Martin RA: Diagnostic criteria and tumor screening for individuals with isolated hemihyperplasia. Genet Med 11 (3): 220-2, 2009.
- Algar EM, St Heaps L, Darmanian A, et al.: Paternally inherited submicroscopic duplication at 11p15.5 implicates insulin-like growth factor II in overgrowth and Wilms' tumorigenesis. Cancer Res 67 (5): 2360-5, 2007.
- Steenman M, Westerveld A, Mannens M: Genetics of Beckwith-Wiedemann syndrome-associated tumors: common genetic pathways. Genes Chromosomes Cancer 28 (1): 1-13, 2000.
- Albrecht S, Hartmann W, Houshdaran F, et al.: Allelic loss but absence of mutations in the polyspecific transporter gene BWR1A on 11p15.5 in hepatoblastoma. Int J Cancer 111 (4): 627-32, 2004.
- Clericuzio CL, Chen E, McNeil DE, et al.: Serum alpha-fetoprotein screening for hepatoblastoma in children with Beckwith-Wiedemann syndrome or isolated hemihyperplasia. J Pediatr 143 (2): 270-2, 2003.
- Mussa A, Molinatto C, Baldassarre G, et al.: Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J Pediatr 176: 142-149.e1, 2016.
- Aretz S, Koch A, Uhlhaas S, et al.: Should children at risk for familial adenomatous polyposis be screened for hepatoblastoma and children with apparently sporadic hepatoblastoma be screened for APC germline mutations? Pediatr Blood Cancer 47 (6): 811-8, 2006.
- Hirschman BA, Pollock BH, Tomlinson GE: The spectrum of APC mutations in children with hepatoblastoma from familial adenomatous polyposis kindreds. J Pediatr 147 (2): 263-6, 2005.
- Koch A, Denkhaus D, Albrecht S, et al.: Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res 59 (2): 269-73, 1999.
- Kalish JM, Doros L, Helman LJ, et al.: Surveillance Recommendations for Children with Overgrowth Syndromes and Predisposition to Wilms Tumors and Hepatoblastoma. Clin Cancer Res 23 (13): e115-e122, 2017.
- Eichenmüller M, Trippel F, Kreuder M, et al.: The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J Hepatol 61 (6): 1312-20, 2014.
- Jia D, Dong R, Jing Y, et al.: Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology 60 (5): 1686-96, 2014.
- Sumazin P, Chen Y, Treviño LR, et al.: Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology 65 (1): 104-121, 2017.
- Carrillo-Reixach J, Torrens L, Simon-Coma M, et al.: Epigenetic footprint enables molecular risk stratification of hepatoblastoma with clinical implications. J Hepatol 73 (2): 328-341, 2020.
- Gröbner SN, Worst BC, Weischenfeldt J, et al.: The landscape of genomic alterations across childhood cancers. Nature 555 (7696): 321-327, 2018.
- Sekiguchi M, Seki M, Kawai T, et al.: Integrated multiomics analysis of hepatoblastoma unravels its heterogeneity and provides novel druggable targets. NPJ Precis Oncol 4: 20, 2020.
- Cairo S, Armengol C, Maibach R, et al.: A combined clinical and biological risk classification improves prediction of outcome in hepatoblastoma patients. Eur J Cancer 141: 30-39, 2020.
- Cancer Genome Atlas Research Network. Electronic address: wheeler@bcm.edu, Cancer Genome Atlas Research Network: Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 169 (7): 1327-1341.e23, 2017.
- Albrecht S, von Schweinitz D, Waha A, et al.: Loss of maternal alleles on chromosome arm 11p in hepatoblastoma. Cancer Res 54 (19): 5041-4, 1994.
- Cairo S, Armengol C, De Reyniès A, et al.: Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 14 (6): 471-84, 2008.
- Yoon JM, Burns RC, Malogolowkin MH, et al.: Treatment of infantile choriocarcinoma of the liver. Pediatr Blood Cancer 49 (1): 99-102, 2007.
- Boman F, Bossard C, Fabre M, et al.: Mesenchymal hamartomas of the liver may be associated with increased serum alpha foetoprotein concentrations and mimic hepatoblastomas. Eur J Pediatr Surg 14 (1): 63-6, 2004.
- Blohm ME, Vesterling-Hörner D, Calaminus G, et al.: Alpha 1-fetoprotein (AFP) reference values in infants up to 2 years of age. Pediatr Hematol Oncol 15 (2): 135-42, 1998 Mar-Apr.
- Wu JT, Book L, Sudar K: Serum alpha fetoprotein (AFP) levels in normal infants. Pediatr Res 15 (1): 50-2, 1981.
- Schneider DT, Calaminus G, Göbel U: Diagnostic value of alpha 1-fetoprotein and beta-human chorionic gonadotropin in infancy and childhood. Pediatr Hematol Oncol 18 (1): 11-26, 2001 Jan-Feb.
- Nakagawara A, Ikeda K, Tsuneyoshi M, et al.: Hepatoblastoma producing both alpha-fetoprotein and human chorionic gonadotropin. Clinicopathologic analysis of four cases and a review of the literature. Cancer 56 (7): 1636-42, 1985.
- Perilongo G, Malogolowkin M, Feusner J: Hepatoblastoma clinical research: lessons learned and future challenges. Pediatr Blood Cancer 59 (5): 818-21, 2012.
- Trobaugh-Lotrario AD, Katzenstein HM: Chemotherapeutic approaches for newly diagnosed hepatoblastoma: past, present, and future strategies. Pediatr Blood Cancer 59 (5): 809-12, 2012.
- Trobaugh-Lotrario AD, Chaiyachati BH, Meyers RL, et al.: Outcomes for patients with congenital hepatoblastoma. Pediatr Blood Cancer 60 (11): 1817-25, 2013.
- Meyers RL, Rowland JR, Krailo M, et al.: Predictive power of pretreatment prognostic factors in children with hepatoblastoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 53 (6): 1016-22, 2009.
- Ortega JA, Douglass EC, Feusner JH, et al.: Randomized comparison of cisplatin/vincristine/fluorouracil and cisplatin/continuous infusion doxorubicin for treatment of pediatric hepatoblastoma: A report from the Children's Cancer Group and the Pediatric Oncology Group. J Clin Oncol 18 (14): 2665-75, 2000.
- Malogolowkin MH, Katzenstein HM, Meyers RL, et al.: Complete surgical resection is curative for children with hepatoblastoma with pure fetal histology: a report from the Children's Oncology Group. J Clin Oncol 29 (24): 3301-6, 2011.
- De Ioris M, Brugieres L, Zimmermann A, et al.: Hepatoblastoma with a low serum alpha-fetoprotein level at diagnosis: the SIOPEL group experience. Eur J Cancer 44 (4): 545-50, 2008.
- Pritchard J, Brown J, Shafford E, et al.: Cisplatin, doxorubicin, and delayed surgery for childhood hepatoblastoma: a successful approach--results of the first prospective study of the International Society of Pediatric Oncology. J Clin Oncol 18 (22): 3819-28, 2000.
- Perilongo G, Brown J, Shafford E, et al.: Hepatoblastoma presenting with lung metastases: treatment results of the first cooperative, prospective study of the International Society of Paediatric Oncology on childhood liver tumors. Cancer 89 (8): 1845-53, 2000.
- Perilongo G, Shafford E, Maibach R, et al.: Risk-adapted treatment for childhood hepatoblastoma. final report of the second study of the International Society of Paediatric Oncology--SIOPEL 2. Eur J Cancer 40 (3): 411-21, 2004.
- Zsíros J, Maibach R, Shafford E, et al.: Successful treatment of childhood high-risk hepatoblastoma with dose-intensive multiagent chemotherapy and surgery: final results of the SIOPEL-3HR study. J Clin Oncol 28 (15): 2584-90, 2010.
- Czauderna P, Haeberle B, Hiyama E, et al.: The Children's Hepatic tumors International Collaboration (CHIC): Novel global rare tumor database yields new prognostic factors in hepatoblastoma and becomes a research model. Eur J Cancer 52: 92-101, 2016.
- Katzenstein HM, Furman WL, Malogolowkin MH, et al.: Upfront window vincristine/irinotecan treatment of high-risk hepatoblastoma: A report from the Children's Oncology Group AHEP0731 study committee. Cancer 123 (12): 2360-2367, 2017.
- O'Neill AF, Towbin AJ, Krailo MD, et al.: Characterization of Pulmonary Metastases in Children With Hepatoblastoma Treated on Children's Oncology Group Protocol AHEP0731 (The Treatment of Children With All Stages of Hepatoblastoma): A Report From the Children's Oncology Group. J Clin Oncol 35 (30): 3465-3473, 2017.
- Fuchs J, Rydzynski J, Von Schweinitz D, et al.: Pretreatment prognostic factors and treatment results in children with hepatoblastoma: a report from the German Cooperative Pediatric Liver Tumor Study HB 94. Cancer 95 (1): 172-82, 2002.
- Aronson DC, Schnater JM, Staalman CR, et al.: Predictive value of the pretreatment extent of disease system in hepatoblastoma: results from the International Society of Pediatric Oncology Liver Tumor Study Group SIOPEL-1 study. J Clin Oncol 23 (6): 1245-52, 2005.
- Meyers RL, Maibach R, Hiyama E, et al.: Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children's Hepatic tumors International Collaboration. Lancet Oncol 18 (1): 122-131, 2017.
- Haeberle B, Rangaswami A, Krailo M, et al.: The importance of age as prognostic factor for the outcome of patients with hepatoblastoma: Analysis from the Children's Hepatic tumors International Collaboration (CHIC) database. Pediatr Blood Cancer 67 (8): e28350, 2020.
- Dall'Igna P, Brugieres L, Christin AS, et al.: Hepatoblastoma in children aged less than six months at diagnosis: A report from the SIOPEL group. Pediatr Blood Cancer 65 (1): , 2018.
- Ortega JA, Krailo MD, Haas JE, et al.: Effective treatment of unresectable or metastatic hepatoblastoma with cisplatin and continuous infusion doxorubicin chemotherapy: a report from the Childrens Cancer Study Group. J Clin Oncol 9 (12): 2167-76, 1991.
- Douglass EC, Reynolds M, Finegold M, et al.: Cisplatin, vincristine, and fluorouracil therapy for hepatoblastoma: a Pediatric Oncology Group study. J Clin Oncol 11 (1): 96-9, 1993.
- Meyers RL, Czauderna P, Otte JB: Surgical treatment of hepatoblastoma. Pediatr Blood Cancer 59 (5): 800-8, 2012.
- Hiyama E, Hishiki T, Watanabe K, et al.: Resectability and tumor response after preoperative chemotherapy in hepatoblastoma treated by the Japanese Study Group for Pediatric Liver Tumor (JPLT)-2 protocol. J Pediatr Surg 51 (12): 2053-2057, 2016.
- Becker K, Furch C, Schmid I, et al.: Impact of postoperative complications on overall survival of patients with hepatoblastoma. Pediatr Blood Cancer 62 (1): 24-8, 2015.
- Otte JB, Pritchard J, Aronson DC, et al.: Liver transplantation for hepatoblastoma: results from the International Society of Pediatric Oncology (SIOP) study SIOPEL-1 and review of the world experience. Pediatr Blood Cancer 42 (1): 74-83, 2004.
- McAteer JP, Goldin AB, Healey PJ, et al.: Surgical treatment of primary liver tumors in children: outcomes analysis of resection and transplantation in the SEER database. Pediatr Transplant 17 (8): 744-50, 2013.
- Schnater JM, Aronson DC, Plaschkes J, et al.: Surgical view of the treatment of patients with hepatoblastoma: results from the first prospective trial of the International Society of Pediatric Oncology Liver Tumor Study Group. Cancer 94 (4): 1111-20, 2002.
- Zsiros J, Brugieres L, Brock P, et al.: Dose-dense cisplatin-based chemotherapy and surgery for children with high-risk hepatoblastoma (SIOPEL-4): a prospective, single-arm, feasibility study. Lancet Oncol 14 (9): 834-42, 2013.
- Van Tornout JM, Buckley JD, Quinn JJ, et al.: Timing and magnitude of decline in alpha-fetoprotein levels in treated children with unresectable or metastatic hepatoblastoma are predictors of outcome: a report from the Children's Cancer Group. J Clin Oncol 15 (3): 1190-7, 1997.
- Nguyen R, McCarville MB, Sykes A, et al.: Rapid decrease of serum alpha-fetoprotein and tumor volume predicts outcome in children with hepatoblastoma treated with neoadjuvant chemotherapy. Int J Clin Oncol 23 (5): 900-907, 2018.
- Brown J, Perilongo G, Shafford E, et al.: Pretreatment prognostic factors for children with hepatoblastoma-- results from the International Society of Paediatric Oncology (SIOP) study SIOPEL 1. Eur J Cancer 36 (11): 1418-25, 2000.
- D'Antiga L, Vallortigara F, Cillo U, et al.: Features predicting unresectability in hepatoblastoma. Cancer 110 (5): 1050-8, 2007.
- Czauderna P, Lopez-Terrada D, Hiyama E, et al.: Hepatoblastoma state of the art: pathology, genetics, risk stratification, and chemotherapy. Curr Opin Pediatr 26 (1): 19-28, 2014.
- López-Terrada D, Alaggio R, de Dávila MT, et al.: Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol 27 (3): 472-91, 2014.
- Weinberg AG, Finegold MJ: Primary hepatic tumors of childhood. Hum Pathol 14 (6): 512-37, 1983.
- Haas JE, Muczynski KA, Krailo M, et al.: Histopathology and prognosis in childhood hepatoblastoma and hepatocarcinoma. Cancer 64 (5): 1082-95, 1989.
- Rowland JM: Hepatoblastoma: assessment of criteria for histologic classification. Med Pediatr Oncol 39 (5): 478-83, 2002.
- Trobaugh-Lotrario AD, Tomlinson GE, Finegold MJ, et al.: Small cell undifferentiated variant of hepatoblastoma: adverse clinical and molecular features similar to rhabdoid tumors. Pediatr Blood Cancer 52 (3): 328-34, 2009.
- Gunawan B, Schäfer KL, Sattler B, et al.: Undifferentiated small cell hepatoblastoma with a chromosomal translocation t(22;22)(q11;q13). Histopathology 40 (5): 485-7, 2002.
- Trobaugh-Lotrario A, Katzenstein HM, Ranganathan S, et al.: Small Cell Undifferentiated Histology Does Not Adversely Affect Outcome in Hepatoblastoma: A Report From the Children's Oncology Group (COG) AHEP0731 Study Committee. J Clin Oncol 40 (5): 459-467, 2022.
- Conran RM, Hitchcock CL, Waclawiw MA, et al.: Hepatoblastoma: the prognostic significance of histologic type. Pediatr Pathol 12 (2): 167-83, 1992 Mar-Apr.
- Zhou S, Gomulia E, Mascarenhas L, et al.: Is INI1-retained small cell undifferentiated histology in hepatoblastoma unfavorable? Hum Pathol 46 (4): 620-4, 2015.
- Vokuhl C, Oyen F, Häberle B, et al.: Small cell undifferentiated (SCUD) hepatoblastomas: All malignant rhabdoid tumors? Genes Chromosomes Cancer 55 (12): 925-931, 2016.
- Cornet M, De Lambert G, Pariente D, et al.: Rhabdoid tumor of the liver: Report of 6 pediatric cases treated at a single institute. J Pediatr Surg 53 (3): 567-571, 2018.
- Meyers RL, Tiao G, de Ville de Goyet J, et al.: Hepatoblastoma state of the art: pre-treatment extent of disease, surgical resection guidelines and the role of liver transplantation. Curr Opin Pediatr 26 (1): 29-36, 2014.
- Malogolowkin MH, Katzenstein H, Krailo MD, et al.: Intensified platinum therapy is an ineffective strategy for improving outcome in pediatric patients with advanced hepatoblastoma. J Clin Oncol 24 (18): 2879-84, 2006.
- Katzenstein HM, Langham MR, Malogolowkin MH, et al.: Minimal adjuvant chemotherapy for children with hepatoblastoma resected at diagnosis (AHEP0731): a Children's Oncology Group, multicentre, phase 3 trial. Lancet Oncol 20 (5): 719-727, 2019.
- Hiyama E, Hishiki T, Watanabe K, et al.: Outcome and Late Complications of Hepatoblastomas Treated Using the Japanese Study Group for Pediatric Liver Tumor 2 Protocol. J Clin Oncol 38 (22): 2488-2498, 2020.
- Aronson DC, Weeda VB, Maibach R, et al.: Microscopically positive resection margin after hepatoblastoma resection: what is the impact on prognosis? A Childhood Liver Tumours Strategy Group (SIOPEL) report. Eur J Cancer 106: 126-132, 2019.
- Vasudevan SA, Meyers RL, Finegold MJ, et al.: Outcomes of children with well-differentiated fetal hepatoblastoma treated with surgery only: Report from Children's Oncology Group Trial, AHEP0731. J Pediatr Surg 57 (10): 251-256, 2022.
- Perilongo G, Maibach R, Shafford E, et al.: Cisplatin versus cisplatin plus doxorubicin for standard-risk hepatoblastoma. N Engl J Med 361 (17): 1662-70, 2009.
- Reyes JD, Carr B, Dvorchik I, et al.: Liver transplantation and chemotherapy for hepatoblastoma and hepatocellular cancer in childhood and adolescence. J Pediatr 136 (6): 795-804, 2000.
- Molmenti EP, Wilkinson K, Molmenti H, et al.: Treatment of unresectable hepatoblastoma with liver transplantation in the pediatric population. Am J Transplant 2 (6): 535-8, 2002.
- Czauderna P, Otte JB, Aronson DC, et al.: Guidelines for surgical treatment of hepatoblastoma in the modern era--recommendations from the Childhood Liver Tumour Strategy Group of the International Society of Paediatric Oncology (SIOPEL). Eur J Cancer 41 (7): 1031-6, 2005.
- Austin MT, Leys CM, Feurer ID, et al.: Liver transplantation for childhood hepatic malignancy: a review of the United Network for Organ Sharing (UNOS) database. J Pediatr Surg 41 (1): 182-6, 2006.
- Pham TA, Gallo AM, Concepcion W, et al.: Effect of Liver Transplant on Long-term Disease-Free Survival in Children With Hepatoblastoma and Hepatocellular Cancer. JAMA Surg 150 (12): 1150-8, 2015.
- Khan AS, Brecklin B, Vachharajani N, et al.: Liver Transplantation for Malignant Primary Pediatric Hepatic Tumors. J Am Coll Surg 225 (1): 103-113, 2017.
- Xianliang H, Jianhong L, Xuewu J, et al.: Cure of hepatoblastoma with transcatheter arterial chemoembolization. J Pediatr Hematol Oncol 26 (1): 60-3, 2004.
- Malogolowkin MH, Stanley P, Steele DA, et al.: Feasibility and toxicity of chemoembolization for children with liver tumors. J Clin Oncol 18 (6): 1279-84, 2000.
- Aguado A, Dunn SP, Averill LW, et al.: Successful use of transarterial radioembolization with yttrium-90 (TARE-Y90) in two children with hepatoblastoma. Pediatr Blood Cancer 67 (9): e28421, 2020.
- Madanur MA, Battula N, Davenport M, et al.: Staged resection for a ruptured hepatoblastoma: a 6-year follow-up. Pediatr Surg Int 23 (6): 609-11, 2007.
- Aronson DC, Meyers RL: Malignant tumors of the liver in children. Semin Pediatr Surg 25 (5): 265-275, 2016.
- Venkatramani R, Stein JE, Sapra A, et al.: Effect of neoadjuvant chemotherapy on resectability of stage III and IV hepatoblastoma. Br J Surg 102 (1): 108-13, 2015.
- von Schweinitz D, Hecker H, Harms D, et al.: Complete resection before development of drug resistance is essential for survival from advanced hepatoblastoma--a report from the German Cooperative Pediatric Liver Tumor Study HB-89. J Pediatr Surg 30 (6): 845-52, 1995.
- Fuchs J, Cavdar S, Blumenstock G, et al.: POST-TEXT III and IV Hepatoblastoma: Extended Hepatic Resection Avoids Liver Transplantation in Selected Cases. Ann Surg 266 (2): 318-323, 2017.
- Hemming AW, Reed AI, Fujita S, et al.: Role for extending hepatic resection using an aggressive approach to liver surgery. J Am Coll Surg 206 (5): 870-5; discussion 875-8, 2008.
- Fonseca A, Gupta A, Shaikh F, et al.: Extreme hepatic resections for the treatment of advanced hepatoblastoma: Are planned close margins an acceptable approach? Pediatr Blood Cancer 65 (2): , 2018.
- de Freitas Paganoti G, Tannuri ACA, Dantas Marques AC, et al.: Extensive Hepatectomy as an Alternative to Liver Transplant in Advanced Hepatoblastoma: A New Protocol Used in a Pediatric Liver Transplantation Center. Transplant Proc 51 (5): 1605-1610, 2019.
- Katzenstein HM, Malogolowkin MH, Krailo MD, et al.: Doxorubicin in combination with cisplatin, 5-flourouracil, and vincristine is feasible and effective in unresectable hepatoblastoma: A Children's Oncology Group study. Cancer 128 (5): 1057-1065, 2022.
- Wang S, Yang C, Zhang J, et al.: First experience of high-intensity focused ultrasound combined with transcatheter arterial embolization as local control for hepatoblastoma. Hepatology 59 (1): 170-7, 2014.
- Meyers RL, Katzenstein HM, Krailo M, et al.: Surgical resection of pulmonary metastatic lesions in children with hepatoblastoma. J Pediatr Surg 42 (12): 2050-6, 2007.
- Karski EE, Dvorak CC, Leung W, et al.: Treatment of hepatoblastoma with high-dose chemotherapy and stem cell rescue: the pediatric blood and marrow transplant consortium experience and review of the literature. J Pediatr Hematol Oncol 36 (5): 362-8, 2014.
- Partrick DA, Bensard DD, Teitelbaum DH, et al.: Successful thoracoscopic lung biopsy in children utilizing preoperative CT-guided localization. J Pediatr Surg 37 (7): 970-3; discussion 970-3, 2002.
- Katzenstein HM, Rigsby C, Shaw PH, et al.: Novel therapeutic approaches in the treatment of children with hepatoblastoma. J Pediatr Hematol Oncol 24 (9): 751-5, 2002.
- Palmer RD, Williams DM: Dramatic response of multiply relapsed hepatoblastoma to irinotecan (CPT-11). Med Pediatr Oncol 41 (1): 78-80, 2003.
- Qayed M, Powell C, Morgan ER, et al.: Irinotecan as maintenance therapy in high-risk hepatoblastoma. Pediatr Blood Cancer 54 (5): 761-3, 2010.
- Zsíros J, Brugières L, Brock P, et al.: Efficacy of irinotecan single drug treatment in children with refractory or recurrent hepatoblastoma--a phase II trial of the childhood liver tumour strategy group (SIOPEL). Eur J Cancer 48 (18): 3456-64, 2012.
- Sue K, Ikeda K, Nakagawara A, et al.: Intrahepatic arterial injections of cisplatin-phosphatidylcholine-Lipiodol suspension in two unresectable hepatoblastoma cases. Med Pediatr Oncol 17 (6): 496-500, 1989.
- Habrand JL, Nehme D, Kalifa C, et al.: Is there a place for radiation therapy in the management of hepatoblastomas and hepatocellular carcinomas in children? Int J Radiat Oncol Biol Phys 23 (3): 525-31, 1992.
- Semeraro M, Branchereau S, Maibach R, et al.: Relapses in hepatoblastoma patients: clinical characteristics and outcome--experience of the International Childhood Liver Tumour Strategy Group (SIOPEL). Eur J Cancer 49 (4): 915-22, 2013.
- Shi Y, Geller JI, Ma IT, et al.: Relapsed hepatoblastoma confined to the lung is effectively treated with pulmonary metastasectomy. J Pediatr Surg 51 (4): 525-9, 2016.
- Matsunaga T, Sasaki F, Ohira M, et al.: Analysis of treatment outcome for children with recurrent or metastatic hepatoblastoma. Pediatr Surg Int 19 (3): 142-6, 2003.
- Malogolowkin MH, Katzenstein HM, Krailo M, et al.: Redefining the role of doxorubicin for the treatment of children with hepatoblastoma. J Clin Oncol 26 (14): 2379-83, 2008.
- Trobaugh-Lotrario AD, Meyers RL, Feusner JH: Outcomes of Patients With Relapsed Hepatoblastoma Enrolled on Children's Oncology Group (COG) Phase I and II Studies. J Pediatr Hematol Oncol 38 (3): 187-90, 2016.
- Yevich S, Calandri M, Gravel G, et al.: Reiterative Radiofrequency Ablation in the Management of Pediatric Patients with Hepatoblastoma Metastases to the Lung, Liver, or Bone. Cardiovasc Intervent Radiol 42 (1): 41-47, 2019.
- Liu B, Zhou L, Huang G, et al.: First Experience of Ultrasound-guided Percutaneous Ablation for Recurrent Hepatoblastoma after Liver Resection in Children. Sci Rep 5: 16805, 2015.
Esta información no reemplaza el consejo de un médico. Ignite Healthwise, LLC, niega toda garantía y responsabilidad por el uso de esta información. El uso que usted haga de esta información implica que usted acepta los
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
Page Footer
Quiero...
Audiencia
Sitios seguros para miembros
Información sobre The Cigna Group
Aviso legal
Los planes individuales y familiares de seguro médico y dental están asegurados por Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc. y Cigna HealthCare of Texas, Inc. Los planes de beneficios de salud y de seguro de salud de grupo están asegurados o administrados por CHLIC, Connecticut General Life Insurance Company (CGLIC) o sus afiliadas (puedes ver
Todas las pólizas de seguros y los planes de beneficios de grupo contienen exclusiones y limitaciones. Para conocer la disponibilidad, los costos y detalles completos de la cobertura, comunícate con un agente autorizado o con un representante de ventas de Cigna. Este sitio web no está dirigido a los residentes de New Mexico.