Tratamiento del ependimoma infantil (PDQ®) : Tratamiento - información para profesionales de salud [NCI]
Información general sobre el ependimoma infantil
Los tumores de encéfalo primarios, incluso los ependimomas, son un grupo diverso de enfermedades que, juntas, constituyen el tumor sólido más común en la niñez. Para el diagnóstico y la clasificación de los tumores, se usan cada vez más los análisis inmunohistoquímicos, los hallazgos citogenéticos y genético moleculares, y las mediciones de la actividad mitótica. Los tumores de encéfalo se clasifican según las características histológicas, pero la ubicación del tumor, el grado de diseminación, las características moleculares y la edad son factores importantes que afectan el tratamiento y el pronóstico.
De acuerdo con la revisión de 2021 de la clasificación de tumores del sistema nervioso central (SNC) de la Organización Mundial de la Salud (OMS), los tumores ependimarios se clasifican en los siguientes diez subtipos principales en función de su localización anatómica y de sus características histopatológicas y moleculares:[
- Ependimoma supratentorial.
- Ependimoma supratentorial positivo para fusiones de ZFTA (antes llamado positivo para fusiones de RELA).
- Ependimoma supratentorial positivo para fusiones de YAP1.
- Ependimoma de fosa posterior.
- Ependimoma de fosa posterior, grupo PFA.
- Ependimoma de fosa posterior, grupo PFB.
- Ependimoma medular (también conocido como ependimoma de médula espinal).
- Ependimoma medular con amplificación de MYCN.
- Ependimoma mixopapilar.
- Subependimoma (localización supratentorial, de fosa posterior y de médula espinal).
Los resúmenes de tratamiento del PDQ sobre los tumores de encéfalo infantiles se organizan sobre todo de acuerdo con la clasificación de los tumores del SNC establecida por la OMS.[
Incidencia
El ependimoma infantil abarca alrededor del 9 % de todos los tumores de encéfalo y de médula espinal en los niños, lo que representa cerca de 200 casos por año en los Estados Unidos.[
Características anatómicas
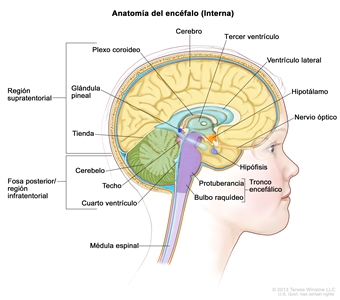
Los ependimomas surgen de los ependimocitos que revisten los ventrículos y los espacios en el encéfalo y el centro de la médula espinal (consultar la
Características clínicas
El cuadro clínico inicial del ependimoma depende de la ubicación del tumor.
- Ependimomas de fosa posterior (infratentoriales): los niños con ependimoma de fosa posterior quizás presenten signos y síntomas de hidrocefalia obstructiva debido a una obstrucción a la altura del cuarto ventrículo. A veces, también tienen ataxia, dolor cervical o parálisis de pares craneales.
- Ependimomas supratentoriales: este tipo de ependimomas producen cefaleas, convulsiones o déficits neurológicos focales que dependen de la ubicación.
- Ependimomas de médula espinal: este tipo de ependimomas a menudo son de la variante mixopapilar, tienden a producir dolor de espalda, debilidad en las extremidades inferiores o disfunción intestinal y vesical.
Evaluación diagnóstica
Todo paciente con diagnóstico de sospecha de ependimoma se evalúa con imágenes diagnósticas de todo el encéfalo y la médula espinal. El método más sensible para evaluar las metástasis subaracnoideas en la médula espinal consiste en las imágenes por resonancia magnética (IRM) de la médula espinal con gadolinio. En condiciones ideales, se obtienen las IRM antes de la cirugía para evitar confusión con la sangre posoperatoria. Si se utilizan IRM, por lo general se obtienen imágenes de toda la columna en al menos dos planos con cortes contiguos de la IRM después del realce con gadolinio.
Si es viable, se hace una evaluación citológica del LCR.[
Factores pronósticos
A continuación, se enumeran los factores desfavorables que afectan el desenlace (excepto cuando se indica):
- Características moleculares.
Los ependimomas de fosa posterior se subdividen en los siguientes dos grupos moleculares primarios a partir de perfiles característicos de expresión génica.[
9 ,10 ,11 ,12 ]- Ependimoma de fosa posterior A (PF-EPN-A).
- Este tipo de ependimoma se presenta sobre todo en niños pequeños y se caracteriza por un perfil genómico que, en su mayor parte, está equilibrado, con un aumento en la frecuencia de ganancia del cromosoma 1q [
13 ,14 ,15 ,16 ] y expresión de genes y proteínas que ya demostraron relación con un pronóstico adverso, como la tenascina C y el receptor del factor de crecimiento epidérmico.[13 ,17 ,18 ] - La ganancia de 1q confiere un pronóstico muy precario a pesar de la resección completa y la radioterapia posoperatoria (tasa de supervivencia sin complicaciones a 5 años, 81,5 % para equilibrio de 1q vs. 35,7 % para ganancia de 1q).[
19 ][Nivel de evidencia B4] - En un análisis retrospectivo combinado de 663 pacientes de 5 cohortes no solapadas se identificó la pérdida de 6q como factor pronóstico precario para pacientes con PF-EPN-A.[
20 ] La pérdida de 6q se observó en el 8,6 % de los casos de PF-EPN-A, y es más frecuente en tumores con ganancia de 1q. El subconjunto de pacientes (n = 22) con ganancia de 1q y pérdida de 6q presentó un pronóstico muy adverso. - En un estudio multicéntrico retrospectivo se compararon las muestras emparejadas de tumores primarios de cada paciente con las de tumores recidivantes. En el estudio se notificó que las características de riesgo alto de ganancia de 1q y pérdida de 6q fueron más frecuentes en los tumores recidivantes que en los tumores primarios, y estas características se siguen relacionando con un pronóstico precario.[
21 ]
- Este tipo de ependimoma se presenta sobre todo en niños pequeños y se caracteriza por un perfil genómico que, en su mayor parte, está equilibrado, con un aumento en la frecuencia de ganancia del cromosoma 1q [
- Ependimoma de fosa posterior B (PF-EPN-B).
- Este tipo de ependimoma se presenta de manera principal en niños mayores y en adultos, y se caracteriza por un pronóstico más favorable y por numerosas anomalías citogenéticas que afectan cromosomas enteros o brazos cromosómicos.[
9 ,12 ,22 ] - Los pacientes con PF-EPN-B tienen un desenlace favorable, en comparación con los pacientes con PF-EPN-A. La tasa de supervivencia sin progresión (SSP) a 5 años de los pacientes con PF-EPN-B es del 73 % y la tasa de supervivencia general (SG) supera el 90 %.[
11 ,12 ] - La ganancia de 1q no es una característica pronóstica en los pacientes con PF-EPN-B, mientras que es posible que la pérdida del cromosoma 13q confiera un pronóstico adverso.[
22 ]
- Este tipo de ependimoma se presenta de manera principal en niños mayores y en adultos, y se caracteriza por un pronóstico más favorable y por numerosas anomalías citogenéticas que afectan cromosomas enteros o brazos cromosómicos.[
Los ependimomas supratentoriales se dividen en los siguientes dos grupos moleculares principales a partir del estado de las fusiones génicas:
- Ependimoma supratentorial positivo para fusiones de ZFTA (ST-EPN-ZFTA) (antes llamado positivo para fusiones de RELA).
- Si bien en un análisis retrospectivo se indicó que la fusión de RELA producía un pronóstico precario,[
11 ] en informes posteriores se indicó que los pacientes con fusiones de RELA que se someten a resección completa y radioterapia posoperatoria tienen tasas de supervivencia relativamente favorables de cerca del 80 % a los 5 años.[11 ,19 ,23 ,24 ] En estudios retrospectivos se indican desenlaces precarios en los pacientes que se someten a resecciones quirúrgicas completas, pero no reciben radioterapia posoperatoria.[11 ] - La deleción homocigota de CDKN2A se ha relacionado con un pronóstico adverso en pacientes con ST-EPN-ZFTA.[
25 ][Nivel de evidencia B4] La deleción de CDKN2A también se ha descrito como un acontecimiento secundario en el ependimoma recidivante.[26 ]
- Si bien en un análisis retrospectivo se indicó que la fusión de RELA producía un pronóstico precario,[
- Ependimoma supratentorial con fusiones de YAP1 (ST-EPN-YAP1).
- Los pacientes con ST-EPN-YAP1 tienen un pronóstico favorable (aunque se basa en pocos casos), con una tasa de supervivencia a 5 años que se acerca al 100 %.[
11 ,23 ,27 ]
- Los pacientes con ST-EPN-YAP1 tienen un pronóstico favorable (aunque se basa en pocos casos), con una tasa de supervivencia a 5 años que se acerca al 100 %.[
Los ependimomas de médula espinal se distinguen mediante estudios del metiloma, pero la clasificación molecular no proporciona ninguna ventaja clinicopatológica sobre la clasificación histopatológica para el ependimoma mixopapilar y el subependimoma. Sin embargo, la clasificación molecular es útil para identificar el ependimoma medular con amplificación de MYCN, que se ha relacionado con un pronóstico adverso. Los datos sobre la estratificación óptima del riesgo del ependimoma medular en niños son escasos, aunque se infiere, a partir de datos de adultos, que una resección completa acarrea un pronóstico favorable.
- Ependimoma medular con amplificación de MYCN (SP-EPN-MYCN).
- Se trata de un ependimoma raro y de crecimiento rápido que afecta sobre todo a adultos jóvenes.
- Por lo general, los tumores SP-EPN-MYCN son de grado 3, y se caracterizan por un comportamiento agresivo, con frecuente diseminación leptomeníngea y alta tasa de recidiva.[
28 ,29 ,30 ,31 ]
- Ependimoma de fosa posterior A (PF-EPN-A).
- Edad más joven en el momento del diagnóstico. Tradicionalmente, una edad más joven en el momento del diagnóstico se consideró un factor de pronóstico precario, aunque es posible que esto se deba en parte a la práctica común de evitar la radiación o diferir este tratamiento en niños menores de 3 años. En un ensayo prospectivo del Children's Oncology Group (COG) (ACNS0121 [NCT00027846]), se administró radioterapia posoperatoria inmediata a todos los niños mayores de 1 año después de una resección macroscópica total o de una resección casi total. En el estudio se demostró ausencia de diferencia significativa en la SSP o la SG a 5 años entre los pacientes de 1 a 3 años de edad y los pacientes de 3 a 21 años.[
19 ] - Tipo histológico anaplásico. El tipo histológico anaplásico se relacionó con un pronóstico adverso.[
32 ][Nivel de evidencia B4]; [33 ,34 ,35 ,36 ]; [37 ][Nivel de evidencia C1]; [38 ][Nivel de evidencia C2] La diferenciación entre la enfermedad de grado 2 y de grado 3 tiene una variabilidad interobservador importante, por lo que se vuelve confuso el uso de la anaplasia como factor pronóstico. [39 ] La clasificación de 2021 de los tumores del SNC establecida por la OMS ya no usa el término ependimoma anaplásico y solo permite un diagnóstico, definido desde el punto de vista histológico, del ependimoma en el diagnóstico integrado. Dentro del informe estratificado, el especialista en anatomía patológica aún puede optar por asignar a un tumor el grado 2 o 3 del SNC de la OMS en función de las características histológicas.[2 ,3 ] - Resección subtotal. La resección subtotal acarrea un pronóstico muy precario.[
19 ,35 ,36 ]; [32 ][Nivel de evidencia B4] - Dosis bajas de radiación. Las dosis bajas de radiación y los protocolos de quimioterapia sola confieren un pronóstico precario.[
12 ,23 ,40 ,41 ]
Seguimiento después del tratamiento
Por lo general, después del tratamiento del ependimoma se recomienda la vigilancia con neuroimágenes y evaluaciones clínicas. En un informe de 198 pacientes con ependimoma, 90 de ellos experimentaron una recaída. Los pacientes cuyo tumor recidivante se detectó por medio de imágenes de vigilancia sistemática tuvieron una segunda SSP superior a la de los pacientes cuyo tumor recidivante se detectó mediante sintomatología clínica. Estos últimos tenían más probabilidad de presentar enfermedad metastásica en el momento de la recaída. Se desconoce si estos pacientes también presentaban enfermedad más agresiva en cuanto a las características biológicas, aunque la mediana del tiempo transcurrido hasta la recaída y la mediana del tiempo desde la última imagen de vigilancia fueron las mismas para ambos grupos.[
La mayoría de los médicos obtienen IRM del encéfalo o la médula espinal durante los siguientes intervalos:[
- Primeros 2 a 3 años después del tratamiento: cada 3 a 4 meses.
- De 4 a 5 años después del tratamiento: cada 6 meses.
- Más de 5 años después del tratamiento: cada año debido a una incidencia alta de recidivas tardías.
Referencias:
- Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016.
- Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
- WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
- Gurney JG, Smith MA, Bunin GR: CNS and miscellaneous intracranial and intraspinal neoplasms. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, Chapter 3, pp 51-63.
Also available online . Last accessed February 9, 2024. - Ostrom QT, Gittleman H, Truitt G, et al.: CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro Oncol 20 (suppl_4): iv1-iv86, 2018.
- Andreiuolo F, Puget S, Peyre M, et al.: Neuronal differentiation distinguishes supratentorial and infratentorial childhood ependymomas. Neuro Oncol 12 (11): 1126-34, 2010.
- Moreno L, Pollack IF, Duffner PK, et al.: Utility of cerebrospinal fluid cytology in newly diagnosed childhood ependymoma. J Pediatr Hematol Oncol 32 (6): 515-8, 2010.
- Benesch M, Mynarek M, Witt H, et al.: Newly Diagnosed Metastatic Intracranial Ependymoma in Children: Frequency, Molecular Characteristics, Treatment, and Outcome in the Prospective HIT Series. Oncologist 24 (9): e921-e929, 2019.
- Wani K, Armstrong TS, Vera-Bolanos E, et al.: A prognostic gene expression signature in infratentorial ependymoma. Acta Neuropathol 123 (5): 727-38, 2012.
- Witt H, Mack SC, Ryzhova M, et al.: Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 20 (2): 143-57, 2011.
- Pajtler KW, Witt H, Sill M, et al.: Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 27 (5): 728-43, 2015.
- Ramaswamy V, Hielscher T, Mack SC, et al.: Therapeutic Impact of Cytoreductive Surgery and Irradiation of Posterior Fossa Ependymoma in the Molecular Era: A Retrospective Multicohort Analysis. J Clin Oncol 34 (21): 2468-77, 2016.
- Mendrzyk F, Korshunov A, Benner A, et al.: Identification of gains on 1q and epidermal growth factor receptor overexpression as independent prognostic markers in intracranial ependymoma. Clin Cancer Res 12 (7 Pt 1): 2070-9, 2006.
- Korshunov A, Witt H, Hielscher T, et al.: Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol 28 (19): 3182-90, 2010.
- Kilday JP, Mitra B, Domerg C, et al.: Copy number gain of 1q25 predicts poor progression-free survival for pediatric intracranial ependymomas and enables patient risk stratification: a prospective European clinical trial cohort analysis on behalf of the Children's Cancer Leukaemia Group (CCLG), Societe Francaise d'Oncologie Pediatrique (SFOP), and International Society for Pediatric Oncology (SIOP). Clin Cancer Res 18 (7): 2001-11, 2012.
- Godfraind C, Kaczmarska JM, Kocak M, et al.: Distinct disease-risk groups in pediatric supratentorial and posterior fossa ependymomas. Acta Neuropathol 124 (2): 247-57, 2012.
- Korshunov A, Golanov A, Timirgaz V: Immunohistochemical markers for intracranial ependymoma recurrence. An analysis of 88 cases. J Neurol Sci 177 (1): 72-82, 2000.
- Andreiuolo F, Le Teuff G, Bayar MA, et al.: Integrating Tenascin-C protein expression and 1q25 copy number status in pediatric intracranial ependymoma prognostication: A new model for risk stratification. PLoS One 12 (6): e0178351, 2017.
- Merchant TE, Bendel AE, Sabin ND, et al.: Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma. J Clin Oncol 37 (12): 974-983, 2019.
- Baroni LV, Sundaresan L, Heled A, et al.: Ultra high-risk PFA ependymoma is characterized by loss of chromosome 6q. Neuro Oncol 23 (8): 1360-1370, 2021.
- Donson AM, Bertrand KC, Riemondy KA, et al.: Significant increase of high-risk chromosome 1q gain and 6q loss at recurrence in posterior fossa group A ependymoma: A multicenter study. Neuro Oncol 25 (10): 1854-1867, 2023.
- Cavalli FMG, Hübner JM, Sharma T, et al.: Heterogeneity within the PF-EPN-B ependymoma subgroup. Acta Neuropathol 136 (2): 227-237, 2018.
- Upadhyaya SA, Robinson GW, Onar-Thomas A, et al.: Molecular grouping and outcomes of young children with newly diagnosed ependymoma treated on the multi-institutional SJYC07 trial. Neuro Oncol 21 (10): 1319-1330, 2019.
- Fukuoka K, Kanemura Y, Shofuda T, et al.: Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol Commun 6 (1): 134, 2018.
- Jünger ST, Andreiuolo F, Mynarek M, et al.: CDKN2A deletion in supratentorial ependymoma with RELA alteration indicates a dismal prognosis: a retrospective analysis of the HIT ependymoma trial cohort. Acta Neuropathol 140 (3): 405-407, 2020.
- Milde T, Pfister S, Korshunov A, et al.: Stepwise accumulation of distinct genomic aberrations in a patient with progressively metastasizing ependymoma. Genes Chromosomes Cancer 48 (3): 229-38, 2009.
- Andreiuolo F, Varlet P, Tauziède-Espariat A, et al.: Childhood supratentorial ependymomas with YAP1-MAMLD1 fusion: an entity with characteristic clinical, radiological, cytogenetic and histopathological features. Brain Pathol 29 (2): 205-216, 2019.
- Ghasemi DR, Sill M, Okonechnikov K, et al.: MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol 138 (6): 1075-1089, 2019.
- Swanson AA, Raghunathan A, Jenkins RB, et al.: Spinal Cord Ependymomas With MYCN Amplification Show Aggressive Clinical Behavior. J Neuropathol Exp Neurol 78 (9): 791-797, 2019.
- Scheil S, Brüderlein S, Eicker M, et al.: Low frequency of chromosomal imbalances in anaplastic ependymomas as detected by comparative genomic hybridization. Brain Pathol 11 (2): 133-43, 2001.
- Raffeld M, Abdullaev Z, Pack SD, et al.: High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathol Commun 8 (1): 101, 2020.
- Massimino M, Miceli R, Giangaspero F, et al.: Final results of the second prospective AIEOP protocol for pediatric intracranial ependymoma. Neuro Oncol 18 (10): 1451-60, 2016.
- Merchant TE, Jenkins JJ, Burger PC, et al.: Influence of tumor grade on time to progression after irradiation for localized ependymoma in children. Int J Radiat Oncol Biol Phys 53 (1): 52-7, 2002.
- Korshunov A, Golanov A, Sycheva R, et al.: The histologic grade is a main prognostic factor for patients with intracranial ependymomas treated in the microneurosurgical era: an analysis of 258 patients. Cancer 100 (6): 1230-7, 2004.
- Tamburrini G, D'Ercole M, Pettorini BL, et al.: Survival following treatment for intracranial ependymoma: a review. Childs Nerv Syst 25 (10): 1303-12, 2009.
- Massimino M, Barretta F, Modena P, et al.: Second series by the Italian Association of Pediatric Hematology and Oncology of children and adolescents with intracranial ependymoma: an integrated molecular and clinical characterization with a long-term follow-up. Neuro Oncol 23 (5): 848-857, 2021.
- Amirian ES, Armstrong TS, Aldape KD, et al.: Predictors of survival among pediatric and adult ependymoma cases: a study using Surveillance, Epidemiology, and End Results data from 1973 to 2007. Neuroepidemiology 39 (2): 116-24, 2012.
- Tihan T, Zhou T, Holmes E, et al.: The prognostic value of histological grading of posterior fossa ependymomas in children: a Children's Oncology Group study and a review of prognostic factors. Mod Pathol 21 (2): 165-77, 2008.
- Ellison DW, Kocak M, Figarella-Branger D, et al.: Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. J Negat Results Biomed 10: 7, 2011.
- Vaidya K, Smee R, Williams JR: Prognostic factors and treatment options for paediatric ependymomas. J Clin Neurosci 19 (9): 1228-35, 2012.
- Zapotocky M, Beera K, Adamski J, et al.: Survival and functional outcomes of molecularly defined childhood posterior fossa ependymoma: Cure at a cost. Cancer 125 (11): 1867-1876, 2019.
- Klawinski D, Indelicato DJ, Hossain J, et al.: Surveillance imaging in pediatric ependymoma. Pediatr Blood Cancer 67 (11): e28622, 2020.
- Massimino M, Barretta F, Modena P, et al.: Pediatric intracranial ependymoma: correlating signs and symptoms at recurrence with outcome in the second prospective AIEOP protocol follow-up. J Neurooncol 140 (2): 457-465, 2018.
Características moleculares del ependimoma infantil
Subgrupos moleculares del ependimoma
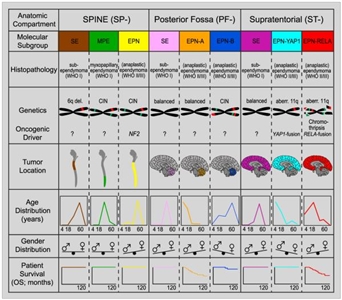
En los estudios de caracterización molecular iniciales se identificaron 9 subgrupos moleculares de ependimoma, 6 de los cuales predominan en niños. Los subgrupos se identificaron a partir de los perfiles característicos de metilación del DNA y de expresión génica, y también por una variedad exclusiva de alteraciones genómicas (consultar la Figura 2).[
Se añadió un nuevo ependimoma definido a nivel molecular a la clasificación de tumores del sistema nervioso central de la Organización Mundial de la Salud (OMS) de 2021: ependimoma medular con amplificación de MYCN. En la clasificación de 2021 se describió con más detalle los tumores ependimarios definidos por la localización anatómica y las características histológicas, pero no por la alteración molecular. Estos tumores se denominan ependimoma de fosa posterior (PF-EPN), ependimoma supratentorial (ST-EPN) y ependimoma medular (SP-EPN). Estos tumores contienen una alteración molecular exclusiva (sin clasificar), o bien su análisis molecular falló o no se obtuvo (sin especificar).[
- Tumores infratentoriales.
- Ependimoma de fosa posterior (PF-EPN).
- Ependimoma de fosa posterior A (PF-EPN-A) con pérdida de la marca de trimetilación de H3 K27.
- Ependimoma de fosa posterior B (PF-EPN-B) con retención de la marca de trimetilación de H3 K27.
- Tumores supratentoriales.
- Ependimoma supratentorial (ST-EPN).
- Ependimoma supratentorial positivo para fusiones de ZFTA (ST-EPN-ZFTA). Antes se denominaba ependimoma supratentorial positivo para fusiones de RELA (ST-EPN-RELA), pero se ha cambiado el nombre porque ZFTA es la nueva denominación de C11orf95 y ZFTA puede fusionarse con otro gen diferente a RELA.[
6 ] - Ependimoma positivo para fusiones de YAP1 (ST-EPN-YAP1).
- Tumores de médula espinal.
- Ependimoma medular (SP-EPN).
- Ependimoma medular con amplificación de MYCN (SP-EPN-MYCN).
- Ependimoma mixopapilar (SP-EPN-MPE).
El subependimoma (supratentorial, infratentorial o medular) incluye las otras tres variantes moleculares que son bastante infrecuentes en niños.
Tumores infratentoriales
Ependimoma de fosa posterior A
El ependimoma de fosa posterior A (PF-EPN-A) es el subgrupo más común que se caracteriza por los siguientes aspectos:
- Cuadro clínico inicial en niños pequeños (mediana de edad, 3 años).[
1 ,7 ] - Tasas bajas de variantes que afectan la estructura proteica, alrededor de 5 por genoma.[
2 ] - Ganancia del cromosoma 1q, un factor de pronóstico precario para pacientes con ependimoma,[
8 ] en alrededor del 25 % de los casos.[1 ,3 ,9 ] - Pérdida del cromosoma 6q, que se ha notificado como factor de pronóstico precario para los pacientes con PF-EPN-A, en el 8 % al 10 % de los casos.[
10 ] - Perfil cromosómico equilibrado con pocas ganancias o pérdidas cromosómicas.[
1 ,2 ] - Pérdida de la marca de trimetilación de H3 K27 y DNA con hipometilación general.[
11 ] En un estudio multiinstitucional prospectivo se analizó un grupo de 147 pacientes con ependimoma. En el estudio se notificaron sensibilidad y especificidad altas para la detección por análisis inmunohistoquímico de la pérdida de la marca de trimetilación H3 K27 para identificar los casos de PF-EPN-A.[12 ] La pérdida de esta marca se presenta mediante múltiples mecanismos, como los siguientes:- Variantes recurrentes de EZHIP en el 10 % de los casos, con expresión alta de mRNA de EZHIP en casi todos los PF-EPN-A.[
13 ,14 ] La expresión de EZHIP (con alteración o sin esta) produce la inhibición de la metiltransferasa EZH2, que conduce a la pérdida de la marca de trimetilación de H3 K27.[14 ,15 ] - Variantes recurrentes de K27M en genes de la histona H3 en una proporción baja de los casos.[
16 ,17 ] A diferencia de los gliomas de línea media difusos, las variantes de H3.1 (H3C2 y H3C3) son más frecuentes que las variantes de H3.3 (H3-3A).[13 ] Las variantes de las histonas son mutuamente excluyentes de la expresión alta de EZHIP,[13 ] y también conducen a la pérdida de la marca de trimetilación de H3 K27 mediante la inhibición de EZH2.
- Variantes recurrentes de EZHIP en el 10 % de los casos, con expresión alta de mRNA de EZHIP en casi todos los PF-EPN-A.[
En un estudio en el que se incluyeron más de 600 casos de PF-EPN-A, se usaron perfiles de matrices de metilación para dividir a la población en dos subgrupos característicos: PFA-1 y PFA-2.[
Ependimoma de fosa posterior B
En niños, el subgrupo de ependimoma de fosa posterior B (PF-EPN-B) es menos común que el subgrupo PF-EPN-A, representa entre 15 y 20 % de todos los ependimomas de fosa posterior, y se caracteriza por los siguientes aspectos:
- Cuadro clínico inicial predominante en adolescentes y adultos jóvenes (mediana de edad, 30 años).[
1 ,7 ] - Tasas bajas de variantes que afectan la estructura proteica (alrededor de 5 por genoma), sin variantes recurrentes.[
3 ] - Numerosas anomalías citogenéticas, sobre todo ganancias o pérdidas de cromosomas enteros.[
1 ,3 ] - Retención de la trimetilación de H3 K27.[
11 ] - La ganancia de 1q y la pérdida de 6q se producen en el PF-EPN-B, pero no se han notificado como factor pronóstico en este subgrupo (a diferencia del PF-EPN-A).[
18 ]
Tumores supratentoriales
Ependimomas supratentoriales con fusiones deZFTA
El subgrupo de ependimomas supratentoriales con fusiones de ZFTA (ST-EPN-ZFTA) es el tipo más numeroso de ependimoma supratentorial infantil que se caracteriza por fusiones génicas que afectan a RELA,[
- Representa alrededor de 70 % de los ependimomas supratentoriales en niños,[
19 ,20 ] y se presenta a una mediana de edad de 8 años.[1 ] - Presencia de fusiones de ZFTA que se producen por cromotripsis del cromosoma 11q13.1.[
19 ] - Tasas bajas de variantes que afectan la estructura proteica y casi ausencia de variantes recurrentes fuera de las fusiones ZFTA::RELA.[
19 ] - Indicios de activación de la vía NF-κB a nivel proteico o del RNA.[
19 ] - Ganancia del cromosoma 1q en cerca de un cuarto de los casos, y efecto indeterminado en la supervivencia.[
1 ] - El grado de concordancia fue elevado entre la prueba inmunohistoquímica del p65-RelA nuclear, la hibridación fluorescente in situ de ZFTA y RELA, y la clasificación según la metilación del DNA para definir el ST-EPN-ZFTA.[
21 ] - La deleción homocigota de CDKN2A se ha relacionado con un pronóstico precario en pacientes con ependimoma positivo para fusiones de ZFTA.[
22 ][Nivel de evidencia B4] La deleción de CDKN2A también se ha notificado como un evento secundario en el ependimoma recidivante.[23 ]
Ependimomas supratentoriales con fusiones deYAP1
El subgrupo de ependimomas supratentoriales con fusiones de YAP1 (ST-EPN-YAP1) es el segundo tipo menos común de ependimomas supratentoriales y exhibe fusiones que afectan el gen YAP1 en el cromosoma 11; se caracteriza por los siguientes aspectos:
- Mediana de edad en el momento del diagnóstico de 1,4 años.[
1 ] - Presencia de una fusión génica que afecta el gen YAP1, y el compañero de fusión más común es MAMLD1.[
1 ,19 ] - Un genoma relativamente estable con pocos cambios cromosómicos además de la fusión del gen YAP1.[
1 ]
Tumores que se confunden con ependimomas supratentoriales
Los ependimomas supratentoriales sin fusiones de ZFTA o YAP1 (en el cromosoma 11) son una entidad que no se ha definido y no se conoce su importancia. Mediante análisis de metilación del DNA, estas muestras a menudo se agrupan con otras entidades como los gliomas de grado alto y los tumores embrionarios. Por ejemplo, en un análisis de metilación retrospectivo de tumores de encéfalo supratentoriales se identificó un grupo de tumores diferente al de los ependimomas supratentoriales que albergan fusiones de PLAGL1 recurrentes.[
Ependimoma medular con amplificación deMYCN
El ependimoma medular con amplificación de MYCN (SP-EPN-MYCN) es poco frecuente; solo se han notificado 27 casos.[
- La mediana de edad en el momento de la presentación fue de 31 años (intervalo, 12–56 años).
- Se presentó un alto nivel de amplificación de MYCN en el momento del diagnóstico y en la recaída.
- SP-EPN-MYCN tiene un perfil de metilación único en comparación con otros ependimomas de la médula espinal, con amplificación de MYCN parecida a la del glioblastoma de tipo pediátrico y el neuroblastoma.
Referencias:
- Pajtler KW, Witt H, Sill M, et al.: Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 27 (5): 728-43, 2015.
- Witt H, Mack SC, Ryzhova M, et al.: Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 20 (2): 143-57, 2011.
- Mack SC, Witt H, Piro RM, et al.: Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506 (7489): 445-50, 2014.
- Pajtler KW, Mack SC, Ramaswamy V, et al.: The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol 133 (1): 5-12, 2017.
- WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
- Zschernack V, Jünger ST, Mynarek M, et al.: Supratentorial ependymoma in childhood: more than just RELA or YAP. Acta Neuropathol 141 (3): 455-466, 2021.
- Ramaswamy V, Hielscher T, Mack SC, et al.: Therapeutic Impact of Cytoreductive Surgery and Irradiation of Posterior Fossa Ependymoma in the Molecular Era: A Retrospective Multicohort Analysis. J Clin Oncol 34 (21): 2468-77, 2016.
- Korshunov A, Witt H, Hielscher T, et al.: Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol 28 (19): 3182-90, 2010.
- Merchant TE, Bendel AE, Sabin ND, et al.: Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma. J Clin Oncol 37 (12): 974-983, 2019.
- Baroni LV, Sundaresan L, Heled A, et al.: Ultra high-risk PFA ependymoma is characterized by loss of chromosome 6q. Neuro Oncol 23 (8): 1360-1370, 2021.
- Panwalkar P, Clark J, Ramaswamy V, et al.: Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol 134 (5): 705-714, 2017.
- Chapman RJ, Ghasemi DR, Andreiuolo F, et al.: Optimizing biomarkers for accurate ependymoma diagnosis, prognostication, and stratification within International Clinical Trials: A BIOMECA study. Neuro Oncol 25 (10): 1871-1882, 2023.
- Pajtler KW, Wen J, Sill M, et al.: Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol 136 (2): 211-226, 2018.
- Hübner JM, Müller T, Papageorgiou DN, et al.: EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol 21 (7): 878-889, 2019.
- Jain SU, Do TJ, Lund PJ, et al.: PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat Commun 10 (1): 2146, 2019.
- Gessi M, Capper D, Sahm F, et al.: Evidence of H3 K27M mutations in posterior fossa ependymomas. Acta Neuropathol 132 (4): 635-7, 2016.
- Ryall S, Guzman M, Elbabaa SK, et al.: H3 K27M mutations are extremely rare in posterior fossa group A ependymoma. Childs Nerv Syst 33 (7): 1047-1051, 2017.
- Cavalli FMG, Hübner JM, Sharma T, et al.: Heterogeneity within the PF-EPN-B ependymoma subgroup. Acta Neuropathol 136 (2): 227-237, 2018.
- Parker M, Mohankumar KM, Punchihewa C, et al.: C11orf95-RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 506 (7489): 451-5, 2014.
- Pietsch T, Wohlers I, Goschzik T, et al.: Supratentorial ependymomas of childhood carry C11orf95-RELA fusions leading to pathological activation of the NF-κB signaling pathway. Acta Neuropathol 127 (4): 609-11, 2014.
- Pagès M, Pajtler KW, Puget S, et al.: Diagnostics of pediatric supratentorial RELA ependymomas: integration of information from histopathology, genetics, DNA methylation and imaging. Brain Pathol 29 (3): 325-335, 2019.
- Jünger ST, Andreiuolo F, Mynarek M, et al.: CDKN2A deletion in supratentorial ependymoma with RELA alteration indicates a dismal prognosis: a retrospective analysis of the HIT ependymoma trial cohort. Acta Neuropathol 140 (3): 405-407, 2020.
- Milde T, Pfister S, Korshunov A, et al.: Stepwise accumulation of distinct genomic aberrations in a patient with progressively metastasizing ependymoma. Genes Chromosomes Cancer 48 (3): 229-38, 2009.
- Sievers P, Henneken SC, Blume C, et al.: Recurrent fusions in PLAGL1 define a distinct subset of pediatric-type supratentorial neuroepithelial tumors. Acta Neuropathol 142 (5): 827-839, 2021.
- Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.
- Fukuoka K, Kanemura Y, Shofuda T, et al.: Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol Commun 6 (1): 134, 2018.
- Ghasemi DR, Sill M, Okonechnikov K, et al.: MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol 138 (6): 1075-1089, 2019.
- Swanson AA, Raghunathan A, Jenkins RB, et al.: Spinal Cord Ependymomas With MYCN Amplification Show Aggressive Clinical Behavior. J Neuropathol Exp Neurol 78 (9): 791-797, 2019.
- Scheil S, Brüderlein S, Eicker M, et al.: Low frequency of chromosomal imbalances in anaplastic ependymomas as detected by comparative genomic hybridization. Brain Pathol 11 (2): 133-43, 2001.
- Raffeld M, Abdullaev Z, Pack SD, et al.: High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathol Commun 8 (1): 101, 2020.
Esta información no reemplaza el consejo de un médico. Ignite Healthwise, LLC, niega toda garantía y responsabilidad por el uso de esta información. El uso que usted haga de esta información implica que usted acepta los
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
Page Footer
Quiero...
Audiencia
Sitios seguros para miembros
Información sobre The Cigna Group
Aviso legal
Los planes individuales y familiares de seguro médico y dental están asegurados por Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc. y Cigna HealthCare of Texas, Inc. Los planes de beneficios de salud y de seguro de salud de grupo están asegurados o administrados por CHLIC, Connecticut General Life Insurance Company (CGLIC) o sus afiliadas (puedes ver
Todas las pólizas de seguros y los planes de beneficios de grupo contienen exclusiones y limitaciones. Para conocer la disponibilidad, los costos y detalles completos de la cobertura, comunícate con un agente autorizado o con un representante de ventas de Cigna. Este sitio web no está dirigido a los residentes de New Mexico.