Tratamiento del melanoma infantil (PDQ®) : Tratamiento - información para profesionales de salud [NCI]
Incidencia
El melanoma, aunque es un cáncer poco frecuente en los niños, es el cáncer de piel más común durante la niñez, seguido del carcinoma de células basales y el carcinoma de células escamosas.[
En los Estados Unidos, se diagnostican alrededor de 300 casos de melanoma cada año en pacientes menores de 20 años; cifra que representa el 0,3 % de todos los casos nuevos de melanoma.[
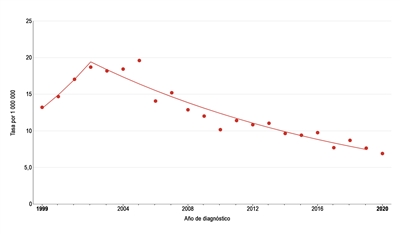
La incidencia anual de melanoma en los Estados Unidos aumenta con la edad, según se indica en la
En la
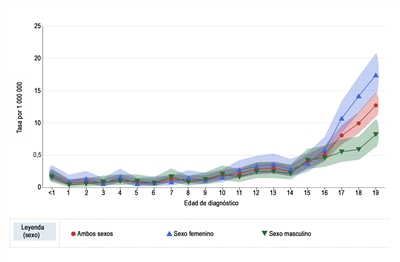
La incidencia de melanoma en pediatría (edad 0–19 años) aumentó en promedio un 1,6 % por año entre 1975 y 1996. Como se observa en la
En un estudio retrospectivo de 22 524 informes patológicos de la piel de pacientes menores de 20 años, se identificaron 38 melanomas; entre ellos, 33 en adolescentes de 15 a 19 años. Los investigadores informaron que fue necesario extirpar 479,8 lesiones para identificar 1 melanoma; esto es 20 veces más que la cantidad necesaria en la población adulta.[
Referencias:
- Sasson M, Mallory SB: Malignant primary skin tumors in children. Curr Opin Pediatr 8 (4): 372-7, 1996.
- Fishman C, Mihm MC, Sober AJ: Diagnosis and management of nevi and cutaneous melanoma in infants and children. Clin Dermatol 20 (1): 44-50, 2002 Jan-Feb.
- Hamre MR, Chuba P, Bakhshi S, et al.: Cutaneous melanoma in childhood and adolescence. Pediatr Hematol Oncol 19 (5): 309-17, 2002 Jul-Aug.
- Ceballos PI, Ruiz-Maldonado R, Mihm MC: Melanoma in children. N Engl J Med 332 (10): 656-62, 1995.
- Schmid-Wendtner MH, Berking C, Baumert J, et al.: Cutaneous melanoma in childhood and adolescence: an analysis of 36 patients. J Am Acad Dermatol 46 (6): 874-9, 2002.
- Pappo AS: Melanoma in children and adolescents. Eur J Cancer 39 (18): 2651-61, 2003.
- Huynh PM, Grant-Kels JM, Grin CM: Childhood melanoma: update and treatment. Int J Dermatol 44 (9): 715-23, 2005.
- Christenson LJ, Borrowman TA, Vachon CM, et al.: Incidence of basal cell and squamous cell carcinomas in a population younger than 40 years. JAMA 294 (6): 681-90, 2005.
- National Cancer Institute: SEER Stat Fact Sheets: Melanoma of the Skin. Bethesda, Md: National Cancer Institute.
Available online . Last accessed December 15, 2023. - National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute.
Available online . Last accessed February 25, 2025. - Paulson KG, Gupta D, Kim TS, et al.: Age-Specific Incidence of Melanoma in the United States. JAMA Dermatol 156 (1): 57-64, 2020.
- Moscarella E, Zalaudek I, Cerroni L, et al.: Excised melanocytic lesions in children and adolescents - a 10-year survey. Br J Dermatol 167 (2): 368-73, 2012.
Esta información no reemplaza el consejo de un médico. Ignite Healthwise, LLC, niega toda garantía y responsabilidad por el uso de esta información. El uso que usted haga de esta información implica que usted acepta los
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
Page Footer
Quiero...
Audiencia
Sitios seguros para miembros
Información sobre The Cigna Group
Aviso legal
Los planes individuales y familiares de seguro médico y dental están asegurados por Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc. y Cigna HealthCare of Texas, Inc. Los planes de beneficios de salud y de seguro de salud de grupo están asegurados o administrados por CHLIC, Connecticut General Life Insurance Company (CGLIC) o sus afiliadas (puedes ver
Todas las pólizas de seguros y los planes de beneficios de grupo contienen exclusiones y limitaciones. Para conocer la disponibilidad, los costos y detalles completos de la cobertura, comunícate con un agente autorizado o con un representante de ventas de Cigna. Este sitio web no está dirigido a los residentes de New Mexico.