Tratamiento del retinoblastoma (PDQ®) : Tratamiento - información para profesionales de salud [NCI]
Información general sobre el retinoblastoma
El retinoblastoma es un cáncer infantil que exige una cuidadosa integración de la atención multidisciplinaria. El objetivo del tratamiento del retinoblastoma es salvar la vida del paciente y conservar la visión útil. En los pacientes que presentan retinoblastoma extraocular, es probable que el tratamiento con quimioterapia sistémica y radioterapia sea curativo. Sin embargo, la enfermedad extraorbitaria exige quimioterapia intensiva y a veces incluye consolidación con dosis altas de quimioterapia y rescate de células madre hematopoyéticas autógenas con radioterapia o sin esta. Aunque la mayoría de los pacientes con metástasis sistémicas fuera del sistema nervioso central (SNC) se pueden curar, el pronóstico de los pacientes con enfermedad intracraneal es muy desalentador.
Incidencia
El retinoblastoma es un tumor infantil relativamente poco frecuente que se origina en la retina. Representa alrededor del 3 % de los cánceres en los niños menores de 15 años.
El retinoblastoma es un cáncer que afecta a niños muy pequeños. Dos tercios de todos los casos de retinoblastoma se diagnostican antes de los 2 años.[
Características anatómicas
El retinoblastoma surge en la retina y suele crecer debajo de la retina y hacia la cavidad vítrea. El compromiso de las membranas oculares y el nervio óptico se produce en secuencia a medida que el tumor progresa.
La invasión focal de la coroides es frecuente, aunque la invasión masiva se produce en casos de enfermedad avanzada. Después de la invasión de la coroides, el tumor llega a la circulación sistémica y crea la posibilidad de metástasis. La progresión adicional a través de las membranas oculares conduce a la invasión de la esclerótica y la órbita. Los tumores que invaden la cámara anterior a veces acceden a la circulación sistémica a través del canal de Schlemm. La progresión por el nervio óptico y más allá de la lámina cribosa aumenta el riesgo de diseminación sistémica y al SNC (consultar la

Exámenes de detección
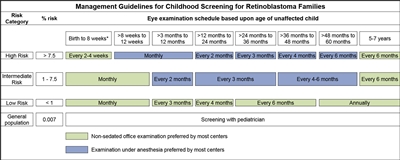
En informes de consenso de la American Association of Ophthalmic Oncologists and Pathologists y el American Association for Cancer Research Childhood Cancer Predisposition Workshop se describen las pautas de detección para niños en riesgo de presentar retinoblastoma.[
En los niños con antecedentes familiares de retinoblastoma se llevan a cabo exámenes de detección al comienzo de la vida mediante oftalmoscopia con anestesia general, a intervalos regulares. Los exámenes se realizan de acuerdo con el cálculo del riesgo absoluto a partir de la identificación de la variante de RB1 en la familia o la presencia de una variante de RB1 en el niño.[
Los hijos de progenitores afectados se someten a una exploración ocular con dilatación pupilar y anestesia general tan pronto como sea posible durante el primer mes de vida, y también se efectúa una evaluación genética. Los lactantes que obtienen resultados positivos en las pruebas genéticas se someten a exámenes mensuales con anestesia. Los lactantes que no presentan la enfermedad continúan con exámenes mensuales durante el primer año de vida. La frecuencia de esos estudios se reduce de manera progresiva durante el segundo año y los siguientes. Los exámenes de detección en los niños con antecedentes familiares de retinoblastoma quizás mejoren el pronóstico, en términos de mantener la integridad del globo ocular gracias al empleo de tratamientos de conservación ocular menos radicales (consultar el
| Familiar del probando (cualquier sexo) | Riesgo previo a la prueba del alelo alterado (%) | |
|---|---|---|
| | Probando bilateral (100) | Probando unilateral (15) |
| a Reproducción de |
||
| b Riesgo previo a la prueba para la variante deRB1en familiares de un niño afectado por |
||
| c Los familiares de tercer y cuarto grado de probandos unilaterales tienen un riesgo estimado de 0,003 y 0,001 %, respectivamente, que es más bajo que el riesgo de la población normal, de 0,007 % (1 en 15 000 nacidos vivos); por lo tanto, el riesgo se establece en 0,007 %. | ||
| Descendiente (lactante) | 50 | 7,5 |
| Progenitor | 5 | 0,8 |
| Hermano | 2,5 | 0,4 |
| Sobrino | 1,3 | 0,2 |
| Tío | 0,1 | 0,007c |
| Primo de primer grado | 0,05 | 0,007c |
| Población general | 0,007 | |
Es una práctica habitual que los padres y hermanos de ambos sexos de pacientes con retinoblastoma se sometan a exámenes de detección oftalmológicos con el fin de excluir una enfermedad familiar inadvertida. Sin embargo, cuando no se dispone de pruebas genéticas, el plan de exámenes de detección para un niño con un progenitor que tenga retinoblastoma unilateral no está bien definido.[
Cuadro clínico inicial
La edad en el momento del cuadro clínico inicial se correlaciona con lateralidad. Los pacientes con enfermedad bilateral exhiben manifestaciones a una edad más temprana, por lo general, en los primeros 12 meses de vida.
La mayoría de los pacientes presentan leucocoria, que en ocasiones se observa por primera vez después de tomar una fotografía con flash. El estrabismo es el segundo signo de presentación más frecuente y, por lo habitual, se correlaciona con compromiso macular. Los tumores intraoculares muy avanzados producen dolor, celulitis orbitaria, glaucoma o buftalmía.
A medida que el tumor progresa, los pacientes a veces presentan enfermedad orbitaria o metastásica. Se encuentran metástasis en los ganglios linfáticos preauriculares y laterocervicales, en el SNC o en forma sistémica (con frecuencia, en los huesos, la médula ósea y el hígado).
En los Estados Unidos, los niños hispanos y aquellos que viven en condiciones socioeconómicas precarias presentan una enfermedad más avanzada.[
Evaluación diagnóstica y estadificación
La evaluación diagnóstica del retinoblastoma incluye los siguientes procedimientos:
- Examen ocular. El diagnóstico del retinoblastoma intraocular por lo general se establece sin confirmación patológica. Para inspeccionar la retina completa, es necesario una exploración con anestesia, una pupila dilatada al máximo e indentación escleral. Se debe documentar en detalle el número, la ubicación y el tamaño de los tumores; la presencia de desprendimiento de retina y de líquido subretiniano; y la presencia de diseminación subretiniana y vítrea.
- Ecografía ocular e imágenes por resonancia magnética (IRM). La ecografía ocular bidimensional y las IRM son útiles para diferenciar el retinoblastoma de otras causas de leucocoria y para evaluar la diseminación fuera de la esclerótica y del ojo en niños con retinoblastoma intraocular avanzado. El realce del nervio óptico en la IRM no indica necesariamente que hay compromiso, de manera que estos hallazgos se deben interpretar con cautela.[
7 ]
Quizás sea necesario evaluar la presencia de enfermedad metastásica en los pacientes en quienes se sospecha diseminación extraocular, por los hallazgos de las imágenes o un análisis patológico de riesgo alto en el ojo enucleado (es decir, invasión masiva de la coroides, compromiso de la esclerótica o del nervio óptico más allá de la lámina cribosa). Los pacientes que presentan estas características patológicas en el ojo enucleado tienen un riesgo alto de metástasis. En estos casos, a veces se hacen los siguientes procedimientos:[
- Centellografía ósea.
- Aspiración de médula ósea y biopsia.
- Punción lumbar.
Características genéticas y genómicas del retinoblastoma
El retinoblastoma es un tumor que se presenta en forma hereditaria (25–30 %) y no hereditaria (70–75 %).
Retinoblastoma hereditario
El retinoblastoma hereditario se define por la presencia de una variante patogénica germinal del gen RB1. Esta variante patogénica germinal se hereda de un progenitor afectado (25 % de los casos), sucede en una célula germinal antes de la concepción o en el útero durante la embriogénesis temprana en pacientes con enfermedad esporádica (75 % de los casos). La presencia de antecedentes familiares de retinoblastoma, o enfermedad bilateral o multifocal puede indicar enfermedad hereditaria.
El retinoblastoma hereditario se manifiesta como enfermedad unilateral o bilateral. Es probable que la penetrancia de la variante de RB1 (lateralidad, edad en el momento del diagnóstico y número de tumores) dependa de modificadores genéticos simultáneos, como los polimorfismos de MDM2 y MDM4.[
En niños con retinoblastoma hereditario, el diagnóstico tiende a hacerse a una edad más temprana que en los niños con la forma no hereditaria de la enfermedad.[
Retinoblastoma no hereditario
El panorama actual de las características genómicas del retinoblastoma se orienta hacia las alteraciones en RB1 que producen inactivación bialélica.[
Los cambios recurrentes en otros genes distintos de RB1 son poco frecuentes en el retinoblastoma, pero se producen. Las variantes o deleciones de BCOR y la amplificación de MYCN son los eventos que se notifican con más frecuencia.[
Asesoramiento genético
El asesoramiento genético es un componente fundamental de la atención de los pacientes con retinoblastoma y sus familias, con independencia del cuadro clínico inicial. El asesoramiento genético incluye una conversación sobre las principales formas de retinoblastoma, lo que ayuda a los padres a comprender las consecuencias genéticas de cada forma de retinoblastoma y permite calcular el riesgo de la enfermedad en la familia.[
Sin embargo, el asesoramiento genético no siempre es sencillo. Alrededor del 10 % de los niños con retinoblastoma exhiben mosaicismo genético somático, lo que dificulta este asesoramiento.[
Pruebas genéticas
Para determinar si un paciente con retinoblastoma tiene una variante germinal o somática del gen RB1, se examinan muestras de sangre o del tumor. Una vez que se identifica la variante genética del paciente, se procede a examinar a otros miembros de la familia para detectar directamente la variante mediante secuenciación dirigida.
Es posible realizar una evaluación genética completa del gen RB1 mediante un análisis de pasos múltiples, como los siguientes:[
- Secuenciación de ADN para identificar variantes de los exones codificantes, en las regiones intrónicas circundantes y en las regiones promotoras.
- Análisis de duplicaciones o deleciones.
- Análisis de metilación de la región promotora de RB1 en ADN aislado del tumor.
En casos de mosaicismo somático o anomalías citogenéticas, es posible que no sea fácil detectar las variantes. Se deben utilizar técnicas más minuciosas, como el cariotipado, la hibridación fluorescente in situ y el análisis de metilación del promotor de RB1. A veces se descubre un mosaicismo de grado bajo mediante la secuenciación masiva (2500 veces) de un amplicón genómico de RB1 obtenido del ADN linfocítico.[
La ausencia de variantes somáticas de RB1 detectables en cerca del 3 % de los casos de retinoblastoma unilateral no hereditario indica que quizás hay mecanismos subyacentes alternos que explican la formación de un retinoblastoma.[
Vigilancia posterior al diagnóstico
Es posible que los niños con una variante patogénica germinal de RB1 continúen presentando tumores nuevos durante unos pocos años después del diagnóstico y tratamiento. Por esta razón, es necesario examinar con frecuencia a estos pacientes. Es una práctica habitual que se examinen cada 2 a 4 meses durante, por lo menos, 28 meses.[
Una proporción de los niños que presentan retinoblastoma unilateral, con el tiempo, presentarán la enfermedad en el ojo opuesto. Por este motivo, se realizan exámenes periódicos del ojo no afectado hasta que se determine el estado en la línea germinal del gen RB1.
Debido al pronóstico precario de los pacientes con retinoblastoma trilateral, es una práctica habitual realizar exámenes de detección con neuroimágenes hasta los 5 años de edad durante la vigilancia de los niños con formas hereditarias de la enfermedad. Para obtener más información, consultar la sección
Causas de la mortalidad relacionada con el retinoblastoma
Si bien el retinoblastoma es una enfermedad muy curable, el reto es conservar la vida y prevenir la pérdida ocular, la ceguera y otros efectos graves del tratamiento que reducen la longevidad o la calidad de vida del paciente. Con las mejoras en el diagnóstico y el tratamiento del retinoblastoma en las últimas décadas, el retinoblastoma metastásico se observa con menor frecuencia en los Estados Unidos y otras naciones desarrolladas. Por este motivo, otras causas de mortalidad relacionada con el retinoblastoma, como el retinoblastoma trilateral y las neoplasias subsiguientes (NS), han cobrado mayor importancia durante los primeros 10 años de vida y los años posteriores. En los Estados Unidos, antes del advenimiento de la quimiorreducción para tratar la enfermedad hereditaria o bilateral, y antes de la implementación de los exámenes de detección con neuroimágenes, el retinoblastoma trilateral era el factor responsable de más del 50 % de la mortalidad relacionada con el retinoblastoma en los primeros 10 años posteriores al diagnóstico.[
Retinoblastoma trilateral
El retinoblastoma trilateral es un síndrome bien reconocido que se presenta en un 5 % a un 15 % de los pacientes con la forma hereditaria del retinoblastoma. Se define por la formación de un tumor neuroblástico en la línea media intracraneal asíncrono que, por lo general, se presenta entre los 20 y 36 meses de vida.[
Puesto que el retinoblastoma trilateral tiene un pronóstico adverso, y que parece que la detección temprana y el tratamiento radical mejoran la supervivencia, los exámenes de detección periódicos con neuroimágenes podrían asegurar la detección de la mayoría de casos dentro de los 2 primeros años del diagnóstico inicial de retinoblastoma.[
Aunque no está claro si el diagnóstico temprano logra mejorar la supervivencia, se ha recomendado la detección con IRM a intervalos de hasta 6 meses durante 5 años en los pacientes en quienes se sospecha una enfermedad hereditaria o que tienen enfermedad unilateral con antecedentes familiares confirmados.[
Una glándula pineal quística, que por lo común se detecta mediante una IRM de vigilancia, se debe diferenciar de una variante quística de pineoblastoma. Se indicó que la incidencia de quistes pineales en niños sin retinoblastoma es del 55,8 %.[
Referencias:
- Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649.
Also available online . Last accessed December 22, 2023. - National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute.
Available online . Last accessed February 25, 2025. - Skalet AH, Gombos DS, Gallie BL, et al.: Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology 125 (3): 453-458, 2018.
- Kamihara J, Bourdeaut F, Foulkes WD, et al.: Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clin Cancer Res 23 (13): e98-e106, 2017.
- Abramson DH: Re: Skalet et al.: Screening children at risk for retinoblastoma: consensus report from the American Association of Ophthalmic Oncologists and Pathologists (Ophthalmology. 2018;125:453-458). Ophthalmology 125 (9): e63-e64, 2018.
- Truong B, Green AL, Friedrich P, et al.: Ethnic, Racial, and Socioeconomic Disparities in Retinoblastoma. JAMA Pediatr 169 (12): 1096-104, 2015.
- Khurana A, Eisenhut CA, Wan W, et al.: Comparison of the diagnostic value of MR imaging and ophthalmoscopy for the staging of retinoblastoma. Eur Radiol 23 (5): 1271-80, 2013.
- Kaliki S, Shields CL, Rojanaporn D, et al.: High-risk retinoblastoma based on international classification of retinoblastoma: analysis of 519 enucleated eyes. Ophthalmology 120 (5): 997-1003, 2013.
- Castéra L, Sabbagh A, Dehainault C, et al.: MDM2 as a modifier gene in retinoblastoma. J Natl Cancer Inst 102 (23): 1805-8, 2010.
- de Oliveira Reis AH, de Carvalho IN, de Sousa Damasceno PB, et al.: Influence of MDM2 and MDM4 on development and survival in hereditary retinoblastoma. Pediatr Blood Cancer 59 (1): 39-43, 2012.
- Shields CL, Dockery P, Ruben M, et al.: Likelihood of Germline Mutation With Solitary Unilateral Retinoblastoma Based on Patient Age at Presentation: Analysis of 482 Consecutive Patients. J Pediatr Ophthalmol Strabismus 58 (6): 355-364, 2021 Nov-Dec.
- Andreoli MT, Chau FY, Shapiro MJ, et al.: Epidemiological trends in 1452 cases of retinoblastoma from the Surveillance, Epidemiology, and End Results (SEER) registry. Can J Ophthalmol 52 (6): 592-598, 2017.
- Zhang J, Benavente CA, McEvoy J, et al.: A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481 (7381): 329-34, 2012.
- Rushlow DE, Mol BM, Kennett JY, et al.: Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol 14 (4): 327-34, 2013.
- McEvoy J, Nagahawatte P, Finkelstein D, et al.: RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget 5 (2): 438-50, 2014.
- Afshar AR, Pekmezci M, Bloomer MM, et al.: Next-Generation Sequencing of Retinoblastoma Identifies Pathogenic Alterations beyond RB1 Inactivation That Correlate with Aggressive Histopathologic Features. Ophthalmology 127 (6): 804-813, 2020.
- Kooi IE, Mol BM, Massink MP, et al.: Somatic genomic alterations in retinoblastoma beyond RB1 are rare and limited to copy number changes. Sci Rep 6: 25264, 2016.
- Francis JH, Richards AL, Mandelker DL, et al.: Molecular Changes in Retinoblastoma beyond RB1: Findings from Next-Generation Sequencing. Cancers (Basel) 13 (1): , 2021.
- Ewens KG, Bhatti TR, Moran KA, et al.: Phosphorylation of pRb: mechanism for RB pathway inactivation in MYCN-amplified retinoblastoma. Cancer Med 6 (3): 619-630, 2017.
- Richter S, Vandezande K, Chen N, et al.: Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet 72 (2): 253-69, 2003.
- Dommering CJ, Mol BM, Moll AC, et al.: RB1 mutation spectrum in a comprehensive nationwide cohort of retinoblastoma patients. J Med Genet 51 (6): 366-74, 2014.
- Reddy MA, Butt M, Hinds AM, et al.: Prognostic Information for Known Genetic Carriers of RB1 Pathogenic Variants (Germline and Mosaic). Ophthalmol Retina 5 (4): 381-387, 2021.
- Eloy P, Dehainault C, Sefta M, et al.: A Parent-of-Origin Effect Impacts the Phenotype in Low Penetrance Retinoblastoma Families Segregating the c.1981C>T/p.Arg661Trp Mutation of RB1. PLoS Genet 12 (2): e1005888, 2016.
- Clark R: Retinoblastoma: genetic testing and counseling. In: Singh A, Damato B: Clinical Ophthalmic Oncology. Saunders Elsevier, 2007, pp 441-6.
- Amitrano S, Marozza A, Somma S, et al.: Next generation sequencing in sporadic retinoblastoma patients reveals somatic mosaicism. Eur J Hum Genet 23 (11): 1523-30, 2015.
- Sagi M, Frenkel A, Eilat A, et al.: Genetic screening in patients with Retinoblastoma in Israel. Fam Cancer 14 (3): 471-80, 2015.
- Chen Z, Moran K, Richards-Yutz J, et al.: Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum Mutat 35 (3): 384-91, 2014.
- Nichols KE, Houseknecht MD, Godmilow L, et al.: Sensitive multistep clinical molecular screening of 180 unrelated individuals with retinoblastoma detects 36 novel mutations in the RB1 gene. Hum Mutat 25 (6): 566-74, 2005.
- Abramson DH, Mendelsohn ME, Servodidio CA, et al.: Familial retinoblastoma: where and when? Acta Ophthalmol Scand 76 (3): 334-8, 1998.
- Broaddus E, Topham A, Singh AD: Survival with retinoblastoma in the USA: 1975-2004. Br J Ophthalmol 93 (1): 24-7, 2009.
- de Jong MC, Kors WA, de Graaf P, et al.: Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol 15 (10): 1157-67, 2014.
- Rodjan F, de Graaf P, Brisse HJ, et al.: Trilateral retinoblastoma: neuroimaging characteristics and value of routine brain screening on admission. J Neurooncol 109 (3): 535-44, 2012.
- Kivelä T: Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol 17 (6): 1829-37, 1999.
- Sirin S, de Jong MC, Galluzzi P, et al.: MRI-based assessment of the pineal gland in a large population of children aged 0-5 years and comparison with pineoblastoma: part II, the cystic gland. Neuroradiology 58 (7): 713-21, 2016.
- Pham TT, Siebert E, Asbach P, et al.: Magnetic resonance imaging based morphologic evaluation of the pineal gland for suspected pineoblastoma in retinoblastoma patients and age-matched controls. J Neurol Sci 359 (1-2): 185-92, 2015.
Esta información no reemplaza el consejo de un médico. Ignite Healthwise, LLC, niega toda garantía y responsabilidad por el uso de esta información. El uso que usted haga de esta información implica que usted acepta los
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
Page Footer
Quiero...
Audiencia
Sitios seguros para miembros
Información sobre The Cigna Group
Aviso legal
Los planes individuales y familiares de seguro médico y dental están asegurados por Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc. y Cigna HealthCare of Texas, Inc. Los planes de beneficios de salud y de seguro de salud de grupo están asegurados o administrados por CHLIC, Connecticut General Life Insurance Company (CGLIC) o sus afiliadas (puedes ver
Todas las pólizas de seguros y los planes de beneficios de grupo contienen exclusiones y limitaciones. Para conocer la disponibilidad, los costos y detalles completos de la cobertura, comunícate con un agente autorizado o con un representante de ventas de Cigna. Este sitio web no está dirigido a los residentes de New Mexico.