Tratamiento de tumores de diferenciación incierta
Hay muchos subtipos de tumores de diferenciación incierta, entre ellos los siguientes:
- Mixoma, sin otra indicación (benigno).
- Sarcoma sinovial, sin otra indicación (poco diferenciado, de células fusiformes y variedades bifásicas).
- Sarcoma epitelioide.
- Sarcoma alveolar de partes blandas.
- Sarcoma de células claras, sin otra indicación.
- Condrosarcoma mixoide extraesquelético.
- Tumor desmoplásico de células redondas pequeñas.
- Tumor rabdoide, sin otra indicación (extrarrenal).
- Tumor epitelioide perivascular (PEComa), maligno.
- Sarcoma indiferenciado.
- Sarcoma pleomórfico indiferenciado.
- Sarcoma de células redondas indiferenciado.
Mixoma, sin otra indicación
Complejo de Carney
El complejo de Carney es un síndrome autosómico dominante causado por variantes del gen PRKAR1A, localizado en el cromosoma 17.[1] El síndrome se caracteriza por mixomas cardíacos y cutáneos, lentigos de color marrón pálido a marrón, nevos azules, enfermedad adrenocortical nodular pigmentada primaria que causa el síndrome de Cushing y una variedad de tumores endocrinos y no endocrinos, incluso adenomas de hipófisis, tumores tiroideos y tumor testicular de células de Sertoli calcificante de células grandes.[1,2,3] Se publicaron directrices de vigilancia para pacientes con complejo de Carney que incluyen ecografías cardíacas, testiculares y tiroideas.
Para los pacientes con complejo de Carney, el pronóstico depende de la frecuencia de las recidivas de los mixomas cardíacos y cutáneos, y otros tumores.
Para obtener más información sobre los tratamientos de las afecciones relacionadas con el complejo de Carney, consultar los siguientes resúmenes:
- Tratamiento de los tumores cardíacos infantiles.
- Tratamiento del cáncer de testículo infantil.
- Tratamiento del cáncer de tiroides infantil.
Sarcoma sinovial, sin otra indicación (poco diferenciado, de células fusiformes y variedades bifásicas)
El sarcoma sinovial representa el 9 % de los sarcomas de tejido blando en pacientes menores de 20 años (consultar el Cuadro 1).
El sarcoma sinovial es uno de los sarcomas de tejido blando no rabdomiosarcomatosos más frecuentes en niños y adolescentes. En una revisión de la base de datos del Surveillance, Epidemiology, and End Results (SEER) Program de 1973 a 2005, se identificaron 1268 pacientes con sarcoma sinovial. Alrededor del 17 % de estos pacientes eran niños y adolescentes, y la mediana de edad en el momento del diagnóstico fue de 34 años.[4] Además, en el protocolo ARST0332 (NCT00346164) del Children's Oncology Group (COG) y el protocolo European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) 2005, el sarcoma sinovial fue el subtipo histológico más común.[5]
Cuadro clínico inicial
Las localizaciones más frecuentes son las extremidades, seguidas del tronco, la cabeza y el cuello.[4] Rara vez, el sarcoma sinovial surge en el corazón o el pericardio y presenta manifestaciones pleuropulmonares.[6,7,8,9]
El sitio más común de metástasis es el pulmón.[10,11] El riesgo de metástasis está más condicionado por el tamaño del tumor. Los pacientes con tumores de más de 5 cm tienen un riesgo estimado 32 veces más alto de presentar metástasis, en comparación con otros pacientes.
El Cooperative Weichteilsarkom Studiengruppe (CWS) notificó 432 pacientes menores de 21 años con diagnóstico de sarcoma sinovial entre 1981 y 2018.[12]En el estudio, se compararon 3 grupos de edad de pacientes: niños (0–12 años; n = 176), adolescentes (13–16 años; n = 178) y adultos jóvenes (17–21 años; n = 78).
- La proporción de tumores invasivos fue significativamente más alta en los pacientes de más edad (niños, 33 %; adolescentes, 39 % y adultos jóvenes, 54 %; P = 0,009).
- La proporción de tumores de más de 10 cm (niños; 13 %; adolescentes, 21 %; adultos jóvenes, 31 %; P = 0,006) y la presencia de metástasis en el diagnóstico inicial también fueron más elevadas en los pacientes de más edad (niños, 6 %; adolescentes, 10 %; adultos jóvenes, 21 %; P = 0,001).
Características histológicas, evaluación diagnóstica y alteraciones genómicas
El sarcoma sinovial se subclasifica en los siguientes tipos:
- Sarcoma sinovial de células fusiformes.
- Sarcoma sinovial bifásico.
- Sarcoma sinovial poco diferenciado.
El diagnóstico del sarcoma sinovial se hace mediante análisis inmunohistoquímico, hallazgos ultraestructurales y comprobación de una translocación cromosómica específica: t(x;18)(p11.2;q11.2). Esta anomalía es específica del sarcoma sinovial y se encuentra en todos los subtipos morfológicos. El sarcoma sinovial se produce como consecuencia de un reordenamiento del gen SS18 en el cromosoma 18 con uno de los subtipos (1, 2 o 4) del gen SSX en el cromosoma X.[13,14] Se cree que el transcrito de fusión SS18::SSX promueve el silenciamiento epigenético de los principales genes supresores de tumores.[15]
En un informe, se observó una reducción de la reactividad nuclear de SMARCB1 en la tinción inmunohistoquímica en 49 casos de sarcoma sinovial, lo que indica que este patrón puede ayudar a distinguir el sarcoma sinovial de otras tipos histológicos.[16]
Factores pronósticos
Factores pronósticos favorables
Los pacientes menores de 10 años tienen desenlaces y características clínicas más favorables que los pacientes de más edad.
Las características clínicas favorables son las siguientes:[4,17,18,19]
- Tumores primarios en las extremidades.
- Tumores más pequeños.
- Enfermedad localizada
- La respuesta a la quimioterapia se correlacionó con una mejora de la supervivencia en un metanálisis.
Factores pronósticos desfavorables
En los siguientes estudios se notificaron múltiples factores relacionados con desenlaces precarios:
- En un análisis retrospectivo de sarcoma sinovial en niños y adolescentes tratados en Alemania e Italia, el tamaño del tumor (>5 cm o ≤5 cm en su mayor dimensión) fue un factor pronóstico importante de la supervivencia sin complicaciones [SSC]).[20] En este análisis, el grado de invasión local confirió una menor probabilidad de SSC, pero el desenlace clínico no se relacionó con los márgenes quirúrgicos.
- En un análisis retrospectivo de una sola institución de 111 pacientes con sarcoma sinovial menores de 22 años en el momento del diagnóstico, se encontró que un tamaño tumoral más grande, una mayor profundidad en el tejido, un mayor grado de invasión local y una ubicación más proximal del tumor se relacionaban con una supervivencia general (SG) más precaria.[21][Nivel de evidencia C1]
- En un análisis multicéntrico de 219 niños de diversos centros de tratamiento, incluso de Alemania, el St. Jude Children's Research Hospital (SJCRH), el Instituto Tumori y el MD Anderson Cancer Center, se notificó una tasa estimada de SG a 5 años del 80 % y una tasa de SSC del 72 %.[19] En este análisis, se observó una interacción entre el tamaño del tumor y la capacidad invasiva. En análisis multivariantes, los pacientes con tumores grandes o invasivos, o con enfermedad del del grupo III (localizada, con resección incompleta o de la que solo se obtuvo una biopsia) y grupo IV (metástasis en el momento del diagnóstico) del Intergroup Rhabdomyosarcoma Study presentaron una SG más precaria. El tratamiento con radioterapia se relacionó con mejora de la SG (cocientes de riesgos instantáneos (CRI), 0,4; intervalo de confianza (IC) 95 %, 0,2–0,7). En los pacientes del grupo III del IRS, la respuesta objetiva a la quimioterapia (18 de 30 [60 %]) se correlacionó con una mejora de la supervivencia.
- Se ha estudiado el uso de firmas pronósticas de expresión e índice genómico del sarcoma sinovial. Los perfiles genómicos complejos, con mayor reordenamiento del genoma, son más comunes en adultos que en pacientes más jóvenes con sarcoma sinovial y se relacionan con un riesgo más alto de metástasis.[22]
- En una revisión de 84 pacientes con sarcoma sinovial localizado, donde se contaba con información sobre el estado de la fusión (SS18::SSX) y la clasificación histológica, no se encontró ninguna diferencia en la SG. Sin embargo, en cuanto al tamaño del tumor en el momento del diagnóstico, se halló que los pacientes con tumores de entre 5 cm y 10 cm presentaron un pronóstico más precario que aquellos con tumores más pequeños (P = 0,02). Los pacientes con tumores de más de 10 cm tuvieron una SG aún más precaria (P = 0,0003).[23][Nivel de evidencia C1]
- El grupo alemán CWS analizó a 27 pacientes evaluables menores de 21 años con metástasis pulmonares entre 296 pacientes con sarcoma sinovial. Todos los pacientes tenían metástasis en los pulmones. La tasa de SSC a 5 años fue del 26 % y la tasa de SG fue del 30 %. El factor pronóstico más importante en el momento de la presentación fue que las metástasis se limitaran a una lesión en un solo pulmón o una lesión en ambos pulmones (grupo que llamaron oligometastásico). Los elementos del tratamiento vinculados con una supervivencia superior fueron la terapia local adecuada del tumor primario y, cuando fue posible, de las metástasis. El uso de irradiación dirigida a todo el pulmón no se correlacionó con mejores desenlaces.[24][Nivel de evidencia C1]
- El EpSSG diseñó un índice genómico para el sarcoma sinovial.[25][Nivel de evidencia C2] El índice genómico se definió como A2 /C, donde A es el número total de alteraciones (ganancias y pérdidas segmentarias) y C es el número de cromosomas afectados en los resultados de la matriz de hibridación genómica comparativa. En un análisis multivariante de 61 pacientes pediátricos, adolescentes y adultos jóvenes (edad <25 años), el índice genómico alto fue un factor de predicción independiente de SSC y SG inferiores.
- En adultos, el pronóstico más precario se relacionó con factores como el estadio III y estadio IVA del International Union Against Cancer/American Joint Committee on Cancer, una escasa necrosis tumoral, la localización en el tronco, un índice mitótico alto, mayor edad y un grado histológico más elevado.[26,27,28]
Tratamiento del sarcoma sinovial
Las opciones de tratamiento del sarcoma sinovial son las siguientes:
- Cirugía sola.
- Cirugía y quimioterapia, con radioterapia o sin esta.[29,30]
Cirugía sola
Evidencia (cirugía sola):
- El COG y el EpSSG informaron sobre un análisis combinado de 60 pacientes menores de 21 años con sarcoma sinovial localizado que se asignaron de manera prospectiva a cirugía sin radioterapia ni quimioterapia adyuvante.[31] La inscripción se limitó a los pacientes con resección completa inicial con márgenes limpios en el análisis histológico y un tumor de grado 2 de cualquier tamaño o un tumor de grado 3 de 5 cm o menos.
- La tasa de SSC a 3 años fue del 90 % (mediana de seguimiento, 5,2 años; intervalo, 1,9–9,1).
- Se presentaron 8 casos de recidiva tumoral; no se observó ningún caso de recidiva metastásica.
- Todos los pacientes con enfermedad recidivante se trataron de manera eficaz con terapia de segunda línea, que produjo una tasa de SG del 100 %.
- Por lo tanto, los autores concluyeron que un abordaje solo quirúrgico fue óptimo para los pacientes que lograron una resección R0 (resección completa con márgenes microscópicos negativos) y tenían tumores de menos de 5 cm, con independencia del grado.
Cirugía y quimioterapia, con radioterapia o sin esta
El sarcoma sinovial es más sensible a la quimioterapia que muchos otros tipos de sarcomas de tejido blando. Los niños con sarcoma sinovial tienen mejor pronóstico que los adultos con este tipo sarcoma.[11,28,32,33,34,35,36,37]
Los regímenes más utilizados para el tratamiento del sarcoma sinovial incorporan ifosfamida y doxorrubicina.[19,35,38] Las tasas de respuesta al régimen de ifosfamida y doxorrubicina son más altas que en otros sarcomas de tejido blando no rabdomiosarcomatoso.[39]
Evidencia (cirugía y quimioterapia con radioterapia o sin esta):
- Varios centros de tratamiento promueven la quimioterapia después de la resección y la radioterapia para niños y adultos jóvenes con sarcoma sinovial.[19,20,40,41,42]
- En los estudios de la International Society of Pediatric Oncology-Malignant Mesenchymal Tumors, se observó que determinados pacientes con sarcoma sinovial no metastásico (edad temprana, tumores resecados de <5 cm) a veces presentan desenlaces excelentes sin radioterapia. Sin embargo, aún no está claro si ese abordaje elimina la ventaja de la radiación para el control local o regional.[41]
- En un ensayo alemán, se indicó un beneficio de la quimioterapia posoperatoria en niños con sarcoma sinovial.[42]
- En un metanálisis también se indicó que la quimioterapia puede proporcionar beneficios.[19]
- El COG notificó un análisis del subgrupo de pacientes con sarcoma sinovial tratados en el ensayo ARST0332 (NCT00346164). Este fue un ensayo prospectivo de asignación de tratamiento para pacientes menores de 30 años con sarcomas de tejido blando no rabdomiosarcomatoso.[43] Se analizaron los desenlaces de 138 pacientes aptos.
- En general, se logró resección R0 o resección R1 (márgenes positivos al microscopio) del tumor primario en 129 pacientes (93,5 %): 69 pacientes (53,5 %) al comienzo del estudio y 60 pacientes (46,5 %) después de la quimioterapia neoadyuvante. De estos pacientes, 104 (80,6 %) presentaron una resección R0: 55 pacientes (53 %) al comienzo del estudio y 49 pacientes (47 %) después de la quimioterapia neoadyuvante.
- La respuesta fue evaluable en 55 pacientes de los 60 que recibieron quimioterapia neoadyuvante. De ellos, 2 pacientes (3,6 %) presentaron respuesta completa, 9 pacientes (16,4 %) presentaron respuesta parcial, 41 pacientes (74,6 %) presentaron enfermedad estable y 3 pacientes (5,5 %) presentaron enfermedad progresiva. El tejido de 57 tumores fue sometido a revisión central después de la resección definitiva. Se encontró que 41 tumores (72 %) exhibían menos del 90 % de necrosis, y 16 tumores (28 %) exhibían el 90 % o más de necrosis.
- En los 46 pacientes del grupo de riesgo bajo, la tasa de SSC a 5 años fue del 81,9 % (IC 95 %, 69–94,8 %) y la tasa de SG fue del 97,7 % (IC 95 %, 92,7–100 %).
- En los 23 pacientes del grupo de riesgo intermedio (grupo de tratamiento C), la tasa de SSC a 5 años fue del 64 % (IC 95 %, 42,4–85,8%) y la tasa de SG fue del 89,5 % (IC 95 %, 75,3–100 %).
- En los 49 pacientes del grupo de riesgo intermedio (grupo de tratamiento D), la tasa de SSC a 5 años fue del 71,2 % (IC 95 %, 56,5–85,9 %) y la tasa de SG fue del 86,5 % (IC 95 %, 75,6–97,3 %).
- En los 21 pacientes del grupo de riesgo alto, la tasa de SSC a 5 años fue del 7,6 % (IC 95 %, 0–22%) y la tasa de SG fue del 12,5 % (IC 95 %, 0–28,7 %).
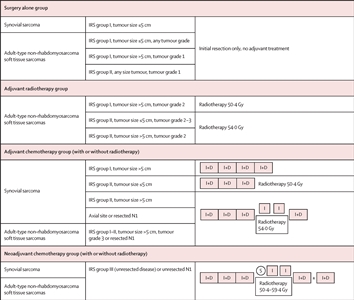
- El EpSSG realizó un estudio prospectivo de pacientes menores de 21 años con sarcoma sinovial (CCLG-EPSSG-NRSTS-2005 [NCT00334854]).[44][Nivel de evidencia C1] Los pacientes se estratificaron en los siguientes 3 grupos de riesgo y se asignaron de forma no aleatoria al tratamiento según el grupo de riesgo:
- Los pacientes de riesgo bajo tenían tumores del grupo I del IRS que medían menos de 5 cm y tumores primarios no axiales.
- Los pacientes de riesgo intermedio no tenían tumores primarios axiales; tenían tumores del grupo I del IRS que medían más de 5 cm o tumores del grupo II del IRS.
- Los pacientes de riesgo alto fueron todos los pacientes con tumores en sitios axiales primarios (cabeza y cuello, pulmones y pleura, tronco, retroperitoneo), tumores del grupo III del IRS o tumores N1.
Los desenlaces de los pacientes tratados en el ensayo CCLG-EPSSG-NRSTS-2005 se describen en el Cuadro 11.
Cuadro 11. Supervivencia sin complicaciones y supervivencia general de pacientes con sarcoma sinovial de riesgo bajo, intermedio y alto tratados en el ensayo CCLG-EPSSG-NRSTS-2005
| Grupo de riesgo |
Tratamiento |
SSC a 3 años (%) |
SG a 3 años (%) |
| IRS = Intergroup Rhabdomyosarcoma Study; RT = radioterapia. |
| a Quimioterapia de ifosfamida y doxorrubicina, sin doxorrubicina durante la radioterapia. |
| b 59,4 Gy en casos sin opción de una resección secundaria; 50,4 Gy como radioterapia preoperatoria; 50,4 Gy, 54 Gy y 59,4 Gy como radioterapia posoperatoria, en el caso de resecciones R0, R1 y R2, respectivamente (sin radioterapia adicional en el caso de resecciones completas secundarias con márgenes limpios en niños menores de 6 años). |
| Bajo |
Cirugía sola |
92 |
100 |
| Intermedio |
Cirugía, 3–6 ciclos de quimioterapiaa, ± RTb |
91 |
100 |
| Alto (grupo III del IRS) |
3 ciclos de quimioterapiaa, cirugía, 3 ciclos adicionales de quimioterapia, ± RTb |
77 |
94 |
| Alto (sitios primarios axiales) |
Cirugía, 6 ciclos de quimioterapiaa, RTb |
78 |
100 |
- El CWS notificó los resultados de un ensayo prospectivo para el tratamiento de pacientes con sarcoma sinovial. La selección de pacientes se restringió a aquellos con tumores localizados con enfermedad residual macroscópica después de la primera cirugía, antes del inicio de la terapia sistémica (IRS III) y sin enfermedad metastásica detectable desde el punto de vista clínico. En el estudio participaron 145 pacientes con una mediana de edad de 14,5 años (intervalo, 0,2–33,2 años). En los protocolos se recomendó, pero no fue un requisito, que se administrara radioterapia antes de la resección definitiva del tumor. Se administró radioterapia a 115 pacientes (79 %) y 23 de ellos no recibieron radioterapia (no se documentó información sobre 7 pacientes). De los 115 pacientes irradiados, 57 se irradiaron antes de la escisión del tumor y 52 después de la escisión del tumor.[45]
- En esta comparación no aleatorizada, la secuenciación de la radioterapia antes de la resección definitiva se relacionó con una mejora estadísticamente significativa en las tasas de supervivencia sin recidiva local, en comparación con la cirugía definitiva antes de la radioterapia.
- La omisión de la radioterapia se relacionó con un desenlace inferior.
- Los desenlaces de los pacientes se describen en el Cuadro 12.
Cuadro 12. Efectos de la programación de la radioterapia en los desenlaces de pacientes con sarcoma sinovial
| Radioterapia |
Número de pacientes |
Tasa de SSC a 5 años |
Tasa de SG a 5 años |
Tasa de supervivencia sin recidiva local a 5 años |
| SSC = supervivencia sin complicaciones; SG = supervivencia general. |
| Sin radioterapia |
23 |
44 % |
57 % |
76 % |
| Radioterapia antes de la cirugía |
57 |
70 % |
83 % |
98 % |
| Radioterapia después de la cirugía. |
52 |
73 % |
82 % |
86 % |
Sarcoma sinovial recidivante, sin otra indicación
Para los pacientes con sarcoma sinovial recidivante, la tasa de supervivencia después de la recaída es precaria (30–40 % a los 5 años). Los factores relacionados con el desenlace después de la recaída incluyen la duración de la primera remisión (> o ≤ 18 meses) y la ausencia de una segunda remisión.[46,47]
En un estudio alemán, la resección quirúrgica de la enfermedad metastásica fue el método más frecuente para lograr una remisión completa.[47] La quimioterapia de mantenimiento por vía oral con trofosfamida, idarrubicina y etopósido o con ciclofosfamida oral y vinblastina intravenosa se administró de forma individual.
Un consorcio de 6 centros de referencia europeos informó sobre una revisión retrospectiva de pacientes menores de 21 años con sarcoma sinovial recidivante. En 41 pacientes, las primeras recidivas se produjeron entre 3 y 132 meses (mediana, 18 meses) después del primer diagnóstico. Las recaídas fueron locales en el 34 % de los pacientes, metastásicas en el 54 %, y locales y metastásicas en el 12 %. Los tratamientos en la primera recaída incluyeron cirugía en el 56 % de los pacientes, radioterapia en el 34 % y terapia sistémica en el 88 %. En total, 36 pacientes recibieron tratamiento médico de segunda línea, que incluyó quimioterapia en 32 pacientes (con 10 regímenes diferentes) y terapia dirigida en 4 pacientes. No se incluyó ningún paciente en ensayos clínicos de fase inicial como terapia de segunda línea. La tasa de respuesta general fue del 42 %. La mediana de SSC fue de 12 meses, y la tasa de SSC a 5 años tras la recaída fue del 15,8 %. La mediana de SG fue de 30 meses, y la tasa de SG a 5 años tras la recaída fue del 22,2 %. En un análisis de regresión multivariantee de Cox, la SG se relacionó de forma significativa con el tiempo y el tipo de recaída.[48]
La radioterapia (radioterapia corporal estereotáctica) se puede utilizar para tratar metástasis pulmonares específicas. Por lo general, se considera su uso después de, por lo menos, una resección para confirmar la presencia de enfermedad metastásica. En particular, la radioterapia es apropiada para pacientes con lesiones que comprometan el intercambio gaseoso por su localización adyacente a los bronquios o que causen dolor al invadir la pared torácica.[49]
Del 70 % al 80 % de los sarcomas sinoviales expresan NY-ESO-1, un antígeno testicular inmunógeno de cáncer.[50] Es posible usar transferencia adoptiva de células T modificadas para que expresen NY-ESO-1c259, un receptor de célula T (TCR) con alta afinidad para NY-ESO-1/LAGE1a, dirigida a NY-ESO-1.[51] El procedimiento para fabricar las células T genomanipuladas restringe su reactividad a un solo tipo de HLA. Todos los ensayos clínicos de este tipo de tecnología han usado HLA-A*02 como diana inicial y han restringido la inscripción a los pacientes cuyos tumores expresaban NY-ESO-1 y que tenían HLA-A*02. En un ensayo multinstitucional, se confirmó la presencia de respuestas antitumorales en el 50 % de los pacientes (6 de 12) caracterizadas por achicamiento del tumor en el transcurso de varios meses. Se encontraron células NY-ESO-1c259T circulantes después de la infusión en todos los pacientes y las células perduraron por lo menos 6 meses en todos los pacientes que respondieron a la infusión.[52]
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
Sarcoma epitelioide
El sarcoma epitelioide es un tumor mesenquimatoso raro con histogénesis incierta que exhibe diferenciación multilinaje.[53]
Cuadro clínico inicial
El sarcoma epitelioide suele presentarse como un nódulo firme de crecimiento lento en el tejido blando profundo. El tipo proximal afecta de manera predominante a adultos y compromete el esqueleto axial y sitios proximales. El tumor es muy maligno y tiene una predisposición a metástasis ganglionares.
Alteraciones genómicas
El sarcoma epitelioide se caracteriza por la inactivación del gen SMARCB1, presente en los tipos convencional y proximal de este sarcoma.[54] Esta anomalía conduce a una mayor dependencia de EZH2 y a la formación de tumores.[55]
Tratamiento del sarcoma epitelioide
Las opciones de tratamiento del sarcoma epitelioide son las siguientes:
- Cirugía con quimioterapia o radioterapia, o sin estas.
- Terapia dirigida.
Cirugía con quimioterapia o radioterapia, o sin estas
El tratamiento más eficaz es la extirpación quirúrgica de los tumores primarios y recidivantes.[56][Nivel de evidencia C1] Debido a que esta enfermedad tiene predisposición a presentar metástasis ganglionar oculta, se recomienda la biopsia de ganglio linfático centinela para el sarcoma epitelioide de extremidades o glúteos en ausencia de comprobación clínica (por imágenes o por examen físico) de ganglios linfáticos agrandados.[57]
Evidencia (cirugía con quimioterapia o radioterapia o sin esta):
- En un análisis retrospectivo del grupo alemán CWS de 67 niños, adolescentes y adultos jóvenes (mediana de edad 14 años) con sarcoma epitelioide, 53 pacientes tenían enfermedad localizada y 14 pacientes tenían enfermedad metastásica.[58][Nivel de evidencia C1] De 67 pacientes, se trató a 58 con resecciones primarias. De acuerdo con el examen microscópico, las resecciones se consideraron completas en 35 pacientes e incompletas en 12 pacientes; en el examen macroscópico las resecciones fueron incompletas en 20 pacientes. Se trató con quimioterapia a 49 pacientes y con radioterapia a 33 pacientes.
- Se logró la remisión completa en 45 de 53 pacientes (85 %) con enfermedad localizada.
- Se observaron recidivas en 27 pacientes después de una mediana de tiempo de 0,9 años (intervalo, 0,1–2,3 años).
- Los pacientes con enfermedad localizada tuvieron una tasa de SSC a 5 años del 35 % (IC 95 %, ±12 %) y una tasa de SG del 48 % (IC 95 % ±14 %).
- Los pacientes con enfermedad metastásica tuvieron una tasa de SSC a 5 años del 7 % (IC 95 %, ±14 %) y una tasa de SG del 9 % (IC 95 %, ±16 %).
- Los factores que se correlacionaron con un pronóstico favorable en los pacientes con enfermedad localizada fueron un tamaño tumoral pequeño, un grupo del IRS más bajo, menor grado de invasión tumoral, un estado negativo para el compromiso ganglionar y una resección completa según el análisis microscópico.
- En un análisis retrospectivo se revisaron los ensayos clínicos prospectivos del COG y el EpSSG en los que participaron pacientes menores de 30 años con sarcoma epitelioide.[59][Nivel de evidencia B4] En el análisis se identificaron 63 pacientes tratados entre julio de 2005 y noviembre de 2015. Los pacientes se estratificaron en 3 grupos de riesgo mediante una combinación de características clínicas y tratamientos recibidos. Los pacientes de riesgo bajo (n = 34) se sometieron a cirugía con radioterapia o sin esta. Este grupo incluyó de modo predominante a pacientes con tumores no metastásicos con resección amplia o marginal que medían 5 cm o menos. El grupo de riesgo intermedio incluyó a pacientes (n = 16) con tumores no metastásicos de grado alto y tumores que medían más de 5 cm o irresecables. Los pacientes de riesgo alto (n = 13) tenían enfermedad ganglionar o metástasis a distancia, sin importar el grado o el tamaño del tumor.
- Se observaron respuestas parciales en 11 de 22 pacientes (50 %) que recibieron terapia neoadyuvante.
- Se presentaron casos de recidiva local (n = 10) y recidiva a distancia (n = 15).
- Las tasas estimadas de SG a 5 años fueron del 86,4 % en los pacientes de riesgo bajo, del 63,5 % en los pacientes de riesgo intermedio y del 0 % en los pacientes de riesgo alto.
- Los factores de predicción de una SSC más precaria en los pacientes sin metástasis fueron un compromiso ganglionar locorregional, un tumor invasor, un grado alto y un menor alcance de la resección.
- En una revisión de 30 pacientes pediátricos con sarcoma epitelioide (mediana de edad en el momento de la presentación, 12 años), se notificaron los siguientes resultados:[60]
- Se observaron respuestas a la quimioterapia en el 40 % de los pacientes que recibieron regímenes de tratamiento propios del sarcoma.
- El 60 % de los pacientes estaban vivos al cabo de 5 años del diagnóstico inicial.
- En una revisión retrospectiva de una sola institución de 20 pacientes, que incluyó niños y adultos (mediana de edad, 27,3 años), se notificó lo siguiente:[56]
- No hubo diferencia en la probabilidad de recidiva entre los pacientes que recibieron quimioterapia y los que no la recibieron.
- Los autores indicaron que la radioterapia quizás sea útil.
Terapia dirigida
Evidencia (tazemetostat):
- En un ensayo de fase II de 62 adultos con sarcoma epitelioide en quienes se documentó por análisis inmunohistioquímico la pérdida de INI1 o las alteraciones bialélicas en SMARCB1 (gen codificador de INI1), el tazemetostat demostró actividad clínica.[61]
- Se presentaron 9 de 62 respuestas parciales confirmadas, la tasa de respuesta objetiva fue del 15 % y la tasa de control de la enfermedad fue del 26 %.
En enero de 2020, la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) otorgó la aprobación acelerada al uso del tazemetostat en pacientes adultos y jóvenes de 16 años o más con sarcoma epitelioide metastásico, o localmente avanzado, que no cumplían los requisitos para una resección completa.
Opciones de tratamiento en evaluación clínica para el sarcoma epitelioide
La información en inglés sobre los ensayos clínicos patrocinados por el NCI se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de un ensayo clínico nacional o institucional en curso:
- PEPN2121 (NCT05286801) (Tiragolumab and Atezolizumab for the Treatment of Relapsed or Refractory SMARCB1- or SMARCA4-Deficient Tumors): en este estudio se evalúa la combinación de un anticuerpo dirigido a PD-L1 (atezolizumab) con un anticuerpo dirigido a TIGIT (tiragolumab) en pacientes que tienen tumores con deficiencia de SMARCB1 o SMARCA4. Es posible que los pacientes con sarcoma epitelioide sean aptos para este estudio.
Sarcoma alveolar de partes blandas
Los sarcomas alveolares de partes blandas representan el 1,4 % de los sarcomas de tejido blando en pacientes menores de 20 años (consultar el Cuadro 1
Cuadro clínico inicial
La mediana de edad de presentación es de 25 años en pacientes con sarcoma alveolar de partes blandas. Este tumor surge de manera más frecuente en las extremidades, pero también puede aparecer en la región oral y maxilofacial.[62,63,64] El sarcoma alveolar de partes blandas en niños a veces se manifiesta con indicios de enfermedad metastásica.[65] Son infrecuentes las metástasis encefálicas y pulmonares tardías.[62]
En una serie de 61 pacientes con sarcoma alveolar de partes blandas que se trataron en 4 ensayos consecutivos del CWS y del Soft Tissue Sarcoma Registry (SoTiSaR), 46 pacientes presentaron enfermedad localizada y 15 pacientes presentaron indicios de metástasis en el momento del diagnóstico.[66]
Entre 1980 y 2014, se trataron 69 pacientes menores de 30 años con sarcoma alveolar de partes blandas en cuatro instituciones importantes. La mediana de edad en el momento del diagnóstico fue de 17 años y el 64 % de los pacientes eran mujeres. El sitio más común de la enfermedad fue la extremidad inferior y 26 pacientes tenían una translocación génica ASPSCR1::TFE3.[67]
Alteraciones genómicas
Este tumor de histogénesis incierta se caracteriza por una translocación cromosómica constante t(X;17)(p11,2;q25) en la que se fusiona el gen ASPSCR1 con el gen TFE3.[68,69]
Pronóstico
En los pacientes con sarcoma alveolar de partes blandas, las metástasis son frecuentes y, a menudo, tienen una evolución poco activa y prolongada.
El sarcoma alveolar de partes blandas en niños a veces también presenta una evolución poco activa.[65] Es posible que los pacientes con sarcoma alveolar de partes blandas presenten una recidiva muchos años después de un largo periodo de remisión aparente.[66,70]
- En un estudio se notificó que en una serie de 19 pacientes con sarcoma alveolar de partes blandas que recibieron tratamiento la tasa de SG a 5 años fue del 80 %. La tasa de SG fue del 91 % en los pacientes con enfermedad localizada, del 100 % en los pacientes con tumores que medían 5 cm o menos y del 31 % en los pacientes con tumores que medían más de 5 cm.[71]
- En otra serie de 33 pacientes, la tasa de SG fue del 68 % a los 5 años del diagnóstico y del 53 % a los 10 años del diagnóstico. La supervivencia fue mejor en los pacientes con tumores más pequeños (≤5 cm) y tumores que se resecaron por completo.[72][Nivel de evidencia C1]
- En una revisión retrospectiva de niños y adultos jóvenes menores de 30 años (mediana de edad, 17 años; intervalo, 1,5–30 años) de 4 instituciones, se identificó a 69 pacientes tratados de manera primaria con cirugía entre 1980 y 2014.[67][Nivel de evidencia C1] Se encontró una translocación ASPSCR1::TFE3 en los 26 pacientes analizados. Hubo 19 pacientes con tumores del grupo I del IRS (28 %), 7 pacientes con tumores del grupo II del IRS (10 %), 5 pacientes con tumores del grupo III del IRS (7 %) y 38 pacientes con tumores del grupo IV del IRS (55 %). En los 31 pacientes con tumores localizados (grupos posquirúrgicos I, II y III del IRS), la tasa de SSC a 5 años fue del 80 % y la tasa de SG fue del 87 %. En los 38 pacientes con tumores metastásicos (grupo IV del IRS), la tasa de SSC a 5 años fue del 7 % y la tasa de SG fue del 61 %.
- En una serie de pacientes tratados en estudios consecutivos de Alemania, 15 de 61 pacientes (25 %) presentaron metástasis, por lo general de índole miliar. A pesar de la falta de respuesta a la quimioterapia, la tasa de SG a 5 años fue del 61 %, con una tasa de SSC del 20 %.[66]
Tratamiento del sarcoma alveolar de partes blandas
Las opciones de tratamiento del sarcoma alveolar de partes blandas son las siguientes:
- Cirugía con radioterapia y quimioterapia o sin estas.[29,30]
- Terapia dirigida (inhibidores de tirosina–cinasas e inhibidores de puntos de control).[73]
Cirugía con radioterapia y quimioterapia o sin estas
El abordaje estándar de tratamiento es la resección completa de la lesión primaria.[71] Si la escisión completa no es posible, se administra radioterapia.
Evidencia (cirugía con quimioterapia o sin esta):
- En un estudio realizado en China, se notificó sobre 18 pacientes con sarcoma alveolar de partes blandas en la región oral y maxilofacial. Entre ellos, 15 pacientes eran menores de 30 años. El tratamiento primario fue la resección quirúrgica con márgenes negativos.[64][Nivel de evidencia C2]
- Todos los pacientes sobrevivieron y solo 1 paciente presentó recidiva de enfermedad metastásica.
- En una serie de pacientes tratados en estudios consecutivos en Alemania, se notificó lo siguiente:[66]
- La supervivencia sin progresión (SSP) de los pacientes sin metástasis en el momento de la presentación mejoró con la resección completa del tumor primario.
- La tasa de SSC a 5 años fue del 100 % en los pacientes con tumores que se resecaron por completo, en comparación con el 50 % en los pacientes con enfermedad residual microscópica o macroscópica.
- En una serie de 51 pacientes pediátricos de 0 a 21 años con sarcoma alveolar de partes blandas, se notificó lo siguiente:[62][Nivel de evidencia C1]
- La tasa de SG fue del 78 % a los 10 años y la tasa de SSC fue de alrededor del 63 %.
- En los pacientes con enfermedad localizada (n = 37) la tasa de SG a 10 años fue del 87 %.
- Los 14 pacientes con metástasis en el momento del diagnóstico presentaron una tasa de SG a 10 años del 44 %, en parte debido a la resección quirúrgica del tumor primario y a las metástasis pulmonares en algunos pacientes.
- Solo 3 de 18 pacientes (17 %) con enfermedad cuantificable presentaron una reacción favorable a la quimioterapia antisarcoma convencional, pero 2 de 4 pacientes tratados con sunitinib presentaron una respuesta parcial.
Terapia dirigida
Se han realizado estudios de terapia dirigida (inhibidores de tirosina–cinasas e inhibidores de puntos de control).
Sunitinib
Evidencia (sunitinib):
- En un estudio retrospectivo pequeño con 9 pacientes adultos con sarcoma alveolar de partes blandas metastásico que recibieron sunitinib, se notificaron respuestas parciales en 5 pacientes y enfermedad estable en 2 pacientes.[74][Nivel de evidencia C3]
- En otro estudio, 15 pacientes adultos con sarcoma alveolar de partes blandas se trataron con sunitinib. De los pacientes, 5 se trataron con sunitinib durante más de 2 años.[75][Nivel de evidencia C1]
- Presentaron respuestas parciales 6 pacientes.
- La mediana de SSP fue de 19 meses y la mediana de SG fue de 56 meses.
- La tasa de SG a 5 años fue del 49 %.
Cediranib
El cediranib es un inhibidor de los 3 receptores del factor de crecimiento epidérmico vascular que se conocen.
Evidencia (cediranib):
- En un ensayo pediátrico de fase II de cediranib en el que se usó el 70 % de la dosis máxima tolerada en adultos para el tratamiento de pacientes menores de 16 años, se notificó lo siguiente:[76][Nivel de evidencia B4]
- De 7 pacientes, 5 presentaron enfermedad estable durante 14 meses o más.
- Un grupo internacional realizó un ensayo aleatorizado controlado con placebo con enmascaramiento doble de fase II sobre cediranib en pacientes adolescentes y adultos con sarcoma alveolar de partes blandas.[77][Nivel de evidencia A1]
- La mediana del cambio porcentual en la suma de los diámetros de la lesión objetivo en la población evaluable fue de un -8,3 % (intervalo intercuartílico [IIC], -26,5 a 5,9) en los pacientes que recibieron cediranib, en comparación con un 13,4 % (IIC, 1,1–21,3) en los pacientes que recibieron el placebo (P unilateral = 0,0010).
- Los autores concluyeron que el cediranib es un fármaco activo en pacientes con sarcoma alveolar de partes blandas.
- En un ensayo de fase II de cediranib, 15 de 43 pacientes adultos (35 %) con sarcoma alveolar de partes blandas metastásico presentaron respuestas parciales.[78][Nivel de evidencia C3]
Pazopanib
Evidencia (pazopanib):
- En un ensayo sin enmascaramiento en el que se evaluó la eficacia del pazopanib en 6 pacientes adultos, 1 paciente logró una respuesta parcial y 5 pacientes presentaron enfermedad estable.[79]
- En otro ensayo se incluyeron 30 pacientes adultos que se trataron con pazopanib.[80]
- Se presentó 1 caso de respuesta completa, 7 pacientes tuvieron respuestas parciales y 17 pacientes, enfermedad estable.
- La mediana de SSP fue de 13,6 meses.
Axitinib y pembrolizumab
El axitinib es un inhibidor de la tirosina–cinasa del receptor del factor de crecimiento endotelial vascular. El pembrolizumab es un inhibidor del punto de control inmunitario de la proteína 1 de muerte celular programada.
Evidencia (axitinib y pembrolizumab):
- En un ensayo, pacientes adultos con sarcomas avanzados se trataron con una combinación de axitinib y pembrolizumab.[73]
- La tasa de SSP a 3 meses en 12 pacientes con sarcoma alveolar de partes blandas fue del 73 %.
- De 11 pacientes con enfermedad evaluable, 6 exhibieron respuestas parciales con axitinib.
Otras terapias
Hay informes esporádicos de respuestas objetivas al tratamiento con interferón α y bevacizumab.[62,81,82]
Debido a que estos son tumores raros, se debe considerar la inscripción en ensayos clínicos prospectivos de todos los niños con sarcoma alveolar de partes blandas. Para obtener más información sobre ensayos clínicos en curso, consultar el portal de Internet del NCI.
Opciones de tratamiento en evaluación clínica para el sarcoma alveolar de partes blandas
La información en inglés sobre los ensayos clínicos patrocinados por el NCI se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
Sarcoma de células claras, sin otra indicación
El sarcoma de células claras (antes llamado melanoma maligno de partes blandas) es un sarcoma de tejido blando raro que suele afectar los tejidos blandos profundos de las extremidades. También se llama sarcoma de células claras de tendones y aponeurosis. El tumor suele afectar a adolescentes y adultos jóvenes.
Cuadro clínico inicial
El tumor afecta con mayor frecuencia la extremidad inferior, en particular el pie, el talón y el tobillo.[83,84] Tiene una predisposición alta a la diseminación ganglionar, en especial las metástasis en los ganglios linfáticos regionales (12–43 %).[84,85]
Es común que el tumor tenga una evolución clínica poco activa. Los pacientes con tumores pequeños localizados con índice mitótico bajo y un grado histológico intermedio presentan los mejores desenlaces.[86]
Alteraciones genómicas
El sarcoma de células claras de tejido blando se caracteriza por las fusiones génicas EWSR1::ATF1 o EWSR1::CREB1.[87,88]
Tratamiento del sarcoma de células claras de tejido blando
Las opciones de tratamiento del sarcoma de células claras de tejido blando son las siguientes:
- Cirugía con radioterapia o sin esta.[29,30]
- Terapia dirigida.
Cirugía con radioterapia o sin esta
La cirugía, con radioterapia o sin esta, es el tratamiento de elección y ofrece la mejor probabilidad de curación.
Evidencia (cirugía con radioterapia o sin esta):
- En una serie de 28 pacientes pediátricos de los Soft Tissue Cooperative Studies en Italia y Alemania, la mediana de edad en el momento del diagnóstico fue de 14 años y la extremidad inferior fue el sitio primario más común (50 %).[89]; [90][Nivel de evidencia C2]
- En esta serie, se curaron 12 de 13 pacientes con tumores resecados por completo.
- En los pacientes con estadio más avanzado de la enfermedad, el desenlace es precario y la quimioterapia casi nunca es eficaz.
Terapia dirigida
Evidencia (terapia dirigida):
- En un estudio de la European Organization for Research and Treatment of Cancer, se trató con crizotinib a 26 pacientes de sarcoma de células claras con enfermedad metastásica y reordenamientos de EWSR1 documentados.[91]
- Uno de los pacientes tuvo una respuesta parcial y 17 tuvieron enfermedad estable.
Condrosarcoma mixoide extraesquelético
El condrosarcoma mixoide extraesquelético es un tipo relativamente infrecuente de sarcoma de tejido blando, y representa solo el 2,3 % de todos los sarcomas de tejido blando.[92] Se ha notificado en niños y adolescentes.[93]
Este tumor se ha considerado tradicionalmente de bajo potencial maligno.[94] Sin embargo, en informes de instituciones grandes se observó que el condrosarcoma mixoide extraesquelético exhibe un potencial maligno importante, en especial si a los pacientes se los somete a seguimiento prolongado.[95,96] Los pacientes suelen presentar una evolución lenta prolongada. Se ha descrito bien el compromiso ganglionar. Además, se ha notificado recidiva local (57 %) y diseminación metastásica a los pulmones (26 %).[96]
Alteraciones genómicas
El condrosarcoma mixoide extraesquelético es una neoplasia multinodular. Las células redondeadas se organizan en cordones y cadenas sobre un fondo mixoide de sulfato de condroitina. Se identificaron varias anomalías citogenéticas (consultar el Cuadro 2); la más frecuente es la fusión génica EWSR1::NR4A3.[97]
Tratamiento del condrosarcoma mixoide extraesquelético
Las opciones de tratamiento del condrosarcoma mixoide extraesquelético son las siguientes:
- Cirugía.
- Radioterapia.
El control local intensivo y la resección de las metástasis produjo tasas de SG del 87 % a los 5 años y del 63 % a los 10 años. Los tumores fueron relativamente resistentes a la radioterapia.[95] No se ha establecido el beneficio terapéutico de la quimioterapia.
Hay posibles dianas genéticas para la acción de moléculas pequeñas, pero se tienen que estudiar en un ensayo clínico. En un estudio de adultos, 6 de 10 pacientes que recibieron sunitinib lograron respuestas parciales.[98]
Sarcoma de Ewing extraesquelético
Casi una quinta parte de los pacientes con sarcoma de Ewing presentarán sitios primarios no óseos (extraóseos). El tratamiento de este tumor es el mismo que el de los tumores primarios óseos.[99] Para obtener más información, consultar Tratamiento del sarcoma de Ewing y los sarcomas indiferenciados de células redondas pequeñas de hueso y tejido blando.
Tumor desmoplásico de células redondas pequeñas
El tumor desmoplásico de células redondas pequeñas es un sarcoma primitivo raro.
Cuadro clínico inicial
El tumor desmoplásico de células redondas pequeñas afecta con mayor frecuencia el peritoneo abdominal, la pelvis o el peritoneo del escroto, pero es posible que se presente en el riñón u otros órganos sólidos.[100,101,102,103,104] Con frecuencia se encuentran de docenas a cientos de implantes intraperitoneales. Se presentan sobre todo en hombres (85 %) y en ocasiones se diseminan a los pulmones y a otros sitios.[104,105]
Evaluación diagnóstica
En una serie grande de 65 pacientes en una sola institución, se compararon las tomografías computadas (TC) (n = 54) y las TC combinadas con tomografías por emisión de positrones (TEP)-TC (n = 11). Las TEP-TC arrojaron muy pocos resultados negativos falsos y permitieron la detección de sitios metastásicos que no se encontraron en las TC convencionales.[105]
Alteraciones genómicas
En estudios citogenéticos de estos tumores, se observó una translocación recurrente t(11;22)(p13;q12), que se ha caracterizado como una fusión de los genes WT1 y EWSR1.[103,106] La fusión EWSR1::WT1 confirma el diagnóstico de tumor desmoplásico de células redondas pequeñas. La carga de variantes del tumor es en promedio baja para el tumor desmoplásico de células redondas pequeñas (<1 variante por megabase), y es infrecuente la presencia de alteraciones génicas recurrentes diferentes a la fusión EWSR1::WT1.[107] Un porcentaje pequeño de los casos (aproximadamente el 3 %) tiene variantes activadoras de FGFR4, y con igual frecuencia se observó la amplificación de FGFR4.[107,108] Se han observado variantes inactivadoras de TP53 y ARID1A en un porcentaje pequeño de los casos de tumores desmoplásicos de células redondas pequeñas.[107,108]
Pronóstico
El pronóstico general de los pacientes con tumor desmoplásico de células redondas pequeñas continúa siendo muy precario y la tasa de mortalidad notificada es del 90 %. Una resección de más del 90 % del tumor, en el momento de la presentación inicial o después de la quimioterapia preoperatoria, a veces es un factor de pronóstico favorable para la SG.[109,110]; [111][Nivel de evidencia C1] La respuesta a la quimioterapia neoadyuvante y la resección completa (cercana al 100 %) se relaciona con un mejor desenlace.[104,112]
Tratamiento del tumor desmoplásico de células redondas pequeñas
No hay un abordaje estándar para el tratamiento del tumor desmoplásico de células redondas pequeñas.
Las opciones de tratamiento del tumor desmoplásico de células redondas pequeñas son las siguientes:
- Terapia multimodal (quimioterapia, cirugía y radioterapia).
- Cirugía con quimioterapia hipertérmica intraperitoneal.
- Otras opciones de tratamiento.
Terapia multimodal
Las resecciones quirúrgicas completas son infrecuentes y a menudo se realizan en centros muy especializados, pero son decisivas para mejorar la supervivencia. Las modalidades de tratamiento exitosas incluyen quimioterapia neoadyuvante de tipo Ewing, seguida de resección quirúrgica completa de los tumores intraabdominales extensos, seguida de radioterapia abdominal total. Con este abordaje multimodal, se puede lograr la supervivencia a los 5 años en el 30 % al 40 % de los pacientes.[100,101,109,113,114,115,116]
Cirugía con quimioterapia hipertérmica intraperitoneal
La quimioterapia hipertérmica intraperitoneal (HIPEC) es un método de tratamiento local que, en ocasiones, proporciona más control de la enfermedad intraabdominal microscópica. La teoría es que la quimioterapia caliente que se introduce en la cavidad abdominal después de la resección quirúrgica (en el momento de la cirugía) produce citotoxicidad sinérgica en las células microscópicas que quedan en el abdomen.[117]
La adición de HIPEC para completar la resección quirúrgica (cirugía citorreductora) es una técnica nueva que se utilizó por primera vez en niños durante un ensayo clínico de fase I realizado en 2006. La cirugía citorreductora y la HIPEC para el tratamiento de tumores desmoplásicos de células redondas pequeñas forman parte de un abordaje multidisciplinario y solo se administran en centros muy especializados. Las cirugías pueden durar más de 12 horas y se deben considerar los aspectos técnicos de esta resección tumoral particular.[117]
Evidencia (cirugía con HIPEC):
- En un estudio de fase II de una sola institución, se observó que la HIPEC es una adición prometedora de la resección quirúrgica completa. En el estudio se inscribieron 14 pacientes con tumor desmoplásico de células redondas pequeñas y 5 pacientes con otros sarcomas. Estos pacientes muy seleccionados tenían tumores confinados a la cavidad abdominal. Estos pacientes exhibieron una respuesta parcial a la quimioterapia neoadyuvante de tipo Ewing, se sometieron a resecciones quirúrgicas completas y recibieron HIPEC con cisplatino. También recibieron radioterapia abdominal total adyuvante seguida de quimioterapia adyuvante.[117]
- Con este abordaje estandarizado, los pacientes con tumores desmoplásicos de células redondas pequeñas tuvieron una tasa de SG del 80 % a los 30 meses y del 40 % a los 50 meses.
- Los pacientes con tumores desmoplásico de células redondas pequeñas sin metástasis hepáticas no presentaron recidivas intraabdominales; sin embargo, el 87 % de los pacientes con metástasis hepáticas o enfermedad portal presentaron recidivas.
- En un estudio retrospectivo en varias instituciones de Francia, se trató a los pacientes con cirugía citorreductora e HIPEC. Se seleccionó a 22 pacientes con una mediana de edad en el momento del diagnóstico de 14,8 años (intervalo, 4,2–17,6 años). Entre estos, 7 pacientes tenían mesoteliomas peritoneales, 7 tenían tumores desmoplásicos de células redondas pequeñas y 8 pacientes exhibían tumores con otros tipos histológicos. Se logró una resección macroscópica completa (CC-0, donde CC indica integridad de la citorreducción) en 16 casos (73 %). De los 7 pacientes con tumores desmoplásicos de células redondas pequeñas, 4 tuvieron resecciones completas.[118][Nivel de evidencia C1]
- Se observó recaída en 16 pacientes (72 %) luego de una mediana de tiempo de 9,6 meses (intervalo, 1,4–86,4 años).
- Al cabo de una mediana de tiempo de 5,3 meses (intervalo, 0,1–36,1 meses), 9 pacientes (41 %) murieron por enfermedad recidivante.
- En otro estudio realizado en Francia, se analizó el uso de cirugía citorreductora con HIPEC para el tratamiento de pacientes con tumores desmoplásicos de células redondas pequeñas con enfermedad limitada al abdomen. De los 107 pacientes con tumores desmoplásicos de células redondas pequeñas, 48, que no tenían metástasis extraperitoneales, se sometieron a cirugía citorreductora. De 48 pacientes (media de edad, 22 años), 38 (79 %) recibieron quimioterapia preoperatoria o posoperatoria y 23 (48 %) recibieron radioterapia abdominopélvica completa posoperatoria. Se administró quimioterapia intraperitoneal a 11 pacientes (23 %); 2 pacientes recibieron quimioterapia intraperitoneal posoperatoria temprana (EPIC) y 9 pacientes recibieron HIPEC.[119]
- Después de una mediana de seguimiento de 30 meses, la mediana de SG de toda la cohorte fue de 42 meses.
- La tasa de SG a 2 años fue del 72 % y la tasa de SG a 5 años fue del 19 %.
- La tasa de supervivencia sin enfermedad (SSE) a 2 años fue del 30 % y la tasa de SSE a 5 años fue del 12 %.
- La radioterapia dirigida a toda la cavidad abdominopélvica fue la única variable relacionada con supervivencia sin recidiva peritoneal y SSE después de la cirugía citorreductora.
- Se utilizaron 6 regímenes diferentes de quimioterapia en los 11 pacientes que recibieron quimioterapia intraperitoneal (HIPEC o EPIC). En el estudio, no se indica la supervivencia o el desenlace para este grupo.
- La repercusión de HIPEC o EPIC sobre la SG y la SSE no fue estadísticamente significativa pero, como no se utilizaron regímenes estandarizados en todos los pacientes, resulta difícil evaluar los resultados.
- En un estudio retrospectivo de una sola institución, se informó sobre 9 pacientes (mediana de edad, 19 años) con tumor desmoplásico de células redondas pequeñas. La mayoría de los pacientes tenían enfermedad generalizada; entre ellos, 4 pacientes con enfermedad extraabdominal y 5 pacientes con compromiso hepático. Estos 9 pacientes se sometieron a 10 tratamientos citorreductores con HIPEC. Además, 7 pacientes también recibieron radioterapia y 3 pacientes se sometieron a trasplante de células madre.[120]
- La tasa de supervivencia sin recaída a 3 años fue del 13 %, y la tasa de SG fue del 55 %.
- El tratamiento a menudo se vinculó con hospitalizaciones prolongadas.
- Fue necesario administrar nutrición parenteral a largo plazo a 8 pacientes durante una mediana de 261 días.
- Otras complicaciones a largo plazo fueron gastroparesia (n = 1), obstrucción del intestino delgado (n = 3) y cistitis hemorrágica (n = 2).
Otras opciones de tratamiento
El Center for International Blood and Marrow Transplant Research analizó a pacientes con tumor desmoplásico de células redondas pequeñas de su registro que recibieron consolidación con dosis altas de quimioterapia y reconstitución de células madre autógenas.[121] Si bien en este análisis retrospectivo del registro se indicó algún beneficio de este abordaje, otros investigadores han dejado de usarlo debido al exceso de efectos tóxicos y la ausencia de eficacia.[109]
En un estudio de una sola institución, se notificó que 5 de 5 pacientes con tumor desmoplásico de células redondas pequeñas recidivante presentaron respuestas parciales al tratamiento con la combinación de vinorelbina, ciclofosfamida y temsirólimus.[122]
Tumor rabdoide, sin otra indicación (extrarrenal)
Los tumores rabdoides malignos en niños con tumores renales se describieron por primera vez en 1981. Después, estos tumores se identificaron en varios sitios extrarrenales. Estos tumores son infrecuentes y muy malignos, en especial en niños menores de 2 años. Para obtener más información, consultar la sección Tumores rabdoides de riñón en Tratamiento del tumor de Wilms y otros tumores renales infantiles.
Los tumores rabdoides extrarrenales (extracraneales) representan el 2 % de los sarcomas de tejido blando en pacientes menores de 20 años (consultar el Cuadro 1).
Alteraciones genéticas y genómicas
La primera serie de tamaño considerable de niños con tumor rabdoide maligno extrarrenal y extracraneal de tejidos blandos se creó a partir de 26 pacientes inscritos en los estudios IRS I a III durante una revisión de material de patología. Solo 5 pacientes (19 %) estaban vivos y sin enfermedad después de 2 años.[123]
Durante la investigación de niños que presentaban tumores teratoides rabdoides atípicos en el encéfalo, así como otros con tumores rabdoides renales y extrarrenales, se encontraron variantes germinales y adquiridas del gen SMARCB1 en 29 tumores analizados.[124] Los tumores rabdoides a veces se relacionan con variantes germinales del gen SMARCB1 y se pueden heredar de un progenitor en apariencia no afectado.[125] Esta observación se extendió a 32 tumores rabdoides malignos en todos los sitios en pacientes cuya mediana de edad en el momento del diagnóstico fue de 12 meses.[126]
Pruebas genéticas y vigilancia
Se debe considerar el análisis de la línea germinal para personas de todas las edades con tumores rabdoides. Se recomienda el asesoramiento genético como parte del plan de tratamiento debido al riesgo bajo, pero real, de que se presenten más casos dentro de una familia. Cuando hay variantes, se deberán considerar los exámenes de detección en los progenitores, aunque estos exámenes de portadores tienen una probabilidad baja de positividad. Es posible realizar un diagnóstico prenatal en situaciones en las que se ha documentado una variante o deleción específica de SMARCB1 en la familia.[125]
Hasta la fecha, hay escasa evidencia sobre la eficacia de la vigilancia de pacientes con un síndrome de predisposición a tumores rabdoides de tipo 1 causado por variantes germinales de pérdida de función en SMARCB1. Sin embargo, se han formulado recomendaciones por consenso, debido a la naturaleza maligna de los tumores, la mortalidad significativa, y el comienzo a una edad temprana en portadores de variantes interruptoras de SMARCB1. Un grupo de expertos en las características genéticas del cáncer infantil (conformado por oncólogos, radiólogos y genetistas) formularon estas recomendaciones. No se ha hecho un estudio oficial para corroborar el beneficio de la vigilancia de los pacientes con variantes germinales de SMARCB1. Se ha postulado que la detección temprana quizás mejore la supervivencia general (SG) por el posible beneficio para la supervivencia al identificar una enfermedad resecable.[127,128,129]
La vigilancia de los pacientes con variantes germinales de SMARCB1 incluye los siguientes aspectos:
- Imágenes por resonancia magnética (IRM) del encéfalo cada 3 meses desde el nacimiento (o el diagnóstico) hasta los 5 años de edad.
- Se recomienda una ecografía abdominal con énfasis en los riñones cada 3 meses.
Pronóstico y cuadro clínico inicial
La edad temprana y la enfermedad metastásica en el momento de la presentación se relacionan con desenlace precario en niños con tumores rabdoides extracraneales.
En un estudio en el que se usaron datos de la National Cancer Database, se identificaron 202 pacientes (menores de 18 años) con tumores rabdoides malignos fuera del sistema nervioso central (SNC). El sitio primario del tumor rabdoide maligno fue el tejido blando (46 %), los riñones (45 %) y el hígado (9 %).[130]
- La tasa de SG a 1 año fue del 48,8 % y la tasa de SG a 5 años fue del 35,9 %.
- En el análisis multivariante se demostró que la edad inferior a 1 año y la presencia de metástasis fueron indicadores de pronóstico precario (P = 0,058).
- En la cohorte de pacientes quirúrgicos (n = 143), se observó cierta relación clínicamente significativa entre la presencia de enfermedad residual y un desenlace más precario (CRI, 1,54; IC 95 %, 0,88–2,69; P = 0,13).
En un estudio del SEER, se examinó a 229 pacientes con tumor rabdoide maligno de riñón, del SNC y extrarrenal. Los factores que tuvieron una repercusión favorable fueron la edad de 2 a 18 años de los pacientes, tumores de extensión limitada y la administración de radioterapia, en comparación con otros pacientes (P < 0,002 para cada comparación). El sitio del tumor primario no tuvo importancia pronóstica. La tasa de SG fue del 33 % a los 5 años.[131]
En un registro europeo de tumores rabdoides extracraneales, se identificaron 100 pacientes de 14 países entre 2009 y 2018.[132] La mitad de los pacientes tenían menos de 1 año en el momento del diagnóstico. En 30 pacientes (30 %), el tumor se ubicó en los riñones. Se encontró un tumor rabdoide maligno extracraneal y extrarrenal en el 70 % de los pacientes (70 de 100), y los sitios más frecuentes fueron la región cervical, la región torácica y el hígado. En 9 pacientes se encontraron tumores sincrónicos. Se presentaron metástasis a distancia en el momento del diagnóstico en el 35 % de los pacientes (35 de 100). Se detectaron variantes germinales de SMARCB1 en un 21 % de los pacientes (17 de 81 pacientes evaluables). La tasa de SG a 5 años fue del 45,8 % (± 5,4 %) y la tasa de SSC fue del 35,2 % (± 5,1 %). En un modelo multivariante ajustado, los factores de predicción más significativos para un desenlace negativo fueron la presencia de una variante germinal, metástasis y la ausencia de resección macroscópica total.
Tratamiento del tumor rabdoide extrarrenal (extracraneal)
Las opciones de tratamiento del tumor rabdoide extrarrenal (extracraneal) son las siguientes:[133,134,135][Nivel de evidencia C1]
- Resección quirúrgica siempre que sea posible.
- Régimen de quimioterapia que se usa para los sarcomas de tejido blando (aunque no haya un régimen único que se acepte como el mejor en la actualidad).
- Radioterapia.
Se documentaron respuestas favorables al alisertib en 4 pacientes con tumores teratoides rabdoides atípicos del SNC.[136] Para obtener más información sobre los tumores teratoides rabdoides atípicos del SNC, consultar Tratamiento del tumor teratoide rabdoide atípico del sistema nervioso central infantil.
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el NCI se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de un ensayo clínico nacional o institucional en curso:
- PEPN2121 (NCT05286801) (Tiragolumab and Atezolizumab for the Treatment of Relapsed or Refractory SMARCB1- or SMARCA4-Deficient Tumors): en este estudio se evalúa la combinación de un anticuerpo dirigido a PD-L1 (atezolizumab) con un anticuerpo dirigido a TIGIT (tiragolumab) en pacientes que tienen tumores con deficiencia de SMARCB1 o SMARCA4. Es posible que los pacientes con tumores rabdoides extrarrenales (extracraneales) sean aptos para participar en este estudio.
Tumor epitelioide perivascular (PEcoma) maligno
Cuadro clínico inicial
Los PEComas se presentan en varios sitios infrecuentes: gastrointestinales, pulmonares, ginecológicos y genitourinarios. Los PEComas de tejido blando, visceral y ginecológico se observan con mayor frecuencia en mujeres de mediana edad y, por lo general, no se relacionan con el complejo de la esclerosis tuberosa.[137] La evolución de la enfermedad puede ser lenta.
Factores de riesgo y características moleculares
Los PEComas benignos son frecuentes en los pacientes con esclerosis tuberosa, un síndrome autosómico dominante que también predispone al cáncer de células renales y los tumores de encéfalo. La causa de la esclerosis tuberosa es la inactivación germinal de TSC1 (9q34) o TSC2 (16p13.3); en los PEComas esporádicos se encuentra inactivación somática de los mismos genes supresores de tumores.[138] La inactivación de cualquiera de estos genes produce una estimulación de la vía de mTOR, lo que fundamenta el tratamiento de los tumores curables no quirúrgicos que tienen una inactivación genética similar (linfangioleiomiomatosis y angiomiolipoma) con inhibidores de mTOR.[139,140] Una proporción baja de los PEComas tiene reordenamientos de TFE3 con fusiones que comprometen varios genes; entre ellos, SFPQ y RAD51B.[141]
Pronóstico
La mayoría de los PEComas tiene un curso clínico benigno, pero se ha informado de comportamiento maligno predecible a partir del tamaño tumoral, el índice mitótico y la presencia de necrosis.[142]
Tratamiento del PEComa
No hay opciones de tratamiento estándar. El tratamiento incluye cirugía y observación seguida de cirugía cuando el tumor es voluminoso.[143]
En tumores con indicios de activación de mTORC1 y pérdida de TSC1 o TSC2, como la linfangioleiomiomatosis y el angiomiolipoma,[139] se ha documentado bien la actividad clínica de los inhibidores de mTOR, como el sirólimus. En una serie de casos pequeña, 3 pacientes adultos con PEComas respondieron al sirólimus.[144]
En un ensayo de fase II, 34 pacientes con PEComas malignos metastásicos o localmente avanzados recibieron tratamiento con partículas de sirólimus unidas a proteínas para suspensión inyectable (unidas a albúmina) (nab-sirólimus). De los 31 pacientes aptos para el análisis de eficacia, 12 (39 %) tuvieron respuesta (1 respuesta completa y 11 respuestas parciales), 16 (52 %) presentaron enfermedad estable y 3 (10 %) enfermedad progresiva. Las respuestas fueron rápidas y duraderas. La mediana de duración de la respuesta no se logró después de una mediana de seguimiento de 2,5 años. De 12 pacientes que respondieron al tratamiento, 7 aún continuaban con el tratamiento (intervalo, de 5,6 meses a más de 47,2 meses). Se realizó el perfil de variantes del tumor en 25 muestras. De 9 pacientes con variantes de TSC2, 8 respondieron al tratamiento, mientras que, de 16 pacientes sin variantes de TSC2, solo 2 respondieron. Además, se observaron respuestas en 10 de 17 pacientes con expresión de fosfo-S6 (pS6). No se observó ninguna respuesta en 8 pacientes sin expresión de pS6. La ausencia de expresión de pS6 refleja la falta de activación de mTORC1.[145][Nivel de evidencia C1] En 2021, la FDA aprobó el nab-sirólimus para pacientes adultos con PEComas.
Sarcoma indiferenciado
Desde 1972 hasta 2006, los pacientes con sarcoma indiferenciado de tejido blando fueron aptos para participar en los ensayos de rabdomiosarcoma coordinados por el grupo IRS y el COG. La justificación fue que los pacientes con sarcoma indiferenciado de tejido blando y los pacientes con rabdomiosarcoma alveolar tienen sitios de enfermedad y desenlaces similares. En los ensayos terapéuticos de adultos con sarcoma de tejido blando, se incluyen a pacientes con sarcoma indiferenciado de tejido blando y otros tipos histológicos, quienes reciben tratamientos semejantes, con ifosfamida y doxorrubicina y, en ocasiones, con otros fármacos quimioterapéuticos, cirugía y radioterapia.
En el ensayo ARST0332 (NCT00346164) del COG, los pacientes con sarcoma indiferenciado de grado alto se trataron con un régimen a base de ifosfamida y doxorrubicina. En el ensayo se notificaron los resultados de los pacientes con sarcoma indiferenciado de grado alto y de los pacientes con todos los sarcomas de tejido blando de grado alto. La tasa estimada de SSC a 5 años fue del 64 % y la tasa de SG fue de 77 % para los sarcomas clasificados como de grado alto por la Fédération Nationale des Centres de Lutte Contre Le Cancer (FNCLC).[5][Nivel de evidencia C1]
En un informe de 32 pacientes con sarcomas indiferenciados de tejido blando inscritos en el ensayo ARST0332 (NCT00346164), la mediana de edad en el momento de la inscripción fue de 13,6 años y 2 tercios de los pacientes eran hombres. Los sitios primarios más frecuentes fueron la región paravertebral y las extremidades. Cinco pacientes presentaron enfermedad metastásica.[146]
- La tasa de SSC a 5 años fue del 71 %, y la tasa de SG fue del 83 %.
- De los 9 niños con enfermedad de riesgo bajo (enfermedad localizada de grado bajo resecada o enfermedad localizada de grado alto <5 cm resecada con márgenes negativos) que se trataron solo con cirugía o radioterapia, la tasa de SSC a 5 años fue del 65 % y la tasa de SG fue del 100 %, lo que indica que los pacientes con enfermedad de riesgo bajo se pueden rescatar si la enfermedad recidiva.
- Los 23 pacientes restantes que tenían enfermedad de riesgo intermedio (tumor de grado alto >5 cm resecado, tumor de grado alto >5 cm sin resecar) o enfermedad de riesgo alto (metástasis a ganglios linfáticos o sitios lejanos) se trataron con quimiorradioterapia y se retrasó la cirugía cuando fue posible. La tasa de SSC a 5 años fue del 73 % y la SG estimada fue del 77 %.
- Las alteraciones en el número de copias fueron frecuentes, por lo general con pérdida de 1p (25 %), ganancia de 1q (25 %), ganancia del cromosoma 8 (25 %) y ganancia del cromosoma 2 (16 %). Estas anomalías se observaron en su mayoría en pacientes con tumores de riesgo intermedio o riesgo alto, y hubo una relación clara entre la pérdida del cromosoma 1p o la ganancia del cromosoma 1q y desenlaces clínicos más precarios. La presencia simultánea de la ganancia de 1q y la pérdida de 1p se relacionó con un desenlace clínico muy desfavorable (tasas de SSC a 5 años y SG del 20 %). Con secuenciación de última generación se identificaron fusiones oncogénicas en 8 de 10 muestras que incluyeron reordenamientos de BCOR y CIC, así como fusiones génicas COL1A1::PDGFB, KIAA1549::BRAF y SAMD5::SASH1.
Sarcoma pleomórfico indiferenciado (histiocitoma fibroso maligno)
En un momento determinado, el histiocitoma fibroso maligno fue el tipo histológico más común en los adultos con sarcomas de tejido blando. Sin embargo, desde que se identificó en los primeros años de la década de 1960, ha persistido la controversia sobre el histiocitoma fibroso maligno en términos de su histogénesis y validez como entidad clinicopatológica. La clasificación de la Organización Mundial de la Salud (OMS) ya no incluye el histiocitoma fibroso maligno como una categoría diagnóstica diferenciada, sino como un subtipo de sarcoma pleomórfico indiferenciado.[147,148]
Esta entidad representa del 2 % al 6 % de todos los sarcomas de tejido blando en los niños.[149]
Cuadro clínico inicial
Estos tumores suelen presentarse entre los 10 y 20 años. En una serie de10 pacientes, la mediana de edad fue de 10 años y, por lo general, el tumor se encontraba en las extremidades. En esta serie, todos los tumores eran localizados y 5 de 9 pacientes (a quienes se les dio seguimiento) estaban vivos y en primera remisión.[149]
En otra serie de 17 pacientes pediátricos con histiocitoma fibroso maligno, la mediana de edad en el momento del diagnóstico fue de 5 años y se presentó compromiso de las extremidades en 8 casos.[150] Todos los pacientes con enfermedad metastásica murieron y 2 pacientes exhibieron una respuesta clínica al régimen de doxorrubicina.
Para obtener más información sobre el tratamiento del histiocitoma fibroso maligno óseo, consultar Tratamiento del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo.
Factores de riesgo
Estos tumores pueden surgir en sitios previamente irradiados o como una neoplasia secundaria en pacientes con retinoblastoma.[151]
Características moleculares
En un análisis de 70 pacientes diagnosticados con histiocitosis fibrosa maligna sin tipo específico, histiocitoma fibroso maligno estoriforme o pleomórfico, sarcoma pleomórfico o sarcoma pleomórfico indiferenciado se observó un cariotipo muy complejo sin anomalías recurrentes específicas.[152]
Los sarcomas indiferenciados con la amplificación 12q13–15, como MDM2 y CDK4, se clasifican mejor como liposarcomas desdiferenciados.[152] No ha se ha definido bien la relación entre este tumor y la familia de tumores indiferenciados o sin clasificar con características morfológicas de células fusiformes.
Tratamiento del sarcoma pleomórfico recién diagnosticado
Para obtener información sobre el tratamiento del sarcoma pleomórfico indiferenciado óseo, consultar Tratamiento del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo.
Tratamiento del sarcoma pleomórfico recidivante o resistente al tratamiento
Las opciones de tratamiento del sarcoma pleomórfico recidivante o resistente al tratamiento son las siguientes:
- Pembrolizumab.
La Sarcoma Alliance for Research through Collaboration dirigió un ensayo de fase II sobre el inhibidor de puntos de control pembrolizumab en pacientes de 18 años o más con sarcoma de tejido blando recidivante.[153][Nivel de evidencia C3]
- De 40 pacientes con sarcoma de tejido blando, 7 (18 %) presentaron una respuesta objetiva.
- De 10 pacientes con sarcoma pleomórfico indiferenciado, 4 (40 %) presentaron una respuesta objetiva, al igual que 2 (20 %) de 10 pacientes con liposarcoma y 1 (10 %) de 10 pacientes con sarcoma sinovial.
- Ninguno de los pacientes con leiomiosarcoma (n = 10) exhibió una respuesta objetiva.
Sarcoma de células redondas indiferenciado
Sarcomas indiferenciados de células redondas pequeñas con alteraciones genéticas enBCOR
Consultar las secciones Sarcomas indiferenciados de células redondas pequeñas con alteraciones genéticas en BCOR y Características genómicas del sarcoma de Ewing en Tratamiento del sarcoma de Ewing y los sarcomas indiferenciados de células redondas pequeñas de hueso y tejido blando.
Sarcomas indiferenciados de células redondas pequeñas con alteraciones genéticas enCIC
Consultar las secciones Sarcomas indiferenciados de células redondas pequeñas con alteraciones genéticas en CIC y Características genómicas del sarcoma de Ewing en Tratamiento del sarcoma de Ewing y los sarcomas indiferenciados de células redondas pequeñas de hueso y tejido blando.
Sarcomas indiferenciados de células redondas pequeñas con fusiones deEWSR1y genes diferentes a ETS
Consultar la sección Sarcomas indiferenciados de células redondas pequeñas con fusiones de EWSR1 y genes diferentes a ETS en Tratamiento del sarcoma de Ewing y los sarcomas indiferenciados de células redondas pequeñas de hueso y tejido blando.
Referencias:
- Wilkes D, Charitakis K, Basson CT: Inherited disposition to cardiac myxoma development. Nat Rev Cancer 6 (2): 157-65, 2006.
- Carney JA, Young WF: Primary pigmented nodular adrenocortical disease and its associated conditions. Endocrinologist 2: 6-21, 1992.
- Ryan MW, Cunningham S, Xiao SY: Maxillary sinus melanoma as the presenting feature of Carney complex. Int J Pediatr Otorhinolaryngol 72 (3): 405-8, 2008.
- Sultan I, Rodriguez-Galindo C, Saab R, et al.: Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer 115 (15): 3537-47, 2009.
- Spunt SL, Million L, Chi YY, et al.: A risk-based treatment strategy for non-rhabdomyosarcoma soft-tissue sarcomas in patients younger than 30 years (ARST0332): a Children's Oncology Group prospective study. Lancet Oncol 21 (1): 145-161, 2020.
- Wang JG, Li NN: Primary cardiac synovial sarcoma. Ann Thorac Surg 95 (6): 2202-9, 2013.
- Chirmade PC, Parikh S, Anand A, et al.: Primary pleuropulmonary synovial sarcoma with brain metastases in a paediatric patient: an unusual presentation. Adv Respir Med 85 (4): 206-210, 2017.
- Frazier AA, Franks TJ, Pugatch RD, et al.: From the archives of the AFIP: Pleuropulmonary synovial sarcoma. Radiographics 26 (3): 923-40, 2006 May-Jun.
- Essary LR, Vargas SO, Fletcher CD: Primary pleuropulmonary synovial sarcoma: reappraisal of a recently described anatomic subset. Cancer 94 (2): 459-69, 2002.
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994.
- Ferrari A, De Salvo GL, Oberlin O, et al.: Synovial sarcoma in children and adolescents: a critical reappraisal of staging investigations in relation to the rate of metastatic involvement at diagnosis. Eur J Cancer 48 (9): 1370-5, 2012.
- Scheer M, Blank B, Bauer S, et al.: Synovial sarcoma disease characteristics and primary tumor sites differ between patient age groups: a report of the Cooperative Weichteilsarkom Studiengruppe (CWS). J Cancer Res Clin Oncol 146 (4): 953-960, 2020.
- van de Rijn M, Barr FG, Collins MH, et al.: Absence of SYT-SSX fusion products in soft tissue tumors other than synovial sarcoma. Am J Clin Pathol 112 (1): 43-9, 1999.
- Krsková L, Sumerauer D, Stejskalová E, et al.: A novel variant of SYT-SSX1 fusion gene in a case of spindle cell synovial sarcoma. Diagn Mol Pathol 16 (3): 179-83, 2007.
- Su L, Sampaio AV, Jones KB, et al.: Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell 21 (3): 333-47, 2012.
- Arnold MA, Arnold CA, Li G, et al.: A unique pattern of INI1 immunohistochemistry distinguishes synovial sarcoma from its histologic mimics. Hum Pathol 44 (5): 881-7, 2013.
- Vlenterie M, Ho VK, Kaal SE, et al.: Age as an independent prognostic factor for survival of localised synovial sarcoma patients. Br J Cancer 113 (11): 1602-6, 2015.
- Smolle MA, Parry M, Jeys L, et al.: Synovial sarcoma: Do children do better? Eur J Surg Oncol 45 (2): 254-260, 2019.
- Okcu MF, Munsell M, Treuner J, et al.: Synovial sarcoma of childhood and adolescence: a multicenter, multivariate analysis of outcome. J Clin Oncol 21 (8): 1602-11, 2003.
- Brecht IB, Ferrari A, Int-Veen C, et al.: Grossly-resected synovial sarcoma treated by the German and Italian Pediatric Soft Tissue Sarcoma Cooperative Groups: discussion on the role of adjuvant therapies. Pediatr Blood Cancer 46 (1): 11-7, 2006.
- Stanelle EJ, Christison-Lagay ER, Healey JH, et al.: Pediatric and adolescent synovial sarcoma: multivariate analysis of prognostic factors and survival outcomes. Ann Surg Oncol 20 (1): 73-9, 2013.
- Lagarde P, Przybyl J, Brulard C, et al.: Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol 31 (5): 608-15, 2013.
- Stegmaier S, Leuschner I, Poremba C, et al.: The prognostic impact of SYT-SSX fusion type and histological grade in pediatric patients with synovial sarcoma treated according to the CWS (Cooperative Weichteilsarkom Studie) trials. Pediatr Blood Cancer 64 (1): 89-95, 2017.
- Scheer M, Dantonello T, Hallmen E, et al.: Primary Metastatic Synovial Sarcoma: Experience of the CWS Study Group. Pediatr Blood Cancer 63 (7): 1198-206, 2016.
- Orbach D, Mosseri V, Pissaloux D, et al.: Genomic complexity in pediatric synovial sarcomas (Synobio study): the European pediatric soft tissue sarcoma group (EpSSG) experience. Cancer Med 7 (4): 1384-1393, 2018.
- Trassard M, Le Doussal V, Hacène K, et al.: Prognostic factors in localized primary synovial sarcoma: a multicenter study of 128 adult patients. J Clin Oncol 19 (2): 525-34, 2001.
- Guillou L, Benhattar J, Bonichon F, et al.: Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol 22 (20): 4040-50, 2004.
- Ferrari A, Gronchi A, Casanova M, et al.: Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer 101 (3): 627-34, 2004.
- Bahig H, Roberge D, Bosch W, et al.: Agreement among RTOG sarcoma radiation oncologists in contouring suspicious peritumoral edema for preoperative radiation therapy of soft tissue sarcoma of the extremity. Int J Radiat Oncol Biol Phys 86 (2): 298-303, 2013.
- Baldini EH, Wang D, Haas RL, et al.: Treatment Guidelines for Preoperative Radiation Therapy for Retroperitoneal Sarcoma: Preliminary Consensus of an International Expert Panel. Int J Radiat Oncol Biol Phys 92 (3): 602-12, 2015.
- Ferrari A, Chi YY, De Salvo GL, et al.: Surgery alone is sufficient therapy for children and adolescents with low-risk synovial sarcoma: A joint analysis from the European paediatric soft tissue sarcoma Study Group and the Children's Oncology Group. Eur J Cancer 78: 1-6, 2017.
- McGrory JE, Pritchard DJ, Arndt CA, et al.: Nonrhabdomyosarcoma soft tissue sarcomas in children. The Mayo Clinic experience. Clin Orthop (374): 247-58, 2000.
- Van Glabbeke M, van Oosterom AT, Oosterhuis JW, et al.: Prognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: an analysis of 2,185 patients treated with anthracycline-containing first-line regimens--a European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 17 (1): 150-7, 1999.
- Koscielniak E, Harms D, Henze G, et al.: Results of treatment for soft tissue sarcoma in childhood and adolescence: a final report of the German Cooperative Soft Tissue Sarcoma Study CWS-86. J Clin Oncol 17 (12): 3706-19, 1999.
- Pappo AS, Devidas M, Jenkins J, et al.: Phase II trial of neoadjuvant vincristine, ifosfamide, and doxorubicin with granulocyte colony-stimulating factor support in children and adolescents with advanced-stage nonrhabdomyosarcomatous soft tissue sarcomas: a Pediatric Oncology Group Study. J Clin Oncol 23 (18): 4031-8, 2005.
- Cecchetto G, Alaggio R, Dall'Igna P, et al.: Localized unresectable non-rhabdo soft tissue sarcomas of the extremities in pediatric age: results from the Italian studies. Cancer 104 (9): 2006-12, 2005.
- Pappo AS, Rao BN, Jenkins JJ, et al.: Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: the St. Jude Children's Research Hospital experience. Med Pediatr Oncol 33 (2): 76-82, 1999.
- Brennan B, Stevens M, Kelsey A, et al.: Synovial sarcoma in childhood and adolescence: a retrospective series of 77 patients registered by the Children's Cancer and Leukaemia Group between 1991 and 2006. Pediatr Blood Cancer 55 (1): 85-90, 2010.
- Ferrari A, Miceli R, Rey A, et al.: Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Cancer 47 (5): 724-31, 2011.
- Raney RB: Synovial sarcoma in young people: background, prognostic factors, and therapeutic questions. J Pediatr Hematol Oncol 27 (4): 207-11, 2005.
- Orbach D, Mc Dowell H, Rey A, et al.: Sparing strategy does not compromise prognosis in pediatric localized synovial sarcoma: experience of the International Society of Pediatric Oncology, Malignant Mesenchymal Tumors (SIOP-MMT) Working Group. Pediatr Blood Cancer 57 (7): 1130-6, 2011.
- Ladenstein R, Treuner J, Koscielniak E, et al.: Synovial sarcoma of childhood and adolescence. Report of the German CWS-81 study. Cancer 71 (11): 3647-55, 1993.
- Venkatramani R, Xue W, Randall RL, et al.: Synovial Sarcoma in Children, Adolescents, and Young Adults: A Report From the Children's Oncology Group ARST0332 Study. J Clin Oncol 39 (35): 3927-3937, 2021.
- Ferrari A, De Salvo GL, Brennan B, et al.: Synovial sarcoma in children and adolescents: the European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann Oncol 26 (3): 567-72, 2015.
- Scheer M, Hallmen E, Vokuhl C, et al.: Pre-operative radiotherapy is associated with superior local relapse-free survival in advanced synovial sarcoma. J Cancer Res Clin Oncol 149 (5): 1717-1731, 2023.
- Ferrari A, De Salvo GL, Dall'Igna P, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with initially localised synovial sarcoma. Eur J Cancer 48 (18): 3448-55, 2012.
- Scheer M, Dantonello T, Hallmen E, et al.: Synovial Sarcoma Recurrence in Children and Young Adults. Ann Surg Oncol 23 (Suppl 5): 618-626, 2016.
- Ferrari A, Orbach D, Bergamaschi L, et al.: Treatment at relapse for synovial sarcoma of children and adolescents: A multi-institutional European retrospective analysis. Pediatr Blood Cancer 71 (7): e31038, 2024.
- Dhakal S, Corbin KS, Milano MT, et al.: Stereotactic body radiotherapy for pulmonary metastases from soft-tissue sarcomas: excellent local lesion control and improved patient survival. Int J Radiat Oncol Biol Phys 82 (2): 940-5, 2012.
- Lai JP, Robbins PF, Raffeld M, et al.: NY-ESO-1 expression in synovial sarcoma and other mesenchymal tumors: significance for NY-ESO-1-based targeted therapy and differential diagnosis. Mod Pathol 25 (6): 854-8, 2012.
- Robbins PF, Kassim SH, Tran TL, et al.: A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 21 (5): 1019-27, 2015.
- D'Angelo SP, Melchiori L, Merchant MS, et al.: Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 c259T Cells in Synovial Sarcoma. Cancer Discov 8 (8): 944-957, 2018.
- Chbani L, Guillou L, Terrier P, et al.: Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French sarcoma group. Am J Clin Pathol 131 (2): 222-7, 2009.
- Hornick JL, Dal Cin P, Fletcher CD: Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 33 (4): 542-50, 2009.
- Knutson SK, Warholic NM, Wigle TJ, et al.: Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110 (19): 7922-7, 2013.
- Guzzetta AA, Montgomery EA, Lyu H, et al.: Epithelioid sarcoma: one institution's experience with a rare sarcoma. J Surg Res 177 (1): 116-22, 2012.
- Hawkins DS, Spunt SL, Skapek SX, et al.: Children's Oncology Group's 2013 blueprint for research: Soft tissue sarcomas. Pediatr Blood Cancer 60 (6): 1001-8, 2013.
- Sparber-Sauer M, Koscielniak E, Vokuhl C, et al.: Epithelioid sarcoma in children, adolescents, and young adults: Localized, primary metastatic and relapsed disease. Treatment results of five Cooperative Weichteilsarkom Studiengruppe (CWS) trials and one registry. Pediatr Blood Cancer 66 (9): e27879, 2019.
- Spunt SL, Francotte N, De Salvo GL, et al.: Clinical features and outcomes of young patients with epithelioid sarcoma: an analysis from the Children's Oncology Group and the European paediatric soft tissue Sarcoma Study Group prospective clinical trials. Eur J Cancer 112: 98-106, 2019.
- Casanova M, Ferrari A, Collini P, et al.: Epithelioid sarcoma in children and adolescents: a report from the Italian Soft Tissue Sarcoma Committee. Cancer 106 (3): 708-17, 2006.
- Gounder M, Schöffski P, Jones RL, et al.: Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open-label, phase 2 basket study. Lancet Oncol 21 (11): 1423-1432, 2020.
- Orbach D, Brennan B, Casanova M, et al.: Paediatric and adolescent alveolar soft part sarcoma: A joint series from European cooperative groups. Pediatr Blood Cancer 60 (11): 1826-32, 2013.
- Ferrari A, Sultan I, Huang TT, et al.: Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer 57 (6): 943-9, 2011.
- Wang HW, Qin XJ, Yang WJ, et al.: Alveolar soft part sarcoma of the oral and maxillofacial region: clinical analysis in a series of 18 patients. Oral Surg Oral Med Oral Pathol Oral Radiol 119 (4): 396-401, 2015.
- Kayton ML, Meyers P, Wexler LH, et al.: Clinical presentation, treatment, and outcome of alveolar soft part sarcoma in children, adolescents, and young adults. J Pediatr Surg 41 (1): 187-93, 2006.
- Sparber-Sauer M, Seitz G, von Kalle T, et al.: Alveolar soft-part sarcoma: Primary metastatic disease and metastatic relapse occurring during long-term follow-up: Treatment results of four Cooperative Weichteilsarkom Studiengruppe (CWS) trials and one registry. Pediatr Blood Cancer 65 (12): e27405, 2018.
- Flores RJ, Harrison DJ, Federman NC, et al.: Alveolar soft part sarcoma in children and young adults: A report of 69 cases. Pediatr Blood Cancer 65 (5): e26953, 2018.
- Ladanyi M, Lui MY, Antonescu CR, et al.: The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 20 (1): 48-57, 2001.
- Williams A, Bartle G, Sumathi VP, et al.: Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: useful diagnostic tools in cases with unusual histological features. Virchows Arch 458 (3): 291-300, 2011.
- Lieberman PH, Brennan MF, Kimmel M, et al.: Alveolar soft-part sarcoma. A clinico-pathologic study of half a century. Cancer 63 (1): 1-13, 1989.
- Casanova M, Ferrari A, Bisogno G, et al.: Alveolar soft part sarcoma in children and adolescents: A report from the Soft-Tissue Sarcoma Italian Cooperative Group. Ann Oncol 11 (11): 1445-9, 2000.
- Pennacchioli E, Fiore M, Collini P, et al.: Alveolar soft part sarcoma: clinical presentation, treatment, and outcome in a series of 33 patients at a single institution. Ann Surg Oncol 17 (12): 3229-33, 2010.
- Wilky BA, Trucco MM, Subhawong TK, et al.: Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: a single-centre, single-arm, phase 2 trial. Lancet Oncol 20 (6): 837-848, 2019.
- Stacchiotti S, Negri T, Zaffaroni N, et al.: Sunitinib in advanced alveolar soft part sarcoma: evidence of a direct antitumor effect. Ann Oncol 22 (7): 1682-90, 2011.
- Jagodzińska-Mucha P, Świtaj T, Kozak K, et al.: Long-term results of therapy with sunitinib in metastatic alveolar soft part sarcoma. Tumori 103 (3): 231-235, 2017.
- Cohen JW, Widemann BC, Derdak J, et al.: Cediranib phase-II study in children with metastatic alveolar soft-part sarcoma (ASPS). Pediatr Blood Cancer 66 (12): e27987, 2019.
- Judson I, Morden JP, Kilburn L, et al.: Cediranib in patients with alveolar soft-part sarcoma (CASPS): a double-blind, placebo-controlled, randomised, phase 2 trial. Lancet Oncol 20 (7): 1023-1034, 2019.
- Kummar S, Allen D, Monks A, et al.: Cediranib for metastatic alveolar soft part sarcoma. J Clin Oncol 31 (18): 2296-302, 2013.
- Kim M, Kim TM, Keam B, et al.: A Phase II Trial of Pazopanib in Patients with Metastatic Alveolar Soft Part Sarcoma. Oncologist 24 (1): 20-e29, 2019.
- Stacchiotti S, Mir O, Le Cesne A, et al.: Activity of Pazopanib and Trabectedin in Advanced Alveolar Soft Part Sarcoma. Oncologist 23 (1): 62-70, 2018.
- Roozendaal KJ, de Valk B, ten Velden JJ, et al.: Alveolar soft-part sarcoma responding to interferon alpha-2b. Br J Cancer 89 (2): 243-5, 2003.
- Conde N, Cruz O, Albert A, et al.: Antiangiogenic treatment as a pre-operative management of alveolar soft-part sarcoma. Pediatr Blood Cancer 57 (6): 1071-3, 2011.
- Meis-Kindblom JM: Clear cell sarcoma of tendons and aponeuroses: a historical perspective and tribute to the man behind the entity. Adv Anat Pathol 13 (6): 286-92, 2006.
- Dim DC, Cooley LD, Miranda RN: Clear cell sarcoma of tendons and aponeuroses: a review. Arch Pathol Lab Med 131 (1): 152-6, 2007.
- Blazer DG, Lazar AJ, Xing Y, et al.: Clinical outcomes of molecularly confirmed clear cell sarcoma from a single institution and in comparison with data from the Surveillance, Epidemiology, and End Results registry. Cancer 115 (13): 2971-9, 2009.
- Coindre JM, Hostein I, Terrier P, et al.: Diagnosis of clear cell sarcoma by real-time reverse transcriptase-polymerase chain reaction analysis of paraffin embedded tissues: clinicopathologic and molecular analysis of 44 patients from the French sarcoma group. Cancer 107 (5): 1055-64, 2006.
- Fujimura Y, Siddique H, Lee L, et al.: EWS-ATF-1 chimeric protein in soft tissue clear cell sarcoma associates with CREB-binding protein and interferes with p53-mediated trans-activation function. Oncogene 20 (46): 6653-9, 2001.
- Hisaoka M, Ishida T, Kuo TT, et al.: Clear cell sarcoma of soft tissue: a clinicopathologic, immunohistochemical, and molecular analysis of 33 cases. Am J Surg Pathol 32 (3): 452-60, 2008.
- Ferrari A, Casanova M, Bisogno G, et al.: Clear cell sarcoma of tendons and aponeuroses in pediatric patients: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Cancer 94 (12): 3269-76, 2002.
- Karita M, Tsuchiya H, Yamamoto N, et al.: Caffeine-potentiated chemotherapy for clear cell sarcoma: a report of five cases. Int J Clin Oncol 18 (1): 33-7, 2013.
- Schöffski P, Wozniak A, Stacchiotti S, et al.: Activity and safety of crizotinib in patients with advanced clear-cell sarcoma with MET alterations: European Organization for Research and Treatment of Cancer phase II trial 90101 'CREATE'. Ann Oncol 28 (12): 3000-3008, 2017.
- Tsuneyoshi M, Enjoji M, Iwasaki H, et al.: Extraskeletal myxoid chondrosarcoma--a clinicopathologic and electron microscopic study. Acta Pathol Jpn 31 (3): 439-47, 1981.
- Hachitanda Y, Tsuneyoshi M, Daimaru Y, et al.: Extraskeletal myxoid chondrosarcoma in young children. Cancer 61 (12): 2521-6, 1988.
- Enzinger FM, Shiraki M: Extraskeletal myxoid chondrosarcoma. An analysis of 34 cases. Hum Pathol 3 (3): 421-35, 1972.
- McGrory JE, Rock MG, Nascimento AG, et al.: Extraskeletal myxoid chondrosarcoma. Clin Orthop Relat Res (382): 185-90, 2001.
- Drilon AD, Popat S, Bhuchar G, et al.: Extraskeletal myxoid chondrosarcoma: a retrospective review from 2 referral centers emphasizing long-term outcomes with surgery and chemotherapy. Cancer 113 (12): 3364-71, 2008.
- Hisaoka M, Ishida T, Imamura T, et al.: TFG is a novel fusion partner of NOR1 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer 40 (4): 325-8, 2004.
- Stacchiotti S, Pantaleo MA, Astolfi A, et al.: Activity of sunitinib in extraskeletal myxoid chondrosarcoma. Eur J Cancer 50 (9): 1657-64, 2014.
- Leavey PJ, Laack NN, Krailo MD, et al.: Phase III Trial Adding Vincristine-Topotecan-Cyclophosphamide to the Initial Treatment of Patients With Nonmetastatic Ewing Sarcoma: A Children's Oncology Group Report. J Clin Oncol 39 (36): 4029-4038, 2021.
- Leuschner I, Radig K, Harms D: Desmoplastic small round cell tumor. Semin Diagn Pathol 13 (3): 204-12, 1996.
- Kushner BH, LaQuaglia MP, Wollner N, et al.: Desmoplastic small round-cell tumor: prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol 14 (5): 1526-31, 1996.
- Saab R, Khoury JD, Krasin M, et al.: Desmoplastic small round cell tumor in childhood: the St. Jude Children's Research Hospital experience. Pediatr Blood Cancer 49 (3): 274-9, 2007.
- Wang LL, Perlman EJ, Vujanic GM, et al.: Desmoplastic small round cell tumor of the kidney in childhood. Am J Surg Pathol 31 (4): 576-84, 2007.
- Hayes-Jordan A, LaQuaglia MP, Modak S: Management of desmoplastic small round cell tumor. Semin Pediatr Surg 25 (5): 299-304, 2016.
- Arora VC, Price AP, Fleming S, et al.: Characteristic imaging features of desmoplastic small round cell tumour. Pediatr Radiol 43 (1): 93-102, 2013.
- Gerald WL, Ladanyi M, de Alava E, et al.: Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round-cell tumor and its variants. J Clin Oncol 16 (9): 3028-36, 1998.
- Slotkin EK, Bowman AS, Levine MF, et al.: Comprehensive Molecular Profiling of Desmoplastic Small Round Cell Tumor. Mol Cancer Res 19 (7): 1146-1155, 2021.
- Chow WA, Yee JK, Tsark W, et al.: Recurrent secondary genomic alterations in desmoplastic small round cell tumors. BMC Med Genet 21 (1): 101, 2020.
- Lal DR, Su WT, Wolden SL, et al.: Results of multimodal treatment for desmoplastic small round cell tumors. J Pediatr Surg 40 (1): 251-5, 2005.
- Philippe-Chomette P, Kabbara N, Andre N, et al.: Desmoplastic small round cell tumors with EWS-WT1 fusion transcript in children and young adults. Pediatr Blood Cancer 58 (6): 891-7, 2012.
- Sedig L, Geiger J, Mody R, et al.: Paratesticular desmoplastic small round cell tumors: A case report and review of the literature. Pediatr Blood Cancer 64 (12): , 2017.
- Subbiah V, Lamhamedi-Cherradi SE, Cuglievan B, et al.: Multimodality Treatment of Desmoplastic Small Round Cell Tumor: Chemotherapy and Complete Cytoreductive Surgery Improve Patient Survival. Clin Cancer Res 24 (19): 4865-4873, 2018.
- Schwarz RE, Gerald WL, Kushner BH, et al.: Desmoplastic small round cell tumors: prognostic indicators and results of surgical management. Ann Surg Oncol 5 (5): 416-22, 1998 Jul-Aug.
- Goodman KA, Wolden SL, La Quaglia MP, et al.: Whole abdominopelvic radiotherapy for desmoplastic small round-cell tumor. Int J Radiat Oncol Biol Phys 54 (1): 170-6, 2002.
- Osborne EM, Briere TM, Hayes-Jordan A, et al.: Survival and toxicity following sequential multimodality treatment including whole abdominopelvic radiotherapy for patients with desmoplastic small round cell tumor. Radiother Oncol 119 (1): 40-4, 2016.
- Atallah V, Honore C, Orbach D, et al.: Role of Adjuvant Radiation Therapy After Surgery for Abdominal Desmoplastic Small Round Cell Tumors. Int J Radiat Oncol Biol Phys 95 (4): 1244-53, 2016.
- Hayes-Jordan AA, Coakley BA, Green HL, et al.: Desmoplastic Small Round Cell Tumor Treated with Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy: Results of a Phase 2 Trial. Ann Surg Oncol 25 (4): 872-877, 2018.
- Scalabre A, Philippe-Chomette P, Passot G, et al.: Cytoreductive surgery and hyperthermic intraperitoneal perfusion with chemotherapy in children with peritoneal tumor spread: A French nationwide study over 14 years. Pediatr Blood Cancer 65 (4): , 2018.
- Honoré C, Atallah V, Mir O, et al.: Abdominal desmoplastic small round cell tumor without extraperitoneal metastases: Is there a benefit for HIPEC after macroscopically complete cytoreductive surgery? PLoS One 12 (2): e0171639, 2017.
- Stiles ZE, Murphy AJ, Anghelescu DL, et al.: Desmoplastic Small Round Cell Tumor: Long-Term Complications After Cytoreduction and Hyperthermic Intraperitoneal Chemotherapy. Ann Surg Oncol 27 (1): 171-178, 2020.
- Cook RJ, Wang Z, Arora M, et al.: Clinical outcomes of patients with desmoplastic small round cell tumor of the peritoneum undergoing autologous HCT: a CIBMTR retrospective analysis. Bone Marrow Transplant 47 (11): 1455-8, 2012.
- Tarek N, Hayes-Jordan A, Salvador L, et al.: Recurrent desmoplastic small round cell tumor responding to an mTOR inhibitor containing regimen. Pediatr Blood Cancer 65 (1): , 2018.
- Kodet R, Newton WA, Sachs N, et al.: Rhabdoid tumors of soft tissues: a clinicopathologic study of 26 cases enrolled on the Intergroup Rhabdomyosarcoma Study. Hum Pathol 22 (7): 674-84, 1991.
- Biegel JA, Zhou JY, Rorke LB, et al.: Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59 (1): 74-9, 1999.
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011.
- Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012.
- Teplick A, Kowalski M, Biegel JA, et al.: Educational paper: screening in cancer predisposition syndromes: guidelines for the general pediatrician. Eur J Pediatr 170 (3): 285-94, 2011.
- Mitchell SG, Pencheva B, Porter CC: Germline Genetics and Childhood Cancer: Emerging Cancer Predisposition Syndromes and Psychosocial Impacts. Curr Oncol Rep 21 (10): 85, 2019.
- Foulkes WD, Kamihara J, Evans DGR, et al.: Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin Cancer Res 23 (12): e62-e67, 2017.
- Morgan KM, Siow VS, Strotmeyer S, et al.: Characteristics and Outcomes in Pediatric Non-Central Nervous System Malignant Rhabdoid Tumors: A Report from the National Cancer Database. Ann Surg Oncol 29 (1): 671-678, 2022.
- Sultan I, Qaddoumi I, Rodríguez-Galindo C, et al.: Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr Blood Cancer 54 (1): 35-40, 2010.
- Nemes K, Bens S, Kachanov D, et al.: Clinical and genetic risk factors define two risk groups of extracranial malignant rhabdoid tumours (eMRT/RTK). Eur J Cancer 142: 112-122, 2021.
- Puri DR, Meyers PA, Kraus DH, et al.: Radiotherapy in the multimodal treatment of extrarenal extracranial malignant rhabdoid tumors. Pediatr Blood Cancer 50 (1): 167-9, 2008.
- Madigan CE, Armenian SH, Malogolowkin MH, et al.: Extracranial malignant rhabdoid tumors in childhood: the Childrens Hospital Los Angeles experience. Cancer 110 (9): 2061-6, 2007.
- Bourdeaut F, Fréneaux P, Thuille B, et al.: Extra-renal non-cerebral rhabdoid tumours. Pediatr Blood Cancer 51 (3): 363-8, 2008.
- Wetmore C, Boyett J, Li S, et al.: Alisertib is active as single agent in recurrent atypical teratoid rhabdoid tumors in 4 children. Neuro Oncol 17 (6): 882-8, 2015.
- Folpe A, Inwards C, eds.: Bone and Soft Tissue Pathology: A Volume in the Foundations in Diagnostic Pathology. WB Saunders Co, 2010.
- Martignoni G, Pea M, Reghellin D, et al.: Molecular pathology of lymphangioleiomyomatosis and other perivascular epithelioid cell tumors. Arch Pathol Lab Med 134 (1): 33-40, 2010.
- Bissler JJ, McCormack FX, Young LR, et al.: Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 358 (2): 140-51, 2008.
- Davies DM, Johnson SR, Tattersfield AE, et al.: Sirolimus therapy in tuberous sclerosis or sporadic lymphangioleiomyomatosis. N Engl J Med 358 (2): 200-3, 2008.
- Agaram NP, Sung YS, Zhang L, et al.: Dichotomy of Genetic Abnormalities in PEComas With Therapeutic Implications. Am J Surg Pathol 39 (6): 813-25, 2015.
- Armah HB, Parwani AV: Perivascular epithelioid cell tumor. Arch Pathol Lab Med 133 (4): 648-54, 2009.
- Alaggio R, Cecchetto G, Martignoni G, et al.: Malignant perivascular epithelioid cell tumor in children: description of a case and review of the literature. J Pediatr Surg 47 (6): e31-40, 2012.
- Wagner AJ, Malinowska-Kolodziej I, Morgan JA, et al.: Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol 28 (5): 835-40, 2010.
- Wagner AJ, Ravi V, Riedel RF, et al.: nab-Sirolimus for Patients With Malignant Perivascular Epithelioid Cell Tumors. J Clin Oncol 39 (33): 3660-3670, 2021.
- Laetsch TW, Roy A, Xu L, et al.: Undifferentiated Sarcomas in Children Harbor Clinically Relevant Oncogenic Fusions and Gene Copy-Number Alterations: A Report from the Children's Oncology Group. Clin Cancer Res 24 (16): 3888-3897, 2018.
- Randall RL, Albritton KH, Ferney BJ, et al.: Malignant fibrous histiocytoma of soft tissue: an abandoned diagnosis. Am J Orthop 33 (12): 602-8, 2004.
- Fletcher CDM, Bridge JA, Hogendoorn P, et al., eds.: WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. IARC Press, 2013.
- Alaggio R, Collini P, Randall RL, et al.: Undifferentiated high-grade pleomorphic sarcomas in children: a clinicopathologic study of 10 cases and review of literature. Pediatr Dev Pathol 13 (3): 209-17, 2010 May-Jun.
- Daw NC, Billups CA, Pappo AS, et al.: Malignant fibrous histiocytoma and other fibrohistiocytic tumors in pediatric patients: the St. Jude Children's Research Hospital experience. Cancer 97 (11): 2839-47, 2003.
- Bjerkehagen B, Smeland S, Walberg L, et al.: Radiation-induced sarcoma: 25-year experience from the Norwegian Radium Hospital. Acta Oncol 47 (8): 1475-82, 2008.
- Le Guellec S, Chibon F, Ouali M, et al.: Are peripheral purely undifferentiated pleomorphic sarcomas with MDM2 amplification dedifferentiated liposarcomas? Am J Surg Pathol 38 (3): 293-304, 2014.
- Tawbi HA, Burgess M, Bolejack V, et al.: Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol 18 (11): 1493-1501, 2017.