Birt-Hogg-Dubé Syndrome (PDQ®): Genetics - Health Professional Information [NCI]
Clinical Manifestations
The three major features of Birt-Hogg-Dubé syndrome (BHD) include fibrofolliculomas /trichodiscomas, pulmonary cysts and spontaneous pneumothorax, and renal tumors.[
Cutaneous Lesions
Individuals with BHD usually present with multiple, small, skin-colored, dome-shaped papules distributed over the face, neck, and upper trunk. The characteristic dermatologic manifestation is a fibrofolliculoma or trichodiscoma (hamartoma of the hair follicle).[
Histologically, fibrofolliculomas/trichodiscomas are characterized by multiple anastomosing epithelial strands emanating from a central follicle. Mucin-rich or thick connective tissue stroma may encapsulate the epithelial component.[
Pulmonary Cysts and Spontaneous Pneumothorax
Computed tomography (CT) imaging results showed that lung cysts are present in 85% to 87% of patients with BHD.[
In a study of 198 patients with BHD, spontaneous pneumothorax occurrences were comparable between men (20%) and women (29%). Initial pneumothoraxes occurred between the ages of 22 and 75 years.[
The clinical presentation of spontaneous pneumothorax ranges from asymptomatic to dyspnea and chest pain. Clinical findings include tachypnea or decreased-to-absent breath sounds. Radiographic investigation may require a high-resolution chest CT to confirm the diagnosis because a chest x-ray may not be sensitive enough to detect a loculated pneumothorax. Up to 75% of patients with a history of spontaneous pneumothorax experience a second one. Differences in reported spontaneous pneumothorax recurrence may reflect the efficacy of different treatment modalities.
Histologic findings of pleuropulmonary lesions associated with BHD patients include thin-walled pleural and subpleural cysts and bullae, intraparenchymal air cysts, pleural blebs and changes consistent with spontaneous pneumothorax, and underlying emphysematous changes in lung tissue parenchyma adjacent to the bullae.[
Renal Tumors
Approximately 25% to 35% of individuals with BHD develop renal tumors,[
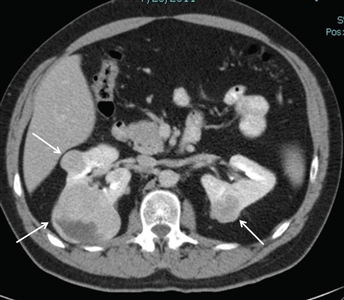
Figure 1. Birt-Hogg-Dubé syndrome–associated renal tumors are commonly multifocal and bilateral. Arrows indicate the locations of the tumors.
The most common tumors are a hybrid of oncocytoma and chromophobe histologic cell types, so-called oncocytic hybrid tumors, chromophobe RCC, and renal oncocytoma. Only renal oncocytoma is considered a benign tumor.[
Among 70 BHD patients with renal tumors and an FLCN pathogenic variant seen at the National Institutes of Health and identified through a literature review, 5 (7%) reportedly died from metastatic RCC.[
Other Manifestations
Bilateral multifocal parotid oncocytomas [
It should be noted that germline FLCNvariants were also found in patients without cutaneous findings but suspected of having BHD because of their specific renal and pulmonary manifestations.[
Lipomas, angiolipomas,[
Although initial epidemiologic observations linked BHD to an increased risk for colon polyps, subsequent epidemiologic studies did not confirm this association.[
References:
- Toro JR, Wei MH, Glenn GM, et al.: BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 45 (6): 321-31, 2008.
- Schmidt LS, Nickerson ML, Warren MB, et al.: Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet 76 (6): 1023-33, 2005.
- Schmidt LS, Linehan WM: Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol 12 (10): 558-69, 2015.
- Toro JR, Glenn G, Duray P, et al.: Birt-Hogg-Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol 135 (10): 1195-202, 1999.
- Graham RB, Nolasco M, Peterlin B, et al.: Nonsense mutations in folliculin presenting as isolated familial spontaneous pneumothorax in adults. Am J Respir Crit Care Med 172 (1): 39-44, 2005.
- Painter JN, Tapanainen H, Somer M, et al.: A 4-bp deletion in the Birt-Hogg-Dubé gene (FLCN) causes dominantly inherited spontaneous pneumothorax. Am J Hum Genet 76 (3): 522-7, 2005.
- Vernooij M, Claessens T, Luijten M, et al.: Birt-Hogg-Dubé syndrome and the skin. Fam Cancer 12 (3): 381-5, 2013.
- Toro JR, Pautler SE, Stewart L, et al.: Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dubé syndrome. Am J Respir Crit Care Med 175 (10): 1044-53, 2007.
- Matsumoto K, Lim D, Pharoah PD, et al.: A systematic review assessing the existence of pneumothorax-only variants of FLCN. Implications for lifelong surveillance of renal tumours. Eur J Hum Genet 29 (11): 1595-1600, 2021.
- Zbar B, Alvord WG, Glenn G, et al.: Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomarkers Prev 11 (4): 393-400, 2002.
- Pavlovich CP, Walther MM, Eyler RA, et al.: Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol 26 (12): 1542-52, 2002.
- Pavlovich CP, Grubb RL, Hurley K, et al.: Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J Urol 173 (5): 1482-6, 2005.
- Shuch B, Vourganti S, Ricketts CJ, et al.: Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 32 (5): 431-7, 2014.
- Sattler EC, Reithmair M, Steinlein OK: Kidney cancer characteristics and genotype-phenotype-correlations in Birt-Hogg-Dubé syndrome. PLoS One 13 (12): e0209504, 2018.
- Howlader N, Noone AM, Krapcho M, et al.: SEER Cancer Statistics Review (CSR) 1975-2017. Bethesda, Md: National Cancer Institute, 2020.
Available online . Last accessed August 9, 2024. - Vocke CD, Yang Y, Pavlovich CP, et al.: High frequency of somatic frameshift BHD gene mutations in Birt-Hogg-Dubé-associated renal tumors. J Natl Cancer Inst 97 (12): 931-5, 2005.
- da Silva NF, Gentle D, Hesson LB, et al.: Analysis of the Birt-Hogg-Dubé (BHD) tumour suppressor gene in sporadic renal cell carcinoma and colorectal cancer. J Med Genet 40 (11): 820-4, 2003.
- Khoo SK, Kahnoski K, Sugimura J, et al.: Inactivation of BHD in sporadic renal tumors. Cancer Res 63 (15): 4583-7, 2003.
- Liu V, Kwan T, Page EH: Parotid oncocytoma in the Birt-Hogg-Dubé syndrome. J Am Acad Dermatol 43 (6): 1120-2, 2000.
- Vinit J, Friedel J, Bielefeld P, et al.: [Birt-Hogg-Dubé syndrome and multiple recurrent tumors]. Rev Med Interne 32 (3): e40-2, 2011.
- Maffé A, Toschi B, Circo G, et al.: Constitutional FLCN mutations in patients with suspected Birt-Hogg-Dubé syndrome ascertained for non-cutaneous manifestations. Clin Genet 79 (4): 345-54, 2011.
- Chung JY, Ramos-Caro FA, Beers B, et al.: Multiple lipomas, angiolipomas, and parathyroid adenomas in a patient with Birt-Hogg-Dube syndrome. Int J Dermatol 35 (5): 365-7, 1996.
- Vincent A, Farley M, Chan E, et al.: Birt-Hogg-Dubé syndrome: two patients with neural tissue tumors. J Am Acad Dermatol 49 (4): 717-9, 2003.
- Drummond C, Grigoris I, Dutta B: Birt-Hogg-Dubé syndrome and multinodular goitre. Australas J Dermatol 43 (4): 301-4, 2002.
- Welsch MJ, Krunic A, Medenica MM: Birt-Hogg-Dubé Syndrome. Int J Dermatol 44 (8): 668-73, 2005.
- Tomassetti S, Carloni A, Chilosi M, et al.: Pulmonary features of Birt-Hogg-Dubé syndrome: cystic lesions and pulmonary histiocytoma. Respir Med 105 (5): 768-74, 2011.
- Walter P, Kirchhof B, Korge B, et al.: Flecked chorioretinopathy associated with Birt-Hogg-Dubé syndrome. Graefes Arch Clin Exp Ophthalmol 235 (6): 359-61, 1997.
- Nahorski MS, Lim DH, Martin L, et al.: Investigation of the Birt-Hogg-Dube tumour suppressor gene (FLCN) in familial and sporadic colorectal cancer. J Med Genet 47 (6): 385-90, 2010.
- van de Beek I, Glykofridis IE, Wolthuis RMF, et al.: No evidence for increased prevalence of colorectal carcinoma in 399 Dutch patients with Birt-Hogg-Dubé syndrome. Br J Cancer 122 (4): 590-594, 2020.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
Page Footer
I want to...
Audiences
Secure Member Sites
The Cigna Group Information
Disclaimer
Individual and family medical and dental insurance plans are insured by Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc., and Cigna HealthCare of Texas, Inc. Group health insurance and health benefit plans are insured or administered by CHLIC, Connecticut General Life Insurance Company (CGLIC), or their affiliates (see
All insurance policies and group benefit plans contain exclusions and limitations. For availability, costs and complete details of coverage, contact a licensed agent or Cigna sales representative. This website is not intended for residents of New Mexico.