Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumor Treatment (PDQ®): Treatment - Health Professional Information [NCI]
General Information About Childhood Central Nervous System (CNS) Atypical Teratoid / Rhabdoid Tumor
Primary brain tumors, including atypical teratoid/rhabdoid tumors (AT/RTs), are a diverse group of diseases that together constitute the most common solid tumors of childhood. The PDQ childhood brain tumor treatment summaries are primarily organized according to the World Health Organization classification of nervous system tumors.[
CNS AT/RT is a rare, clinically aggressive tumor that most often affects children aged 3 years and younger but can occur in older children and adults. Approximately one-half of AT/RTs arise in the posterior fossa.[
Based on current biological understanding, AT/RT is part of a larger family of rhabdoid tumors. In this summary, the term AT/RT refers to CNS tumors only, and the term rhabdoid tumor reflects the possibility of both CNS and non-CNS tumors. Unless specifically noted in the text, this summary refers to CNS AT/RT.
Childhood and adolescent cancer survivors require close monitoring because side effects of cancer therapy may persist or develop months or years after treatment. For specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see
Incidence
The exact incidence of childhood CNS AT/RT is difficult to determine because the tumor is rare and has only been recognized since 1996.[
- In two prospective studies performed by the Children's Cancer Group and the Pediatric Oncology Group in North America, retrospective review disclosed that approximately 10% of children aged 3 years or younger at diagnosis with brain tumors had AT/RTs.[
9 ] - A study completed in Taiwan found that AT/RTs account for 26% of primitive or embryonal tumors in children younger than 3 years.[
10 ] - The Austrian Brain Tumor Registry (recruitment period, 1996–2006) confirmed that AT/RTs represented the sixth most common malignant brain tumor among 311 newly diagnosed children (6.1%), with a peak incidence during the first 2 years of life.[
11 ]
The incidence in older patients is unknown. However, in the Central Nervous System Atypical Teratoid/Rhabdoid Tumor Registry (AT/RT Registry), 12 of the 42 patients (29%) were older than 36 months at the time of diagnosis.[
Anatomy
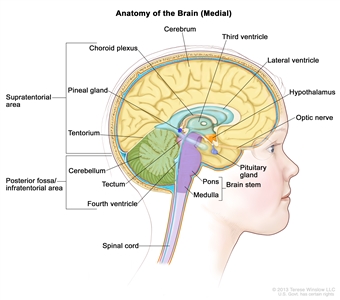
Anatomy of the inside of the brain, showing the pineal and pituitary glands, optic nerve, ventricles (with cerebrospinal fluid shown in blue), and other parts of the brain. The tentorium separates the cerebrum from the cerebellum. The infratentorium (posterior fossa) is the region below the tentorium that contains the brain stem, cerebellum, and fourth ventricle. The supratentorium is the region above the tentorium and denotes the region that contains the cerebrum.
Clinical Presentation
Childhood AT/RT is a clinically aggressive tumor that primarily occurs in children younger than 3 years, but it also can occur in older children and adults.[
Approximately one-half of all AT/RTs arise in the posterior fossa, although they can occur anywhere in the CNS.[
Because AT/RTs grow rapidly, patients often have a fairly short history of progressive symptoms, measured in days to weeks. Signs and symptoms depend on tumor location. Young patients with posterior fossa tumors usually present with symptoms related to hydrocephalus, which include the following:
- Early-morning headaches.
- Vomiting.
- Lethargy.
- Increased head circumference.
They may also develop ataxia, regression of motor skills, or localizing symptoms related to cranial nerve dysfunction.
Registry data suggest that 25% to 30% of patients present with disseminated disease.[
Diagnostic Evaluation
All patients with suspected AT/RT should undergo MRI of the brain and spine. Unless medically contraindicated, the lumbar cerebrospinal fluid should be inspected for evidence of tumor. Patients may also undergo renal ultrasonography to detect synchronous tumors. Germline testing is also indicated.
AT/RTs cannot be reliably distinguished from other malignant brain tumors on the basis of clinical history or radiographic evaluation alone. Surgery is necessary to obtain tissue and confirm the diagnosis. Immunohistochemical staining for loss of SMARCB1 protein expression is also used to confirm the diagnosis.[
Prognosis
Prognostic factors that affect survival for patients with AT/RTs are not fully delineated.
Known factors associated with a poor outcome include the following:
- Germline variant.[
23 ] - Younger age, especially younger than 1 year.[
5 ,24 ] - Metastases at diagnosis.[
24 ] - Subtotal resection.[
25 ] - Specific AT/RT molecular subtypes.[
3 ,5 ,26 ]
Most published data on outcomes of patients with AT/RT are from small series and are retrospective in nature. Initial retrospective studies reported an average survival from diagnosis of only about 12 months.[
There are reports of long-term survivors.[
- Children aged 3 years and older with AT/RT who received postoperative craniospinal irradiation and high-dose, alkylator-based chemotherapy had improved survival compared with patients younger than 3 years. In this report, the incidence of leptomeningeal metastases was also higher in the infant patients.[
29 ] - In one prospective study of 25 children with AT/RT who received intensive multimodality therapy, including radiation and intrathecal chemotherapy, the reported 2-year progression-free survival rate was 53%, and the OS rate was 70%.[
30 ] - For patients in the prospective ACNS0333 (NCT00653068) trial, the 4-year event-free survival rate was 37%, and the 4-year OS rate was 43%.[
31 ] - In the prospective European Rhabdoid Registry series, patients aged 1 year and older with an AT/RT tyrosinase (TYR) subgroup designation demonstrated a 5-year OS rate of 71%, while those younger than 1 year with a non-TYR AT/RT had a very poor survival rate.[
5 ] These data were confirmed in two other trials.[26 ]
References:
- Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
- WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
- Federico A, Thomas C, Miskiewicz K, et al.: ATRT-SHH comprises three molecular subgroups with characteristic clinical and histopathological features and prognostic significance. Acta Neuropathol 143 (6): 697-711, 2022.
- Lu VM, Di L, Eichberg DG, et al.: Age of diagnosis clinically differentiates atypical teratoid/rhabdoid tumors diagnosed below age of 3 years: a database study. Childs Nerv Syst 37 (4): 1077-1085, 2021.
- Frühwald MC, Hasselblatt M, Nemes K, et al.: Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro Oncol 22 (7): 1006-1017, 2020.
- Dho YS, Kim SK, Cheon JE, et al.: Investigation of the location of atypical teratoid/rhabdoid tumor. Childs Nerv Syst 31 (8): 1305-11, 2015.
- Hasselblatt M, Nagel I, Oyen F, et al.: SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128 (3): 453-6, 2014.
- Rorke LB, Packer RJ, Biegel JA: Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 85 (1): 56-65, 1996.
- Packer RJ, Biegel JA, Blaney S, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol 24 (5): 337-42, 2002 Jun-Jul.
- Ho DM, Hsu CY, Wong TT, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol 99 (5): 482-8, 2000.
- Woehrer A, Slavc I, Waldhoer T, et al.: Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian Brain Tumor Registry, 1996-2006. Cancer 116 (24): 5725-32, 2010.
- Hilden JM, Meerbaum S, Burger P, et al.: Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 22 (14): 2877-84, 2004.
- Burger PC, Yu IT, Tihan T, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol 22 (9): 1083-92, 1998.
- Lutterbach J, Liegibel J, Koch D, et al.: Atypical teratoid/rhabdoid tumors in adult patients: case report and review of the literature. J Neurooncol 52 (1): 49-56, 2001.
- Lobón-Iglesias MJ, Andrianteranagna M, Han ZY, et al.: Imaging and multi-omics datasets converge to define different neural progenitor origins for ATRT-SHH subgroups. Nat Commun 14 (1): 6669, 2023.
- Bartelheim K, Nemes K, Seeringer A, et al.: Improved 6-year overall survival in AT/RT - results of the registry study Rhabdoid 2007. Cancer Med 5 (8): 1765-75, 2016.
- Biegel JA, Fogelgren B, Wainwright LM, et al.: Germline INI1 mutation in a patient with a central nervous system atypical teratoid tumor and renal rhabdoid tumor. Genes Chromosomes Cancer 28 (1): 31-7, 2000.
- Bourdeaut F, Lequin D, Brugières L, et al.: Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 17 (1): 31-8, 2011.
- Seeringer A, Reinhard H, Hasselblatt M, et al.: Synchronous congenital malignant rhabdoid tumor of the orbit and atypical teratoid/rhabdoid tumor--feasibility and efficacy of multimodal therapy in a long-term survivor. Cancer Genet 207 (9): 429-33, 2014.
- Nemes K, Clément N, Kachanov D, et al.: The extraordinary challenge of treating patients with congenital rhabdoid tumors-a collaborative European effort. Pediatr Blood Cancer 65 (6): e26999, 2018.
- Bruggers CS, Bleyl SB, Pysher T, et al.: Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatr Blood Cancer 56 (7): 1026-31, 2011.
- Margol AS, Judkins AR: Pathology and diagnosis of SMARCB1-deficient tumors. Cancer Genet 207 (9): 358-64, 2014.
- Kordes U, Gesk S, Frühwald MC, et al.: Clinical and molecular features in patients with atypical teratoid rhabdoid tumor or malignant rhabdoid tumor. Genes Chromosomes Cancer 49 (2): 176-81, 2010.
- Dufour C, Beaugrand A, Le Deley MC, et al.: Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer 118 (15): 3812-21, 2012.
- Lafay-Cousin L, Hawkins C, Carret AS, et al.: Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer 48 (3): 353-9, 2012.
- Upadhyaya SA, Robinson GW, Onar-Thomas A, et al.: Relevance of Molecular Groups in Children with Newly Diagnosed Atypical Teratoid Rhabdoid Tumor: Results from Prospective St. Jude Multi-institutional Trials. Clin Cancer Res 27 (10): 2879-2889, 2021.
- Athale UH, Duckworth J, Odame I, et al.: Childhood atypical teratoid rhabdoid tumor of the central nervous system: a meta-analysis of observational studies. J Pediatr Hematol Oncol 31 (9): 651-63, 2009.
- Olson TA, Bayar E, Kosnik E, et al.: Successful treatment of disseminated central nervous system malignant rhabdoid tumor. J Pediatr Hematol Oncol 17 (1): 71-5, 1995.
- Tekautz TM, Fuller CE, Blaney S, et al.: Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23 (7): 1491-9, 2005.
- Chi SN, Zimmerman MA, Yao X, et al.: Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 27 (3): 385-9, 2009.
- Reddy AT, Strother DR, Judkins AR, et al.: Efficacy of High-Dose Chemotherapy and Three-Dimensional Conformal Radiation for Atypical Teratoid/Rhabdoid Tumor: A Report From the Children's Oncology Group Trial ACNS0333. J Clin Oncol 38 (11): 1175-1185, 2020.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
Page Footer
I want to...
Audiences
Secure Member Sites
The Cigna Group Information
Disclaimer
Individual and family medical and dental insurance plans are insured by Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc., and Cigna HealthCare of Texas, Inc. Group health insurance and health benefit plans are insured or administered by CHLIC, Connecticut General Life Insurance Company (CGLIC), or their affiliates (see
All insurance policies and group benefit plans contain exclusions and limitations. For availability, costs and complete details of coverage, contact a licensed agent or Cigna sales representative. This website is not intended for residents of New Mexico.