Treatment of Tumors of Uncertain Differentiation
Tumors of uncertain differentiation have many subtypes, including the following:
- Myxoma not otherwise specified (NOS) (benign).
- Synovial sarcoma NOS (poorly differentiated, spindle cell, and biphasic varieties).
- Epithelioid sarcoma.
- Alveolar soft part sarcoma.
- Clear cell sarcoma NOS.
- Extraskeletal myxoid chondrosarcoma.
- Desmoplastic small round cell tumor.
- Rhabdoid tumor NOS (extrarenal).
- Perivascular epithelioid tumor (PEComa), malignant.
- Undifferentiated sarcoma.
- Pleomorphic sarcoma, undifferentiated.
- Intracranial mesenchymal tumor.
- Round cell sarcoma, undifferentiated.
Myxoma NOS
Carney complex
Carney complex is an autosomal dominant syndrome caused by variants in the PRKAR1A gene, located on chromosome 17.[1] The syndrome is characterized by cardiac and cutaneous myxomas, pale brown to brown lentigines, blue nevi, primary pigmented nodular adrenocortical disease causing Cushing syndrome, and a variety of endocrine and nonendocrine tumors, including pituitary adenomas, thyroid tumors, and large cell calcifying Sertoli cell tumor of the testis.[1,2,3] There are published surveillance guidelines for patients with Carney complex that include cardiac, testicular, and thyroid ultrasonography.
For patients with the Carney complex, prognosis depends on the frequency of recurrences of cardiac and skin myxomas and other tumors.
For more information about the treatment of conditions related to Carney complex, see the following summaries:
- Childhood Cardiac Tumors Treatment.
- Childhood Testicular Cancer Treatment.
- Childhood Thyroid Cancer Treatment.
Synovial Sarcoma NOS (Poorly Differentiated, Spindle Cell, and Biphasic Varieties)
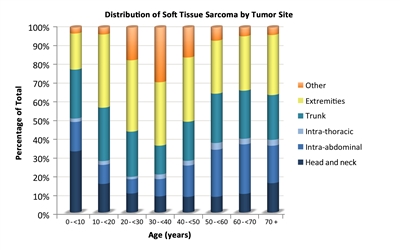
Synovial sarcoma accounts for 9% of soft tissue sarcomas in patients younger than 20 years (see Table 1).
Synovial sarcoma is one of the most common nonrhabdomyosarcomatous soft tissue sarcoma (NRSTS) in children and adolescents. In a review of the Surveillance, Epidemiology, and End Results (SEER) Program database from 1973 to 2005, 1,268 patients with synovial sarcoma were identified. Approximately 17% of these patients were children and adolescents, and the median age at diagnosis was 34 years.[4] In addition, in the Children's Oncology Group (COG) ARST0332 (NCT00346164) and European paediatric Soft Tissue Sarcoma Study Group (EpSSG) 2005 protocols, synovial sarcoma was the single most common histological subtype.[5]
Clinical presentation
The most common primary tumor location is the extremities, followed by trunk and head and neck.[4] Rarely, a synovial sarcoma may arise in the heart or pericardium or appear with a pleuropulmonary presentation.[6,7,8,9]
The most common site of metastasis is the lung.[10,11] The risk of metastases is highly influenced by tumor size. Patients with tumors that are larger than 5 cm have an estimated 32-fold higher risk of developing metastases compared with patients who have tumors smaller than 5 cm.
The Cooperative Weichteilsarkom Studiengruppe (CWS) reported on 432 patients younger than 21 years diagnosed with synovial sarcoma between 1981 and 2018.[12] The study compared three age groups of patients: children (aged 0–12 years; n = 176), adolescents (aged 13–16 years; n = 178), and young adults (aged 17–21 years; n = 78).
- The proportion of invasive tumors was significantly higher in older patients (children, 33%; adolescents, 39%; and young adults, 54%; P = .009).
- The proportion of tumors larger than 10 cm (children, 13%; adolescents, 21%; and young adults, 31%; P = .006) and the presence of metastasis at first diagnosis were also higher in older patients (children, 6%; adolescents, 10%; and young adults, 21%; P = .001).
Histological features, diagnostic evaluation, and genomic alterations
Synovial sarcoma can be subclassified as the following types:
- Synovial sarcoma, spindle cell.
- Synovial sarcoma, biphasic.
- Synovial sarcoma, poorly differentiated.
The diagnosis of synovial sarcoma is made by immunohistochemical analysis, ultrastructural findings, and demonstration of the specific chromosomal translocation t(x;18)(p11.2;q11.2). This abnormality is specific for synovial sarcoma and is found in all morphological subtypes. Synovial sarcoma results in rearrangement of the SS18 gene on chromosome 18 with one of the subtypes (1, 2, or 4) of the SSX gene on chromosome X.[13,14] It is thought that the SS18::SSX fusion transcript promotes epigenetic silencing of key tumor suppressor genes.[15]
In one report, reduced SMARCB1 nuclear reactivity on immunohistochemical staining was seen in 49 cases of synovial sarcoma, suggesting that this pattern may help distinguish synovial sarcoma from other histologies.[16]
Prognostic factors
Favorable prognostic factors
Patients younger than 10 years have more favorable outcomes and clinical features than older patients.
Favorable clinical features include the following:[4,17,18,19]
- Extremity primary tumors.
- Smaller tumors.
- Localized disease.
- Response to chemotherapy was correlated with improved survival in one meta-analysis.
Unfavorable prognostic factors
The following studies have reported multiple factors associated with unfavorable outcomes:
- In a retrospective analysis of synovial sarcoma in children and adolescents who were treated in Germany and Italy, tumor size (>5 cm or ≤5 cm in greatest dimension) was an important predictor of event-free survival (EFS).[20] In this analysis, local invasiveness conferred an inferior probability of EFS, but surgical margins were not associated with clinical outcome.
- In a single-institution retrospective analysis of 111 patients with synovial sarcoma who were younger than 22 years at diagnosis, larger tumor size, greater depth in tissue, greater local invasiveness, and more proximal tumor location were associated with poorer overall survival (OS).[21][Level of evidence C1]
- A multicenter analysis included 219 children from various treating centers, including Germany, St. Jude Children's Research Hospital (SJCRH), Instituto Tumori, and MD Anderson Cancer Center. The study reported an estimated 5-year OS rate of 80% and an EFS rate of 72%.[19] In this analysis, an interaction between tumor size and invasiveness was observed. In multivariate analysis, patients with large or invasive tumors or with Intergroup Rhabdomyosarcoma Study (IRS) group III disease (localized, incompletely resected or with biopsy only) and group IV disease (metastases at diagnosis) had decreased OS. Treatment with radiation therapy was related to improved OS (hazard ratio [HR], 0.4; 95% confidence interval [CI], 0.2–0.7). In patients with IRS group III disease, objective response to chemotherapy (18 of 30 [60%]) correlated with improved survival.
- Expression and genomic index prognostic signatures have been studied in synovial sarcoma. Complex genomic profiles, with greater rearrangement of the genome, are more common in adults than in younger patients with synovial sarcoma and are associated with a higher risk of metastasis.[22]
- A review of 84 patients with localized synovial sarcoma who had information on fusion status (SS18::SSX) and histological grading found no difference in OS according to these criteria. However, for tumor size at diagnosis, the study showed that patients with tumors between 5 cm and 10 cm had a worse prognosis than those with smaller tumors (P = .02). Patients with tumors larger than 10 cm had an even worse OS (P = .0003).[23][Level of evidence C1]
- The German CWS group reviewed 27 evaluable patients younger than 21 years with pulmonary metastases among 296 patients with synovial sarcoma. All patients had metastasis to the lungs. The 5-year EFS rate was 26%, and the OS rate was 30%. The most important prognostic factor at presentation was that the metastases were limited to one lesion in one lung or one lesion in both lungs (a group they termed oligometastatic). Treatment elements associated with superior survival were adequate local therapy of the primary tumor and, if feasible, for the metastases. The use of whole-lung irradiation did not correlate with better outcomes.[24][Level of evidence C1]
- The EpSSG designed a genomic index for synovial sarcoma.[25][Level of evidence C2] Genomic index was defined as A2 /C, where A is the total number of alterations (segmental gains and losses) and C is the number of involved chromosomes on array comparative genomic hybridization results. In a multivariate analysis of 61 pediatric, adolescent, and young adult patients (aged <25 years), high genomic index was an independent predictor of decreased EFS and OS.
- In adults, factors such as International Union Against Cancer/American Joint Committee on Cancer stage III and stage IVA, poor tumor necrosis, truncal location, elevated mitotic rate, older age, and higher histological grade have been associated with a worse prognosis.[26,27,28]
Treatment of synovial sarcoma
Treatment options for synovial sarcoma include the following:
- Surgery alone.
- Surgery and chemotherapy, with or without radiation therapy.[29,30]
Surgery alone
Evidence (surgery alone):
- The COG and the EpSSG reported a combined analysis of 60 patients younger than 21 years with localized synovial sarcoma prospectively assigned to surgery without adjuvant radiation therapy or chemotherapy.[31] Enrollment was limited to patients with initial complete resection with histologically free margins, with a grade 2 tumor of any size or a grade 3 tumor 5 cm or smaller.
- The 3-year EFS rate was 90% (median follow-up, 5.2 years; range, 1.9–9.1 years).
- All eight events were local tumor recurrence; no metastatic recurrences were seen.
- All patients with recurrent disease were effectively treated with second-line therapy, resulting in an OS rate of 100%.
- Therefore, the authors concluded that a surgery-only approach was optimal for patients who achieved an R0 resection (complete resection with negative microscopic margins) and had tumors smaller than 5 cm, regardless of grade.
Surgery and chemotherapy, with or without radiation therapy
Synovial sarcoma appears to be more sensitive to chemotherapy than many other soft tissue sarcomas. Children with synovial sarcoma seem to have a better prognosis than adults with synovial sarcoma.[11,28,32,33,34,35,36,37]
The most commonly used chemotherapy regimens for the treatment of synovial sarcoma incorporate ifosfamide and doxorubicin.[19,35,38] Response rates to the ifosfamide and doxorubicin regimen are higher than in other NRSTS.[39]
Evidence (surgery and chemotherapy with or without radiation therapy):
- Several treatment centers advocate chemotherapy after resection and radiation therapy for children and young adults with synovial sarcoma.[19,20,40,41,42]
- The International Society of Pediatric Oncology-Malignant Mesenchymal Tumors studies showed that select patients (young age, <5 cm resected tumors) with nonmetastatic synovial sarcoma treated with chemotherapy can have excellent outcomes in the absence of radiation therapy. However, it is still unclear whether that approach obviates an advantage of radiation for local or regional control.[41]
- A German trial suggested a benefit for postoperative chemotherapy in children with synovial sarcoma.[42]
- A meta-analysis also suggested that chemotherapy may provide benefit.[19]
- The COG reported an analysis of the subset of patients with synovial sarcoma treated on the ARST0332 (NCT00346164) trial. This was a prospective treatment assignment trial for patients younger than 30 years with NRSTS.[43] They analyzed the outcomes of 138 eligible patients.
- Overall, R0 resection or R1 resection (microscopically positive margins) of the primary tumor was achieved in 129 patients (93.5%): 69 patients (53.5%) at study entry and 60 patients (46.5%) after neoadjuvant chemotherapy. Of these, 104 patients (80.6%) had an R0 resection: 55 patients (53%) at study entry and 49 patients (47%) after neoadjuvant chemotherapy.
- In the 60 patients who received neoadjuvant chemotherapy, response was evaluable in 55 patients. Two patients (3.6%) had complete responses, 9 (16.4%) had partial responses, 41 (74.6%) had stable disease, and 3 (5.5%) had progressive disease. The tissue from 57 tumors was centrally reviewed after definitive resection. Forty-one tumors (72%) had less than 90% necrosis, and 16 tumors (28%) had 90% necrosis or more.
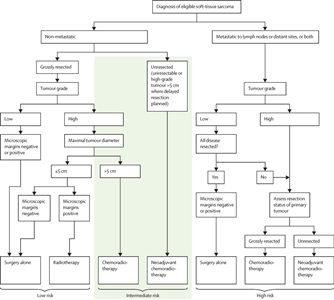
- The study prospectively defined three risk groups:
- Low risk (about 50% of population): Patients with nonmetastatic, grossly resected tumors, except patients who had tumors that were both high grade and >5 cm in maximal diameter.
- Intermediate risk (about 35% of population): Patients with nonmetastatic tumors that were both high grade and >5 cm in maximal diameter and patients with nonmetastatic, nonresectable tumors regardless of grade and size.
- High risk (about 15% of population): Patients with metastatic tumors, including those with metastases restricted to regional lymph nodes.
- For the 46 patients in the low-risk group, the 5-year EFS rate was 81.9% (95% CI, 69%–94.8%), and the OS rate was 97.7% (95% CI, 92.7%–100%).
- For the 23 patients in the intermediate-risk group (treatment arm C), the 5-year EFS rate was 64% (95% CI, 42.4%–85.8%), and the OS rate was 89.5% (95% CI, 75.3%–100%).
- For the 49 patients in the intermediate-risk group (treatment arm D), the 5-year EFS rate was 71.2% (95% CI, 56.5%–85.9%), and the OS rate was 86.5% (95% CI, 75.6%–97.3%).
- For the 21 patients in the high-risk group, the 5-year EFS rate was 7.6% (95% CI, 0%–22%), and the OS rate was 12.5% (95% CI, 0%–28.7%).
- The EpSSG performed a prospective study of patients younger than 21 years with synovial sarcoma (CCLG-EPSSG-NRSTS-2005 [NCT00334854]).[44][Level of evidence C1] Patients were stratified into the following three risk groups and nonrandomly assigned to treatment by risk group:
- Low-risk patients had IRS group I tumors less than 5 cm in size and nonaxial primary tumors.
- Intermediate-risk patients had no axial primary tumors and IRS group I tumors greater than 5 cm or IRS group II tumors.
- High-risk patients included all patients with axial primary sites (head and neck, lung and pleura, trunk, retroperitoneal), IRS group III tumors, or N1 tumors.
Outcomes for patients treated on the CCLG-EPSSG-NRSTS-2005 trial are described in Table 12.
Table 12. Event-Free Survival (EFS) and Overall Survival (OS) in Patients With Low-, Intermediate-, and High-Risk Synovial Sarcoma Treated on the CCLG-EPSSG-NRSTS-2005 Trial
| Risk Group |
Treatment |
3-Year EFS Rate (%) |
3-Year OS Rate (%) |
| IRS = Intergroup Rhabdomyosarcoma Study; RT = radiation therapy. |
| a Chemotherapy was ifosfamide/doxorubicin, with doxorubicin omitted during radiation therapy. |
| b 59.4 Gy in cases without the option of secondary resection; 50.4 Gy as preoperative radiation therapy; 50.4, 54, and 59.4 Gy as postoperative radiation therapy, in the case of R0, R1, and R2 resections, respectively (no additional radiation therapy in the case of secondary complete resections with free margins, in children younger than 6 years). |
| Low |
Surgery alone |
92 |
100 |
| Intermediate |
Surgery, 3–6 cycles chemotherapya, ± RTb |
91 |
100 |
| High (IRS group III) |
3 cycles of chemotherapya, surgery, 3 additional cycles of chemotherapy, ± RTb |
77 |
94 |
| High (axial primary sites) |
Surgery, 6 cycles of chemotherapya, RTb |
78 |
100 |
- The CWS reported results from a prospective trial for the treatment of patients with synovial sarcoma. Eligibility was restricted to patients with localized tumors with macroscopic residual disease after first surgery, before the initiation of systemic therapy (IRS III) and no clinically detectable metastatic disease. There were 145 patients in the study with a median age of 14.5 years (range, 0.2–33.2 years). The protocols recommended but did not require radiation therapy to be given before definitive tumor resection. Radiation therapy was administered to 115 patients (79%), and 23 patients did not receive radiation therapy (no information documented for 7 patients). Of the 115 irradiated patients, 57 were irradiated before tumor excision and 52 after tumor excision.[45]
- In this nonrandomized comparison, the sequencing of radiation therapy before definitive resection was associated with a statistically significant improvement in local recurrence-free survival rates, compared with definitive surgery before radiation therapy.
- Omission of radiation therapy was associated with an inferior outcome.
- Outcomes for patients are described in Table 13.
Table 13. Effects of Radiation Therapy Timing on Outcomes of Patients With Synovial Sarcoma
| Radiation Therapy |
Patients (No.) |
5-Year EFS Rate |
5-Year OS Rate |
5-Year Local Recurrence-Free Survival Rate |
| EFS = event-free survival; OS = overall survival. |
| No radiation therapy |
23 |
44% |
57% |
76% |
| Radiation therapy before surgery |
57 |
70% |
83% |
98% |
| Radiation therapy after surgery |
52 |
73% |
82% |
86% |
Recurrent synovial sarcoma NOS
For patients with recurrent synovial sarcoma, the survival rate after relapse is poor (30%–40% at 5 years). Factors associated with outcome after relapse include duration of first remission (> or ≤ 18 months) and lack of a second remission.[46,47]
In a German experience, surgical resection of metastatic disease was the most common way to achieve a second complete remission.[47] Maintenance chemotherapy with oral trofosfamide, idarubicin, and etoposide or oral cyclophosphamide and intravenous vinblastine was administered on an individual basis.
A consortium of six European referral centers reported a retrospective review of patients younger than 21 years with recurrent synovial sarcoma. Among 41 patients, the first relapses occurred within 3 to 132 months (median, 18 months) after first diagnoses. The relapses were local in 34% of patients, metastatic in 54%, and both in 12%. Treatments at first relapse included surgery in 56% of patients, radiation therapy in 34%, and systemic therapy in 88%. In all, 36 patients received second-line medical treatment, which included chemotherapy in 32 patients (with 10 different regimens) and targeted therapy in 4 patients. No patient was included in early-phase clinical trials as second-line therapy. The overall response rate was 42%. The median EFS was 12 months, and the postrelapse 5-year EFS rate was 15.8%. The median OS was 30 months, and the postrelapse 5-year OS rate was 22.2%. In a Cox multivariable regression analysis, OS was significantly associated with time and type of relapse.[48]
Radiation therapy (stereotactic body radiation therapy) can be used to target select pulmonary metastases. This is usually considered after a minimum of one resection to confirm metastatic disease. Radiation therapy is particularly appropriate for patients with lesions that threaten air exchange because of their location adjacent to bronchi or cause pain by invading the chest wall.[49]
Between 70% to 80% of synovial sarcomas express NY-ESO-1, an immunogenic cancer testis antigen.[50] NY-ESO-1 can be targeted with adoptive transfer of T cells engineered to express NY-ESO-1c259, an affinity-enhanced T-cell receptor (TCR) targeting NY-ESO-1/LAGE1a.[51] The procedure to produce the genetically engineered T cells restricts their reactivity to a single HLA type. All clinical trials of this technology chose HLA-A*02 as the initial target and limited eligibility to patients whose tumors expressed NY-ESO-1 and who had HLA-A*02. In a multi-institutional trial, confirmed antitumor responses occurred in 50% of patients (6 of 12) and were characterized by tumor shrinkage over several months. Circulating NY-ESO-1c259 T cells were present postinfusion in all patients, and the cells persisted for at least 6 months in all responders.[52]
An open-label, international, phase II study enrolled patients with previously treated metastatic or unresectable synovial sarcoma and myxoid round cell liposarcoma.[53] Fifty-two patients with HLA-A*02 and tumors that expressed melanoma-associated antigen A4 (MAGE-A4) received a single intravenous dose of afamitresgene autoleucel (afami-cel) after lymphodepletion. Afami-cel is a MAGE-A4–directed, genetically modified, autologous T-cell immunotherapy. The overall response rate (the primary end point of the study) was 37% (19 of 52; 95% CI, 24%–51%), 39% (17 of 44; 95% CI, 24%–55%) for patients with synovial sarcoma, and 25% (2 of 8; 95% CI, 3%–65%) for patients with myxoid round cell liposarcoma. Cytokine release syndrome and cytopenias were the most frequent side effects. The authors concluded that afami-cel treatment resulted in durable responses in heavily pretreated eligible patients with synovial sarcoma.[53] These data led to FDA approval of afami-cel in adults with unresectable or metastatic synovial sarcoma who have received prior chemotherapy, are HLA-A*02 positive, and whose tumors express the MAGE-A4 antigen.
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Epithelioid Sarcoma
Epithelioid sarcoma is a rare mesenchymal tumor of uncertain histogenesis that displays multilineage differentiation.[54]
Clinical presentation
Epithelioid sarcoma commonly presents as a slowly growing firm nodule based in the deep soft tissue. The proximal type predominantly affects adults and involves the axial skeleton and proximal sites. The tumor is highly aggressive and has a propensity for lymph node metastases.
Genomic alterations
Epithelioid sarcoma is characterized by inactivation of the SMARCB1 gene, which is present in both conventional and proximal types of epithelioid sarcoma.[55] This abnormality leads to increased dependence on EZH2 and tumor formation.[56]
Treatment of epithelioid sarcoma
Treatment options for epithelioid sarcoma include the following:
- Surgery with or without chemotherapy and/or radiation therapy.
- Targeted therapy.
Surgery with or without chemotherapy and/or radiation therapy
Surgical removal of primary and recurrent tumor(s) is the most effective treatment.[57][Level of evidence C1] Because of the propensity of this disease to have occult metastasis to the lymph nodes, sentinel lymph node biopsy is recommended for epithelioid sarcoma of the extremities or buttocks in the absence of clinically (by imaging or physical examination) enlarged lymph nodes.[58]
Evidence (surgery with or without chemotherapy and/or radiation therapy):
- In a German CWS retrospective analysis of 67 children, adolescents, and young adults (median age, 14 years) with epithelioid sarcoma, 53 patients presented with localized disease and 14 patients presented with metastatic disease.[59][Level of evidence C1] Fifty-eight of 67 patients were treated with primary resections. Resections were microscopically complete in 35 patients, microscopically incomplete in 12 patients, and macroscopically incomplete in 20 patients. Forty-nine patients received chemotherapy, and 33 patients received radiation therapy.
- Complete remission was achieved in 45 of 53 patients (85%) with localized disease.
- Twenty-seven patients with localized disease had local (n = 16), metastatic (n = 6), or combined (n = 4) relapses after a median time of 0.9 years (range, 0.1–2.3 years) after complete response of disease (45 of 63).
- Patients with localized disease had a 5-year EFS rate of 35% (95% CI, ±12%) and an OS rate of 48% (95% CI, ±14%).
- Patients with metastatic disease had a 5-year EFS rate of 7% (95% CI, ±14%) and an OS rate of 9% (95% CI, ±16%).
- Smaller tumor size, lower IRS group, less tumor invasiveness, negative nodal status, and microscopically complete resection correlated with a favorable prognosis in patients with localized disease.
- A retrospective analysis reviewed COG and EpSSG prospective clinical trials that enrolled patients younger than 30 years with epithelioid sarcoma.[60][Level of evidence B4] The analysis identified 63 patients who were treated between July 2005 and November 2015. Patients were stratified into three risk groups using a combination of clinical features and treatment received. Low-risk patients (n = 34) underwent surgery with or without radiation therapy and included predominantly patients with nonmetastatic widely or marginally resected tumors 5 cm or smaller. The intermediate-risk group included patients (n = 16) with nonmetastatic, high-grade, and larger than 5 cm tumors or unresectable tumors. Patients with nodal or distant metastatic disease were at high risk (n = 13) , regardless of tumor grade or size.
- Partial responses were observed in 11 of 22 patients (50%) who received neoadjuvant therapy.
- Events were local recurrence (n = 10) and distant recurrence (n = 15).
- The estimated 5-year OS rates were 86.4% for low-risk patients, 63.5% for intermediate-risk patients, and 0% for high-risk patients.
- Locoregional nodal involvement, invasive tumor, high grade, and lesser extent of resection predicted poorer EFS in patients without metastases.
- A review of 30 pediatric patients with epithelioid sarcoma (median age at presentation, 12 years) reported the following results:[61]
- Responses to chemotherapy were reported in 40% of patients using sarcoma-based treatment regimens.
- Sixty percent of patients were alive at 5 years after initial diagnosis.
- A single-institution retrospective review of 20 patients, which included children and adults (median age, 27.3 years), reported the following:[57]
- There was no difference in the probability of recurrence between patients who received chemotherapy and those who did not receive chemotherapy.
- The authors suggested that radiation therapy may be useful.
Targeted therapy
Evidence (tazemetostat):
- In a phase II trial of 62 adult patients with epithelioid sarcoma and documented loss of INI1 by immunohistochemistry or biallelic SMARCB1 (the gene that encodes INI1) alterations, tazemetostat showed clinical activity.[62]
- There were 9 of 62 confirmed partial responses, with an objective response rate of 15% and a disease control rate of 26%.
In January 2020, the U.S. Food and Drug Administration (FDA) granted accelerated approval to tazemetostat for adult and pediatric patients aged 16 years and older with metastatic or locally advanced epithelioid sarcoma who were not eligible for complete resection.
Treatment options under clinical evaluation for epithelioid sarcoma
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- PEPN2121 (NCT05286801) (Tiragolumab and Atezolizumab for the Treatment of Relapsed or Refractory SMARCB1- or SMARCA4-Deficient Tumors): This study is evaluating the combination of a PD-L1 targeting antibody (atezolizumab) with a TIGIT targeting antibody (tiragolumab) for patients with SMARCB1- or SMARCA4-deficient tumors. Patients with epithelioid sarcoma may be eligible for this study.
Alveolar Soft Part Sarcoma
Alveolar soft part sarcomas account for 1.4% of soft tissue sarcomas in patients younger than 20 years (see Table 1).
Clinical presentation
The median age at presentation is 25 years for patients with alveolar soft part sarcoma. This tumor most commonly arises in the extremities but can occur in the oral and maxillofacial region.[63,64,65] Alveolar soft part sarcoma in children can present with evidence of metastatic disease.[66] Delayed metastases to the brain and lung are uncommon.[63]
In a series of 61 patients with alveolar soft part sarcoma who were treated in four consecutive CWS trials and the Soft Tissue Sarcoma Registry (SoTiSaR), 46 patients presented with localized disease and 15 patients had evidence of metastasis at diagnosis.[67]
Sixty-nine patients younger than 30 years with alveolar soft part sarcoma were treated between 1980 and 2014 at four major institutions. The median age at diagnosis was 17 years, and 64% of patients were female. The most common site of disease was the lower extremity, and 26 patients had an ASPSCR1::TFE3 gene translocation.[68]
Genomic alterations
This tumor of uncertain histogenesis is characterized by a consistent chromosomal translocation t(X;17)(p11.2;q25) that fuses the ASPSCR1 gene with the TFE3 gene.[69,70]
Prognosis
Alveolar soft part sarcoma in children may have an indolent course.[66] Patients with alveolar soft part sarcoma may relapse several years after a prolonged period of apparent remission.[67,71]
- In a series of 19 treated patients with alveolar soft part sarcoma, one study reported a 5-year OS rate of 80%. The OS rate was 91% for patients with localized disease, 100% for patients with tumors 5 cm or smaller, and 31% for patients with tumors larger than 5 cm.[72]
- In another series of 33 patients, the OS rate was 68% at 5 years from diagnosis and 53% at 10 years from diagnosis. Survival was better for patients with smaller tumors (≤5 cm) and completely resected tumors.[73][Level of evidence C1]
- A retrospective review of children and young adults younger than 30 years (median age, 17 years; range, 1.5–30 years) from four institutions identified 69 patients treated primarily with surgery between 1980 and 2014.[68][Level of evidence C1] The ASPSCR1::TFE3 translocation was present in all 26 patients tested. There were 19 patients with IRS group I tumors (28%), 7 patients with IRS group II tumors (10%), 5 patients with IRS group III tumors (7%), and 38 patients with IRS group IV tumors (55%). The 5-year EFS rate was 80%, and the OS rate was 87% for the 31 patients with localized tumors (IRS postsurgical groups I, II, and III). The 5-year EFS rate was 7%, and the OS rate was 61% for the 38 patients with metastatic tumors (IRS group IV).
- In a series of patients treated on consecutive studies from Germany, 15 of 61 patients (25%) presented with metastases, often miliary in nature. Despite lack of response to chemotherapy, the 5-year OS rate was 61%, with an EFS rate of 20%.[67]
Treatment of alveolar soft part sarcoma
Treatment options for alveolar soft part sarcoma include the following:
- Surgery with or without radiation therapy and chemotherapy.[29,30]
- Targeted therapy (tyrosine kinase inhibitors and checkpoint inhibitors).[74]
Surgery with or without radiation therapy and chemotherapy
The standard treatment approach is complete resection of the primary lesion.[72] If complete excision is not feasible, radiation therapy is administered.
Evidence (surgery with or without chemotherapy):
- A study from China reported on 18 patients with alveolar soft part sarcoma of the oral and maxillofacial region. Fifteen patients were younger than 30 years. Surgical removal with negative margins was the primary treatment.[65][Level of evidence C2]
- All patients survived, and only one patient had metastatic disease recurrence.
- In a series of patients treated on consecutive studies from Germany, the following was reported:[67]
- Progression-free survival (PFS) for patients without metastases on presentation appeared to improve with complete resection of the primary tumor.
- The 5-year EFS rate was 100% for patients with completely resected tumors, compared with 50% for patients with microscopic or gross residual disease.
- In a series of 51 pediatric patients aged 0 to 21 years with alveolar soft part sarcoma, the following was reported:[63][Level of evidence C1]
- The OS rate was 78% at 10 years, and the EFS rate was about 63%.
- Patients with localized disease (n = 37) had a 10-year OS rate of 87%.
- The 14 patients with metastases at diagnosis had a 10-year OS rate of 44%, partly resulting from the surgical removal of the primary tumor and lung metastases in some patients.
- Only 3 of 18 patients (17%) with measurable disease had a response to conventional antisarcoma chemotherapy, but two of four patients treated with sunitinib had a partial response.
Targeted therapy
Studies of targeted therapy (tyrosine kinase inhibitors and checkpoint inhibitors) have been done.
Sunitinib
Evidence (sunitinib):
- A small retrospective study of nine adult patients with metastatic alveolar soft part sarcoma treated with sunitinib reported partial responses in five patients and stable disease in two patients.[75][Level of evidence C3]
- In another study, 15 adult patients with alveolar soft part sarcoma were treated with sunitinib. Five patients were treated with sunitinib for longer than 2 years.[76][Level of evidence C1]
- Six patients experienced partial responses.
- The median PFS was 19 months, and the median OS was 56 months.
- The 5-year OS rate was 49%.
Cediranib
Cediranib is an inhibitor of all three known vascular epidermal growth factor receptors.
Evidence (cediranib):
- In a pediatric phase II trial of cediranib, using 70% of the adult maximum tolerated dose in patients younger than 16 years, the following was reported:[77][Level of evidence B4]
- Five of seven patients had stable disease for 14 months or longer.
- An international group performed a double-blind, placebo-controlled, randomized, phase II trial of cediranib in adolescent and adult patients with alveolar soft part sarcoma.[78][Level of evidence A1]
- Median percentage change in sum of target marker lesion diameters for the evaluable population was -8.3% (interquartile range [IQR], -26.5 to 5.9) for patients who received cediranib therapy, compared with 13.4% (IQR, 1.1–21.3) for patients who received the placebo (one-sided P = .0010).
- The authors concluded that cediranib is an active agent in patients with alveolar soft part sarcoma.
- In a phase II trial of cediranib, 15 of 43 adult patients (35%) with metastatic alveolar soft part sarcoma had partial responses.[79][Level of evidence C3]
Pazopanib
Evidence (pazopanib):
- In an open-label trial that evaluated the efficacy of pazopanib in six adult patients, one patient achieved a partial response and five patients had stable disease.[80]
- Another trial included 30 adult patients who were treated with pazopanib.[81]
- One patient experienced a complete response, seven patients experienced partial responses, and 17 patients had stable disease.
- The median PFS was 13.6 months.
Axitinib and pembrolizumab
Axitinib is a vascular endothelial growth factor receptor tyrosine kinase inhibitor. Pembrolizumab is an anti–PD-1 immune checkpoint inhibitor.
Evidence (axitinib and pembrolizumab):
- In one trial, adult patients with advanced sarcomas were treated with a combination of axitinib and pembrolizumab.[74]
- For the 12 patients with alveolar soft part sarcoma, the 3-month PFS rate was 73%.
- Six of eleven patients with evaluable disease had partial responses to axitinib.
Atezolizumab
Atezolizumab is a monoclonal antibody directed against PD-1 and PD-L1.
Evidence (atezolizumab):
- In a phase II trial, 52 patients older than 2 years with advanced alveolar soft part sarcoma were treated with atezolizumab.[82]
- Nineteen patients experienced a response, 18 had partial responses and 1 had a complete response.
- Based on these data, the FDA approved the use of atezolizumab in children older than 2 years with unresectable or metastatic alveolar soft part sarcoma.
Other therapies
There have been sporadic reports of objective responses to treatment with interferon-alpha and bevacizumab.[63,83,84]
Because these tumors are rare, all children with alveolar soft part sarcoma should be considered for enrollment in prospective clinical trials. Information about ongoing clinical trials is available from the NCI website.
Treatment options under clinical evaluation for alveolar soft part sarcoma
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Clear Cell Sarcoma NOS
Clear cell sarcoma (formerly called malignant melanoma of soft parts) is a rare soft tissue sarcoma that typically involves the deep soft tissues of the extremities. It is also called clear cell sarcoma of tendons and aponeuroses. The tumor often affects adolescents and young adults.
Clinical presentation
The tumor most commonly affects the lower extremity, particularly the foot, heel, and ankle.[85,86] It has a high propensity for nodal dissemination, especially metastases to regional lymph nodes (12%–43%).[86,87]
The tumor typically has an indolent clinical course. Patients who have small, localized tumors with low mitotic rate and intermediate histological grade have the best outcomes.[88]
Genomic alterations
Clear cell sarcoma of soft tissue is characterized by EWSR1::ATF1 or EWSR1::CREB1 gene fusions.[89,90]
Treatment of clear cell sarcoma of soft tissue
Treatment options for clear cell sarcoma of soft tissue include the following:
- Surgery with or without radiation therapy.[29,30]
- Targeted therapy.
Surgery with or without radiation therapy
Surgery with or without radiation therapy is the treatment of choice and offers the best chance for cure.
Evidence (surgery with or without radiation therapy):
- In a series of 28 pediatric patients reported by the Italian and German Soft Tissue Cooperative Studies, the median age at diagnosis was 14 years and the lower extremity was the most common primary site (50%).[91]; [92][Level of evidence C2]
- In this series, 12 of 13 patients with completely resected tumors were cured.
- For patients with more advanced disease, the outcome is poor and chemotherapy is rarely effective.
Targeted therapy
Evidence (targeted therapy):
- In a study by the European Organization for Research and Treatment of Cancer, 26 patients with clear cell sarcoma who had metastatic disease and documented EWSR1 rearrangements were treated with crizotinib.[93]
- One patient had a partial response, and 17 patients had stable disease.
Extraskeletal Myxoid Chondrosarcoma
Extraskeletal myxoid chondrosarcoma is relatively rare among soft tissue sarcomas, representing only 2.3% of all soft tissue sarcomas.[94] It has been reported in children and adolescents.[95]
The tumor has traditionally been considered to have low-grade malignant potential.[96] However, reports from large institutions showed that extraskeletal myxoid chondrosarcoma has significant malignant potential, especially if patients are monitored for a long time.[97,98] Patients tend to have slow protracted courses. Nodal involvement has been well described. Local recurrence (57%) and metastatic spread to lungs (26%) have been reported.[98]
Genomic alterations
Extraskeletal myxoid chondrosarcoma is a multinodular neoplasm. The rounded cells are arranged in cords and strands in a chondroitin sulfate myxoid background. Several cytogenetic abnormalities have been identified (see Table 2), with the most frequent being the EWSR1::NR4A3 gene fusion.[99]
Treatment of extraskeletal myxoid chondrosarcoma
Treatment options for extraskeletal myxoid chondrosarcoma include the following:
- Surgery.
- Radiation therapy.
Aggressive local control and resection of metastases led to OS rates of 87% at 5 years and 63% at 10 years. Tumors were relatively resistant to radiation therapy.[97] The therapeutic benefit of chemotherapy has not been established.
There may be potential genetic targets for small molecules, but these need to be studied as part of a clinical trial. In an adult study, six of ten patients who received sunitinib achieved partial responses.[100]
Extraskeletal Ewing Sarcoma
Almost one-fifth of patients with Ewing sarcoma will present with nonbone primary sites (extraosseous). Treatment for this tumor is the same as it is for patients with bone primary tumors.[101] For more information, see Ewing Sarcoma and Undifferentiated Small Round Cell Sarcomas of Bone and Soft Tissue Treatment.
Desmoplastic Small Round Cell Tumor
Desmoplastic small round cell tumor is a rare primitive sarcoma.
Clinical presentation
Desmoplastic small round cell tumor most frequently involves the peritoneum in the abdomen, pelvis, and/or peritoneum into the scrotal sac, but it may occur in the kidney or other solid organs.[102,103,104,105,106] Dozens to hundreds of intraperitoneal implants are often found. The tumor occurs predominantly in males (85%) and may spread to the lungs and elsewhere.[106,107]
Diagnostic evaluation
A large single-institution series of 65 patients compared computed tomography (CT) scans (n = 54) with positron emission tomography (PET)-CT scans (n = 11). PET-CT scans produced very few false-negative results and detected metastatic sites missed on conventional CT scans.[107]
Genomic alterations
Cytogenetic studies of these tumors have demonstrated the recurrent translocation t(11;22)(p13;q12), which has been characterized as a fusion of the WT1 and EWSR1 genes.[105,108] The EWSR1::WT1 fusion confirms the diagnosis of desmoplastic small round cell tumor. The average tumor variant burden is low for desmoplastic small round cell tumor (<1 variant per megabase), and recurring gene alterations other than the EWSR1::WT1 fusion are uncommon.[109] A small percentage of cases (approximately 3%) have activating variants in FGFR4, with amplification of FGFR4 observed at similar frequency.[109,110] Inactivating variants in TP53 and ARID1A are observed in a small percentage of desmoplastic small round cell tumor cases.[109,110]
Prognosis
The overall prognosis for patients with desmoplastic small round cell tumor remains extremely poor, with reported rates of death at 90%. Greater than 90% tumor resection either at presentation or after preoperative chemotherapy may be a favorable prognostic factor for OS.[111,112]; [113][Level of evidence C1] Response to neoadjuvant chemotherapy and complete resection (near 100%) is associated with improved outcome.[106,114]
Treatment of desmoplastic small round cell tumor
There is no standard approach to the treatment of desmoplastic small round cell tumor.
Treatment options for desmoplastic small round cell tumor include the following:
- Multimodality therapy (chemotherapy, surgery, and radiation therapy).
- Surgery with hyperthermic intraperitoneal chemotherapy (HIPEC).
- Other treatment options.
Multimodality therapy
Complete surgical resections are rare and usually performed in highly specialized centers, but are critical for any improved survival. Successful treatment modalities include neoadjuvant Ewing-type chemotherapy, followed by complete surgical resection of the extensive intra-abdominal tumors, followed by total abdominal radiation therapy. With this multimodality approach, survival can be achieved in 30% to 40% of patients at 5 years.[102,103,111,115,116,117,118]
Surgery with HIPEC
HIPEC is a local treatment method that may control more of the microscopic intra-abdominal disease. The theory is that the heated chemotherapy that is instilled in the abdominal cavity after surgical resection (at the time of surgery) provides synergistic cytotoxicity to any microscopic cells remaining in the abdomen.[119]
The addition of HIPEC to complete surgical resection (cytoreductive surgery) is a new technique first applied to children in 2006 in a phase I clinical trial. Cytoreductive surgery and HIPEC for desmoplastic small round cell tumors is part of a multidisciplinary approach and is only being done in highly specialized centers. Surgeries can last more than 12 hours, and technical aspects of this unique tumor resection should be considered.[119]
Evidence (surgery with HIPEC):
- A single-institution phase II study showed HIPEC to be a potentially promising addition to complete surgical resection. Fourteen patients with desmoplastic small round cell tumor and five patients with other sarcomas were enrolled. These highly selected patients had tumor limited to the abdominal cavity. They demonstrated a partial response to neoadjuvant Ewing-type chemotherapy, had complete surgical resections and received HIPEC using cisplatin. They also received adjuvant total-abdominal radiation therapy followed by adjuvant chemotherapy.[119]
- With this standardized approach, patients with desmoplastic small round cell tumors had an OS rate of 80% at 30 months and 40% at 50 months.
- Patients with desmoplastic small round cell tumors without liver metastasis had no intra-abdominal recurrences, whereas 87% of patients with liver metastasis or portal disease had a recurrence.
- In a retrospective study from centers in France, patients were treated with cytoreductive surgery and HIPEC. Twenty-two patients were selected, and the median age at diagnosis was 14.8 years (range, 4.2–17.6 years). Seven patients had peritoneal mesotheliomas, seven patients had desmoplastic small round cells tumors, and eight patients had other histological tumor types. A complete macroscopic resection (CC-0, where CC is completeness of cytoreduction) was achieved in 16 cases (73%). Four of the seven patients with desmoplastic small round cell tumors had complete resections.[120][Level of evidence C1]
- Sixteen patients (72%) experienced relapses after a median time of 9.6 months (range, 1.4–86.4 months).
- Nine patients (41%) died of relapsed disease after a median time of 5.3 months (range, 0.1–36.1 months).
- Another study from France reviewed the use of cytoreductive surgery and HIPEC for the treatment of patients with desmoplastic small round cell tumors who had disease limited to the abdomen. In 107 patients with desmoplastic small round cell tumors, 48 had no extraperitoneal metastasis and underwent cytoreductive surgery. Of 48 patients (mean age, 22 years), 38 (79%) received preoperative and/or postoperative chemotherapy, and 23 (48%) received postoperative whole-abdominopelvic radiation therapy. Intraperitoneal chemotherapy was administered to 11 patients (23%), 2 of whom received early postoperative intraperitoneal chemotherapy (EPIC) and 9 of whom received HIPEC.[121]
- After a median follow-up of 30 months, the median OS of the entire cohort was 42 months.
- The 2-year OS rate was 72%, and the 5-year OS rate was 19%.
- The 2-year disease-free survival (DFS) rate was 30%, and the 5-year DFS rate was 12%.
- Whole-abdominopelvic radiation therapy was the only variable associated with longer peritoneal recurrence-free survival and DFS after cytoreductive surgery.
- Of 11 patients who received intraperitoneal chemotherapy (HIPEC or EPIC), six different chemotherapy regimens were used. The survival or outcome of this group is not reported in the manuscript.
- The influence of HIPEC/EPIC on OS and DFS was not statistically significant, but standardized regimens were not used in all patients, making results difficult to determine.
- A single-institutional retrospective study reported on nine patients (median age, 19 years) with desmoplastic small round cell tumor. Most patients had widespread disease, including four patients with extra-abdominal disease and five patients with liver involvement. These nine patients underwent ten cytoreductive and HIPEC treatments. Additionally, seven patients also received radiation therapy, and three patients underwent stem cell transplant.[122]
- The 3-year relapse-free survival rate was 13%, and the OS rate was 55%.
- Therapy was often associated with prolonged hospitalizations.
- Long-term parenteral nutrition was required in eight patients for a median of 261 days.
- Other long-term complications included gastroparesis (n = 1), small bowel obstruction (n = 3), and hemorrhagic cystitis (n = 2).
Other treatment options
The Center for International Blood and Marrow Transplant Research analyzed patients with desmoplastic small round cell tumor in their registry who received consolidation with high-dose chemotherapy and autologous stem cell reconstitution.[123] While this retrospective registry analysis suggested some benefit to this approach, other investigators have abandoned the approach because of excessive toxicity and lack of efficacy.[111]
A single-institution study reported that five of five patients with recurrent desmoplastic small round cell tumor had partial responses to treatment with the combination of vinorelbine, cyclophosphamide, and temsirolimus.[124]
Rhabdoid Tumor NOS (Extrarenal)
Malignant rhabdoid tumors were first described in children with renal tumors in 1981. These tumors were later found in a variety of extrarenal sites. These tumors are uncommon and highly malignant, especially in children younger than 2 years. For more information, see the Rhabdoid Tumors of the Kidney section in Wilms Tumor and Other Childhood Kidney Tumors Treatment.
Extrarenal (extracranial) rhabdoid tumors account for 2% of soft tissue sarcomas in patients younger than 20 years (see Table 1).
Genetic and genomic alterations
The first sizeable series of children with extrarenal extracranial malignant rhabdoid tumor of soft tissues came from 26 patients enrolled on the IRS I through III studies during a review of pathology material. Only five patients (19%) were alive without disease beyond 2 years.[125]
Investigation of children with atypical teratoid/rhabdoid tumors of the brain, as well as those with renal and extrarenal malignant rhabdoid tumors, found germline pathogenic variants and acquired variants of the SMARCB1 gene in all 29 tumors tested.[126] Rhabdoid tumors may be associated with germline pathogenic variants of the SMARCB1 gene and may be inherited from an apparently unaffected parent.[127] This observation was extended to 32 malignant rhabdoid tumors at all sites in patients whose mean age at diagnosis was 12 months.[128]
Genetic testing and surveillance
Germline analysis should be considered for individuals of all ages with rhabdoid tumors. Genetic counseling is also part of the treatment plan, given the low-but-actual risk of familial recurrence. In cases of variants, parental screening should be considered, although such screening carries a low probability of positivity. Prenatal diagnosis can be performed in situations where a specific SMARCB1 variant or deletion has been documented in the family.[127]
To date, there is little evidence regarding the effectiveness of surveillance for patients with rhabdoid tumor predisposition syndrome type 1 caused by loss-of-function germline SMARCB1 pathogenic variants. However, because of the aggressive nature of the tumors with significant lethality and young age of onset in SMARCB1 carriers with truncating variants, consensus recommendations have been developed. These recommendations were developed by a group of pediatric cancer genetic experts (including oncologists, radiologists, and geneticists). They have not been formally studied to confirm the benefit of monitoring patients with germline SMARCB1 pathogenic variants. Given the potential survival benefit of surgically resectable disease, it is postulated that early detection might improve OS.[129,130,131]
Surveillance for patients with germline SMARCB1 pathogenic variants includes the following:
- Brain magnetic resonance imaging (MRI) every 3 months from birth (or diagnosis) until age 5 years.
- Abdominal ultrasonography with a focus on the kidneys every 3 months.
For information about SMARCB1 and rhabdoid tumor predisposition syndrome type 1, see Rhabdoid Tumor Predisposition Syndrome Type 1.
Prognosis and clinical presentation
Young age and metastatic disease at presentation are associated with poor outcomes in children with extracranial rhabdoid tumors.
One study that used data from the National Cancer Database identified 202 patients (aged younger than 18 years) with non–central nervous system (CNS) malignant rhabdoid tumors. The primary site of the malignant rhabdoid tumor was soft tissue (46%), kidney (45%), and liver (9%).[132]
- The 1-year OS rate was 48.8%, and the 5-year OS rate was 35.9%.
- The multivariate analysis demonstrated that age younger than 1 year and presence of metastasis were negative prognostic indications (P = .058).
- In the cohort of surgical patients (n = 143), there was a trend for an association between the presence of residual disease and a clinically significant worse outcome (HR, 1.54; 95% CI, 0.88–2.69; P = .13).
A SEER study examined 229 patients with renal, CNS, and extrarenal malignant rhabdoid tumor. Patients aged 2 to 18 years, patients with a limited extent of tumor, and patients who received radiation therapy had favorable outcomes compared with other patients (P < .002 for each comparison). Site of the primary tumor was not prognostically significant. The OS rate was 33% at 5 years.[133]
A European registry for extracranial rhabdoid tumors identified 100 patients from 14 countries between 2009 and 2018.[134] Half of the patients were younger than 1 year at diagnosis. In 30 patients (30%), the tumor was located in the kidneys. Extracranial, extrarenal malignant rhabdoid tumor was found in 70% of patients (70 of 100), and the most common locations were in the cervical region, thoracic region, and liver. Nine patients demonstrated synchronous tumors. Distant metastases at diagnosis were present in 35% of patients (35 of 100). SMARCB1 germline pathogenic variants were detected in 21% of patients (17 of 81 evaluable). The 5-year OS rate was 45.8% (± 5.4%), and the EFS rate was 35.2% (± 5.1%). In an adjusted multivariate model, presence of a germline pathogenic variant, metastasis, and lack of a gross-total resection were the strongest significant negative predictors of outcome.
Treatment of extrarenal (extracranial) rhabdoid tumor
Treatment options for extrarenal (extracranial) rhabdoid tumor include the following:[135,136,137][Level of evidence C1]
- Surgical removal when possible.
- Chemotherapy as used for soft tissue sarcomas (but no single regimen is currently accepted as best).
- Radiation therapy.
Responses to alisertib have been documented in four patients with CNS atypical teratoid/rhabdoid tumors.[138] For more information about CNS atypical teratoid/rhabdoid tumors, see Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumor Treatment.
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- PEPN2121 (NCT05286801) (Tiragolumab and Atezolizumab for the Treatment of Relapsed or Refractory SMARCB1- or SMARCA4-Deficient Tumors): This study is evaluating the combination of a PD-L1 targeting antibody (atezolizumab) with a TIGIT targeting antibody (tiragolumab) for patients with SMARCB1- or SMARCA4-deficient tumors. Patients with extrarenal (extracranial) rhabdoid tumors may be eligible for this study.
PEComa, Malignant
Clinical presentation
PEComas occur in various rare gastrointestinal, pulmonary, gynecological, and genitourinary sites. Soft tissue, visceral, and gynecological PEComas are more commonly seen in middle-aged female patients and are usually not associated with the tuberous sclerosis complex.[139] The disease course may be indolent.
Risk factors and molecular features
Benign PEComas are common in patients with tuberous sclerosis, an autosomal dominant syndrome that also predisposes to renal cell cancer and brain tumors. Tuberous sclerosis is caused by germline pathogenic inactivation of either TSC1 (9q34) or TSC2 (16p13.3), and the same tumor suppressor genes are inactivated somatically in sporadic PEComas.[140] Inactivation of either gene results in stimulation of the mTOR pathway, providing the basis for the treatment of nonsurgically curable tumors with similar genetic inactivation (lymphangioleiomyomatosis and angiomyolipoma) with mTOR inhibitors.[141,142] A small proportion of PEComas have TFE3 rearrangements with fusions involving various genes, including SFPQ and RAD51B.[143]
Prognosis
Most PEComas have a benign clinical course, but malignant behavior has been reported and can be predicted based on the size of the tumor, mitotic rate, and presence of necrosis.[144]
Treatment of PEComas
There are no standard treatment options. Treatment may include surgery or observation followed by surgery when the tumor is large.[145]
In tumors with evidence of mTORC1 activation and TSC1 or TSC2 loss, including lymphangioleiomyomatosis and angiomyolipoma,[141] clinical activity using mTOR inhibitors, such as sirolimus, has been well documented. In a small case series, three adult patients with PEComas responded to sirolimus.[146]
In a phase II trial, 34 patients with metastatic or locally advanced malignant PEComas were treated with sirolimus protein-bound particles for injectable suspension (albumin-bound) (nab-sirolimus). Of the 31 patients eligible for efficacy analysis, 12 (39%) had a response (1 complete response and 11 partial responses), 16 (52%) had stable disease, and 3 (10%) had progressive disease. Responses were rapid and durable. The median duration of response was not reached after a median follow-up of 2.5 years. Treatment was ongoing for 7 of 12 patients who responded to treatment (range, 5.6 months to longer than 47.2 months). Tumor variant profiling was completed for 25 specimens. Eight of nine patients with TSC2 variants responded to treatment, while only 2 of 16 patients without TSC2 variants responded. In addition, responses were noted in 10 of 17 patients with phospho-S6 (pS6) expression. No response was noted in eight patients without pS6 expression. The absence of pS6 expression reflects the lack of mTORC1 activation.[147][Level of evidence C1] In 2021, the FDA approved nab-sirolimus for adult patients with PEComas.
Undifferentiated Sarcoma
From 1972 to 2006, patients with undifferentiated soft tissue sarcoma were eligible for participation in rhabdomyosarcoma trials coordinated by the IRS group and the COG. The rationale was that patients with undifferentiated soft tissue sarcoma had sites of disease and outcomes that were similar to those in patients with alveolar rhabdomyosarcoma. Therapeutic trials for adults with soft tissue sarcoma include patients with undifferentiated soft tissue sarcoma and other histologies, which are treated similarly, using ifosfamide and doxorubicin, and sometimes with other chemotherapy agents, surgery, and radiation therapy.
In the COG ARST0332 (NCT00346164) trial, patients with high-grade undifferentiated sarcoma were treated with an ifosfamide- and doxorubicin-based regimen. Results for the patients with high-grade undifferentiated sarcoma were reported together with all high-grade soft tissue sarcomas in the trial. The estimated 5-year EFS rate was 64% and the OS rate was 77% for sarcomas classified as high grade by the Fédération Nationale des Centres de Lutte Contre Le Cancer (FNCLC).[5][Level of evidence C1]
In a report of 32 patients with undifferentiated soft tissue sarcomas who were enrolled on the ARST0332 (NCT00346164) trial, the median age at enrollment was 13.6 years, and two-thirds of the patients were male. The most common primary sites were the paraspinal region and extremities. Five patients presented with metastatic disease.[148]
- The 5-year EFS rate was 71%, and the OS rate was 83%.
- Of the nine children with low-risk disease (localized low-grade resected disease or localized high-grade disease <5 cm resected with negative margins) who were treated with surgery or radiation therapy only, the 5-year EFS rate was 65% and the OS rate was 100%, suggesting that patients with low-risk disease can be salvaged if the disease recurs.
- The remaining 23 patients had either intermediate-risk disease (resected high-grade tumor >5 cm, unresected high-grade tumor >5 cm) or high-risk disease (metastasis to lymph nodes or distant sites) and were treated with chemoradiation therapy and delayed surgery when feasible. The 5-year EFS rate was 73%, and the OS estimate was 77%.
- Copy number aberrations were common, most frequently involving loss of 1p (25%), gain of 1q (25%), gain of chromosome 8 (25%), and gain of chromosome 2 (16%). These alterations were more commonly seen in patients with intermediate-risk or high-risk tumors, and there was a strong association between loss of chromosome 1p or gain of chromosome 1q and inferior clinical outcomes. Co-occurrence of 1q gain and 1p loss was associated with a particularly poor clinical outcome (5-year EFS and OS rates of 20%). Next-generation sequencing identified oncogenic fusions in eight of ten tumor samples, which included BCOR and CIC rearrangements, as well as COL1A1::PDGFB, KIAA1549::BRAF, and SAMD5::SASH1 gene fusions.
Pleomorphic Sarcoma, Undifferentiated (Malignant Fibrous Histiocytoma)
At one time, malignant fibrous histiocytoma was the single most common histotype among adults with soft tissue sarcomas. Since it was first recognized in the early 1960s, malignant fibrous histiocytoma has been controversial, in terms of both its histogenesis and its validity as a clinico-pathological entity. The World Health Organization (WHO) classification no longer includes malignant fibrous histiocytoma as a distinct diagnostic category but rather as a subtype of an undifferentiated pleomorphic sarcoma.[149,150]
This entity accounts for 2% to 6% of all childhood soft tissue sarcomas.[151]
Clinical presentation
These tumors occur mainly in the second decade of life. In a series of ten patients, the median age was 10 years, and the tumor was most commonly located in the extremities. In this series, all tumors were localized, and five of nine patients (for whom follow-up was available) were alive and in first remission.[151]
In another series of 17 pediatric patients with malignant fibrous histiocytoma (now classified as undifferentiated pleomorphic sarcoma), the median age at diagnosis was 5 years and the extremities were involved in eight cases.[152] All patients with metastatic disease died, and two patients experienced a clinical response to a doxorubicin-based regimen.
For more information about the treatment of malignant fibrous histiocytoma of bone, see Osteosarcoma and Undifferentiated Pleomorphic Sarcoma of Bone Treatment.
Risk factors
These tumors can arise in previously irradiated sites or as a second malignancy in patients with retinoblastoma.[153]
Molecular features
An analysis of 70 patients who were diagnosed with malignant fibrous histiocytosis of no specific type, storiform or pleomorphic malignant fibrous histiocytoma, pleomorphic sarcoma, or undifferentiated pleomorphic sarcoma showed a highly complex karyotype with no specific recurrent aberrations.[154]
Undifferentiated sarcomas with 12q13–15 amplification, including MDM2 and CDK4, are best classified as dedifferentiated liposarcomas.[154] The relationship between this tumor and the family of undifferentiated/unclassified tumors with spindle cell morphology remains relatively undefined.
Treatment of newly diagnosed pleomorphic sarcoma
For information about the treatment of undifferentiated pleomorphic sarcoma of bone, see Osteosarcoma and Undifferentiated Pleomorphic Sarcoma of Bone Treatment.
Treatment of recurrent or refractory pleomorphic sarcoma
Treatment options for recurrent or refractory pleomorphic sarcoma include the following:
- Pembrolizumab.
The Sarcoma Alliance for Research through Collaboration conducted a phase II trial of the checkpoint inhibitor pembrolizumab in patients aged 18 years and older with recurrent soft tissue sarcoma.[155][Level of evidence C3]
- Seven of 40 patients (18%) with soft tissue sarcoma had an objective response.
- Four of ten patients (40%) with undifferentiated pleomorphic sarcoma, two of ten patients (20%) with liposarcoma, and one of ten patients (10%) with synovial sarcoma had objective responses.
- No patients with leiomyosarcoma (n = 10) had an objective response.
Intracranial Mesenchymal Tumor
Intracranial mesenchymal tumor, with the FET::CREB gene fusion, has previously been called angiomatoid fibrous histiocytoma (AFP) or intracranial myxoid mesenchymal tumor. The molecular findings suggest that these tumors are histological variants of intracranial mesenchymal tumor.[156] In one study, the tumors of 20 patients were separated into two epigenetic subgroups. Group A tumors clustered nearest to but independent of solitary fibrous tumor and occurred in adolescents and young adults. Group B tumors clustered nearest to but independent of clear cell sarcoma and occurred in children. Patients with group B tumors had an inferior survival compared with patients with group A tumors (4.5 vs. 49 months; P = .001).[157]
Round Cell Sarcoma, Undifferentiated
Undifferentiated small round cell sarcomas withBCORgenetic alterations
See the sections on Undifferentiated Small Round Cell Sarcomas With BCOR Genetic Alterations and Genomics of Ewing Sarcoma in Ewing Sarcoma and Undifferentiated Small Round Cell Sarcomas of Bone and Soft Tissue Treatment.
Undifferentiated small round cell sarcomas withCICgenetic alterations
See the sections on Undifferentiated Small Round Cell Sarcomas With CIC Genetic Alterations and Genomics of Ewing Sarcoma in Ewing Sarcoma and Undifferentiated Small Round Cell Sarcomas of Bone and Soft Tissue Treatment.
Undifferentiated small round cell sarcomas withEWSR1::non-ETS fusions
See the Undifferentiated Small Round Cell Sarcomas With EWSR1::non-ETS Fusions section in Ewing Sarcoma and Undifferentiated Small Round Cell Sarcomas of Bone and Soft Tissue Treatment.
References:
- Wilkes D, Charitakis K, Basson CT: Inherited disposition to cardiac myxoma development. Nat Rev Cancer 6 (2): 157-65, 2006.
- Carney JA, Young WF: Primary pigmented nodular adrenocortical disease and its associated conditions. Endocrinologist 2: 6-21, 1992.
- Ryan MW, Cunningham S, Xiao SY: Maxillary sinus melanoma as the presenting feature of Carney complex. Int J Pediatr Otorhinolaryngol 72 (3): 405-8, 2008.
- Sultan I, Rodriguez-Galindo C, Saab R, et al.: Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer 115 (15): 3537-47, 2009.
- Spunt SL, Million L, Chi YY, et al.: A risk-based treatment strategy for non-rhabdomyosarcoma soft-tissue sarcomas in patients younger than 30 years (ARST0332): a Children's Oncology Group prospective study. Lancet Oncol 21 (1): 145-161, 2020.
- Wang JG, Li NN: Primary cardiac synovial sarcoma. Ann Thorac Surg 95 (6): 2202-9, 2013.
- Chirmade PC, Parikh S, Anand A, et al.: Primary pleuropulmonary synovial sarcoma with brain metastases in a paediatric patient: an unusual presentation. Adv Respir Med 85 (4): 206-210, 2017.
- Frazier AA, Franks TJ, Pugatch RD, et al.: From the archives of the AFIP: Pleuropulmonary synovial sarcoma. Radiographics 26 (3): 923-40, 2006 May-Jun.
- Essary LR, Vargas SO, Fletcher CD: Primary pleuropulmonary synovial sarcoma: reappraisal of a recently described anatomic subset. Cancer 94 (2): 459-69, 2002.
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994.
- Ferrari A, De Salvo GL, Oberlin O, et al.: Synovial sarcoma in children and adolescents: a critical reappraisal of staging investigations in relation to the rate of metastatic involvement at diagnosis. Eur J Cancer 48 (9): 1370-5, 2012.
- Scheer M, Blank B, Bauer S, et al.: Synovial sarcoma disease characteristics and primary tumor sites differ between patient age groups: a report of the Cooperative Weichteilsarkom Studiengruppe (CWS). J Cancer Res Clin Oncol 146 (4): 953-960, 2020.
- van de Rijn M, Barr FG, Collins MH, et al.: Absence of SYT-SSX fusion products in soft tissue tumors other than synovial sarcoma. Am J Clin Pathol 112 (1): 43-9, 1999.
- Krsková L, Sumerauer D, Stejskalová E, et al.: A novel variant of SYT-SSX1 fusion gene in a case of spindle cell synovial sarcoma. Diagn Mol Pathol 16 (3): 179-83, 2007.
- Su L, Sampaio AV, Jones KB, et al.: Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell 21 (3): 333-47, 2012.
- Arnold MA, Arnold CA, Li G, et al.: A unique pattern of INI1 immunohistochemistry distinguishes synovial sarcoma from its histologic mimics. Hum Pathol 44 (5): 881-7, 2013.
- Vlenterie M, Ho VK, Kaal SE, et al.: Age as an independent prognostic factor for survival of localised synovial sarcoma patients. Br J Cancer 113 (11): 1602-6, 2015.
- Smolle MA, Parry M, Jeys L, et al.: Synovial sarcoma: Do children do better? Eur J Surg Oncol 45 (2): 254-260, 2019.
- Okcu MF, Munsell M, Treuner J, et al.: Synovial sarcoma of childhood and adolescence: a multicenter, multivariate analysis of outcome. J Clin Oncol 21 (8): 1602-11, 2003.
- Brecht IB, Ferrari A, Int-Veen C, et al.: Grossly-resected synovial sarcoma treated by the German and Italian Pediatric Soft Tissue Sarcoma Cooperative Groups: discussion on the role of adjuvant therapies. Pediatr Blood Cancer 46 (1): 11-7, 2006.
- Stanelle EJ, Christison-Lagay ER, Healey JH, et al.: Pediatric and adolescent synovial sarcoma: multivariate analysis of prognostic factors and survival outcomes. Ann Surg Oncol 20 (1): 73-9, 2013.
- Lagarde P, Przybyl J, Brulard C, et al.: Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol 31 (5): 608-15, 2013.
- Stegmaier S, Leuschner I, Poremba C, et al.: The prognostic impact of SYT-SSX fusion type and histological grade in pediatric patients with synovial sarcoma treated according to the CWS (Cooperative Weichteilsarkom Studie) trials. Pediatr Blood Cancer 64 (1): 89-95, 2017.
- Scheer M, Dantonello T, Hallmen E, et al.: Primary Metastatic Synovial Sarcoma: Experience of the CWS Study Group. Pediatr Blood Cancer 63 (7): 1198-206, 2016.
- Orbach D, Mosseri V, Pissaloux D, et al.: Genomic complexity in pediatric synovial sarcomas (Synobio study): the European pediatric soft tissue sarcoma group (EpSSG) experience. Cancer Med 7 (4): 1384-1393, 2018.
- Trassard M, Le Doussal V, Hacène K, et al.: Prognostic factors in localized primary synovial sarcoma: a multicenter study of 128 adult patients. J Clin Oncol 19 (2): 525-34, 2001.
- Guillou L, Benhattar J, Bonichon F, et al.: Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol 22 (20): 4040-50, 2004.
- Ferrari A, Gronchi A, Casanova M, et al.: Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer 101 (3): 627-34, 2004.
- Bahig H, Roberge D, Bosch W, et al.: Agreement among RTOG sarcoma radiation oncologists in contouring suspicious peritumoral edema for preoperative radiation therapy of soft tissue sarcoma of the extremity. Int J Radiat Oncol Biol Phys 86 (2): 298-303, 2013.
- Baldini EH, Wang D, Haas RL, et al.: Treatment Guidelines for Preoperative Radiation Therapy for Retroperitoneal Sarcoma: Preliminary Consensus of an International Expert Panel. Int J Radiat Oncol Biol Phys 92 (3): 602-12, 2015.
- Ferrari A, Chi YY, De Salvo GL, et al.: Surgery alone is sufficient therapy for children and adolescents with low-risk synovial sarcoma: A joint analysis from the European paediatric soft tissue sarcoma Study Group and the Children's Oncology Group. Eur J Cancer 78: 1-6, 2017.
- McGrory JE, Pritchard DJ, Arndt CA, et al.: Nonrhabdomyosarcoma soft tissue sarcomas in children. The Mayo Clinic experience. Clin Orthop (374): 247-58, 2000.
- Van Glabbeke M, van Oosterom AT, Oosterhuis JW, et al.: Prognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: an analysis of 2,185 patients treated with anthracycline-containing first-line regimens--a European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 17 (1): 150-7, 1999.
- Koscielniak E, Harms D, Henze G, et al.: Results of treatment for soft tissue sarcoma in childhood and adolescence: a final report of the German Cooperative Soft Tissue Sarcoma Study CWS-86. J Clin Oncol 17 (12): 3706-19, 1999.
- Pappo AS, Devidas M, Jenkins J, et al.: Phase II trial of neoadjuvant vincristine, ifosfamide, and doxorubicin with granulocyte colony-stimulating factor support in children and adolescents with advanced-stage nonrhabdomyosarcomatous soft tissue sarcomas: a Pediatric Oncology Group Study. J Clin Oncol 23 (18): 4031-8, 2005.
- Cecchetto G, Alaggio R, Dall'Igna P, et al.: Localized unresectable non-rhabdo soft tissue sarcomas of the extremities in pediatric age: results from the Italian studies. Cancer 104 (9): 2006-12, 2005.
- Pappo AS, Rao BN, Jenkins JJ, et al.: Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: the St. Jude Children's Research Hospital experience. Med Pediatr Oncol 33 (2): 76-82, 1999.
- Brennan B, Stevens M, Kelsey A, et al.: Synovial sarcoma in childhood and adolescence: a retrospective series of 77 patients registered by the Children's Cancer and Leukaemia Group between 1991 and 2006. Pediatr Blood Cancer 55 (1): 85-90, 2010.
- Ferrari A, Miceli R, Rey A, et al.: Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Cancer 47 (5): 724-31, 2011.
- Raney RB: Synovial sarcoma in young people: background, prognostic factors, and therapeutic questions. J Pediatr Hematol Oncol 27 (4): 207-11, 2005.
- Orbach D, Mc Dowell H, Rey A, et al.: Sparing strategy does not compromise prognosis in pediatric localized synovial sarcoma: experience of the International Society of Pediatric Oncology, Malignant Mesenchymal Tumors (SIOP-MMT) Working Group. Pediatr Blood Cancer 57 (7): 1130-6, 2011.
- Ladenstein R, Treuner J, Koscielniak E, et al.: Synovial sarcoma of childhood and adolescence. Report of the German CWS-81 study. Cancer 71 (11): 3647-55, 1993.
- Venkatramani R, Xue W, Randall RL, et al.: Synovial Sarcoma in Children, Adolescents, and Young Adults: A Report From the Children's Oncology Group ARST0332 Study. J Clin Oncol 39 (35): 3927-3937, 2021.
- Ferrari A, De Salvo GL, Brennan B, et al.: Synovial sarcoma in children and adolescents: the European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann Oncol 26 (3): 567-72, 2015.
- Scheer M, Hallmen E, Vokuhl C, et al.: Pre-operative radiotherapy is associated with superior local relapse-free survival in advanced synovial sarcoma. J Cancer Res Clin Oncol 149 (5): 1717-1731, 2023.
- Ferrari A, De Salvo GL, Dall'Igna P, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with initially localised synovial sarcoma. Eur J Cancer 48 (18): 3448-55, 2012.
- Scheer M, Dantonello T, Hallmen E, et al.: Synovial Sarcoma Recurrence in Children and Young Adults. Ann Surg Oncol 23 (Suppl 5): 618-626, 2016.
- Ferrari A, Orbach D, Bergamaschi L, et al.: Treatment at relapse for synovial sarcoma of children and adolescents: A multi-institutional European retrospective analysis. Pediatr Blood Cancer 71 (7): e31038, 2024.
- Dhakal S, Corbin KS, Milano MT, et al.: Stereotactic body radiotherapy for pulmonary metastases from soft-tissue sarcomas: excellent local lesion control and improved patient survival. Int J Radiat Oncol Biol Phys 82 (2): 940-5, 2012.
- Lai JP, Robbins PF, Raffeld M, et al.: NY-ESO-1 expression in synovial sarcoma and other mesenchymal tumors: significance for NY-ESO-1-based targeted therapy and differential diagnosis. Mod Pathol 25 (6): 854-8, 2012.
- Robbins PF, Kassim SH, Tran TL, et al.: A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 21 (5): 1019-27, 2015.
- D'Angelo SP, Melchiori L, Merchant MS, et al.: Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 c259T Cells in Synovial Sarcoma. Cancer Discov 8 (8): 944-957, 2018.
- D'Angelo SP, Araujo DM, Abdul Razak AR, et al.: Afamitresgene autoleucel for advanced synovial sarcoma and myxoid round cell liposarcoma (SPEARHEAD-1): an international, open-label, phase 2 trial. Lancet 403 (10435): 1460-1471, 2024.
- Chbani L, Guillou L, Terrier P, et al.: Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French sarcoma group. Am J Clin Pathol 131 (2): 222-7, 2009.
- Hornick JL, Dal Cin P, Fletcher CD: Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 33 (4): 542-50, 2009.
- Knutson SK, Warholic NM, Wigle TJ, et al.: Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110 (19): 7922-7, 2013.
- Guzzetta AA, Montgomery EA, Lyu H, et al.: Epithelioid sarcoma: one institution's experience with a rare sarcoma. J Surg Res 177 (1): 116-22, 2012.
- Hawkins DS, Spunt SL, Skapek SX, et al.: Children's Oncology Group's 2013 blueprint for research: Soft tissue sarcomas. Pediatr Blood Cancer 60 (6): 1001-8, 2013.
- Sparber-Sauer M, Koscielniak E, Vokuhl C, et al.: Epithelioid sarcoma in children, adolescents, and young adults: Localized, primary metastatic and relapsed disease. Treatment results of five Cooperative Weichteilsarkom Studiengruppe (CWS) trials and one registry. Pediatr Blood Cancer 66 (9): e27879, 2019.
- Spunt SL, Francotte N, De Salvo GL, et al.: Clinical features and outcomes of young patients with epithelioid sarcoma: an analysis from the Children's Oncology Group and the European paediatric soft tissue Sarcoma Study Group prospective clinical trials. Eur J Cancer 112: 98-106, 2019.
- Casanova M, Ferrari A, Collini P, et al.: Epithelioid sarcoma in children and adolescents: a report from the Italian Soft Tissue Sarcoma Committee. Cancer 106 (3): 708-17, 2006.
- Gounder M, Schöffski P, Jones RL, et al.: Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open-label, phase 2 basket study. Lancet Oncol 21 (11): 1423-1432, 2020.
- Orbach D, Brennan B, Casanova M, et al.: Paediatric and adolescent alveolar soft part sarcoma: A joint series from European cooperative groups. Pediatr Blood Cancer 60 (11): 1826-32, 2013.
- Ferrari A, Sultan I, Huang TT, et al.: Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer 57 (6): 943-9, 2011.
- Wang HW, Qin XJ, Yang WJ, et al.: Alveolar soft part sarcoma of the oral and maxillofacial region: clinical analysis in a series of 18 patients. Oral Surg Oral Med Oral Pathol Oral Radiol 119 (4): 396-401, 2015.
- Kayton ML, Meyers P, Wexler LH, et al.: Clinical presentation, treatment, and outcome of alveolar soft part sarcoma in children, adolescents, and young adults. J Pediatr Surg 41 (1): 187-93, 2006.
- Sparber-Sauer M, Seitz G, von Kalle T, et al.: Alveolar soft-part sarcoma: Primary metastatic disease and metastatic relapse occurring during long-term follow-up: Treatment results of four Cooperative Weichteilsarkom Studiengruppe (CWS) trials and one registry. Pediatr Blood Cancer 65 (12): e27405, 2018.
- Flores RJ, Harrison DJ, Federman NC, et al.: Alveolar soft part sarcoma in children and young adults: A report of 69 cases. Pediatr Blood Cancer 65 (5): e26953, 2018.
- Ladanyi M, Lui MY, Antonescu CR, et al.: The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 20 (1): 48-57, 2001.
- Williams A, Bartle G, Sumathi VP, et al.: Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: useful diagnostic tools in cases with unusual histological features. Virchows Arch 458 (3): 291-300, 2011.
- Lieberman PH, Brennan MF, Kimmel M, et al.: Alveolar soft-part sarcoma. A clinico-pathologic study of half a century. Cancer 63 (1): 1-13, 1989.
- Casanova M, Ferrari A, Bisogno G, et al.: Alveolar soft part sarcoma in children and adolescents: A report from the Soft-Tissue Sarcoma Italian Cooperative Group. Ann Oncol 11 (11): 1445-9, 2000.
- Pennacchioli E, Fiore M, Collini P, et al.: Alveolar soft part sarcoma: clinical presentation, treatment, and outcome in a series of 33 patients at a single institution. Ann Surg Oncol 17 (12): 3229-33, 2010.
- Wilky BA, Trucco MM, Subhawong TK, et al.: Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: a single-centre, single-arm, phase 2 trial. Lancet Oncol 20 (6): 837-848, 2019.
- Stacchiotti S, Negri T, Zaffaroni N, et al.: Sunitinib in advanced alveolar soft part sarcoma: evidence of a direct antitumor effect. Ann Oncol 22 (7): 1682-90, 2011.
- Jagodzińska-Mucha P, Świtaj T, Kozak K, et al.: Long-term results of therapy with sunitinib in metastatic alveolar soft part sarcoma. Tumori 103 (3): 231-235, 2017.
- Cohen JW, Widemann BC, Derdak J, et al.: Cediranib phase-II study in children with metastatic alveolar soft-part sarcoma (ASPS). Pediatr Blood Cancer 66 (12): e27987, 2019.
- Judson I, Morden JP, Kilburn L, et al.: Cediranib in patients with alveolar soft-part sarcoma (CASPS): a double-blind, placebo-controlled, randomised, phase 2 trial. Lancet Oncol 20 (7): 1023-1034, 2019.
- Kummar S, Allen D, Monks A, et al.: Cediranib for metastatic alveolar soft part sarcoma. J Clin Oncol 31 (18): 2296-302, 2013.
- Kim M, Kim TM, Keam B, et al.: A Phase II Trial of Pazopanib in Patients with Metastatic Alveolar Soft Part Sarcoma. Oncologist 24 (1): 20-e29, 2019.
- Stacchiotti S, Mir O, Le Cesne A, et al.: Activity of Pazopanib and Trabectedin in Advanced Alveolar Soft Part Sarcoma. Oncologist 23 (1): 62-70, 2018.
- Chen AP, Sharon E, O'Sullivan-Coyne G, et al.: Atezolizumab for Advanced Alveolar Soft Part Sarcoma. N Engl J Med 389 (10): 911-921, 2023.
- Roozendaal KJ, de Valk B, ten Velden JJ, et al.: Alveolar soft-part sarcoma responding to interferon alpha-2b. Br J Cancer 89 (2): 243-5, 2003.
- Conde N, Cruz O, Albert A, et al.: Antiangiogenic treatment as a pre-operative management of alveolar soft-part sarcoma. Pediatr Blood Cancer 57 (6): 1071-3, 2011.
- Meis-Kindblom JM: Clear cell sarcoma of tendons and aponeuroses: a historical perspective and tribute to the man behind the entity. Adv Anat Pathol 13 (6): 286-92, 2006.
- Dim DC, Cooley LD, Miranda RN: Clear cell sarcoma of tendons and aponeuroses: a review. Arch Pathol Lab Med 131 (1): 152-6, 2007.
- Blazer DG, Lazar AJ, Xing Y, et al.: Clinical outcomes of molecularly confirmed clear cell sarcoma from a single institution and in comparison with data from the Surveillance, Epidemiology, and End Results registry. Cancer 115 (13): 2971-9, 2009.
- Coindre JM, Hostein I, Terrier P, et al.: Diagnosis of clear cell sarcoma by real-time reverse transcriptase-polymerase chain reaction analysis of paraffin embedded tissues: clinicopathologic and molecular analysis of 44 patients from the French sarcoma group. Cancer 107 (5): 1055-64, 2006.
- Fujimura Y, Siddique H, Lee L, et al.: EWS-ATF-1 chimeric protein in soft tissue clear cell sarcoma associates with CREB-binding protein and interferes with p53-mediated trans-activation function. Oncogene 20 (46): 6653-9, 2001.
- Hisaoka M, Ishida T, Kuo TT, et al.: Clear cell sarcoma of soft tissue: a clinicopathologic, immunohistochemical, and molecular analysis of 33 cases. Am J Surg Pathol 32 (3): 452-60, 2008.
- Ferrari A, Casanova M, Bisogno G, et al.: Clear cell sarcoma of tendons and aponeuroses in pediatric patients: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Cancer 94 (12): 3269-76, 2002.
- Karita M, Tsuchiya H, Yamamoto N, et al.: Caffeine-potentiated chemotherapy for clear cell sarcoma: a report of five cases. Int J Clin Oncol 18 (1): 33-7, 2013.
- Schöffski P, Wozniak A, Stacchiotti S, et al.: Activity and safety of crizotinib in patients with advanced clear-cell sarcoma with MET alterations: European Organization for Research and Treatment of Cancer phase II trial 90101 'CREATE'. Ann Oncol 28 (12): 3000-3008, 2017.
- Tsuneyoshi M, Enjoji M, Iwasaki H, et al.: Extraskeletal myxoid chondrosarcoma--a clinicopathologic and electron microscopic study. Acta Pathol Jpn 31 (3): 439-47, 1981.
- Hachitanda Y, Tsuneyoshi M, Daimaru Y, et al.: Extraskeletal myxoid chondrosarcoma in young children. Cancer 61 (12): 2521-6, 1988.
- Enzinger FM, Shiraki M: Extraskeletal myxoid chondrosarcoma. An analysis of 34 cases. Hum Pathol 3 (3): 421-35, 1972.
- McGrory JE, Rock MG, Nascimento AG, et al.: Extraskeletal myxoid chondrosarcoma. Clin Orthop Relat Res (382): 185-90, 2001.
- Drilon AD, Popat S, Bhuchar G, et al.: Extraskeletal myxoid chondrosarcoma: a retrospective review from 2 referral centers emphasizing long-term outcomes with surgery and chemotherapy. Cancer 113 (12): 3364-71, 2008.
- Hisaoka M, Ishida T, Imamura T, et al.: TFG is a novel fusion partner of NOR1 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer 40 (4): 325-8, 2004.
- Stacchiotti S, Pantaleo MA, Astolfi A, et al.: Activity of sunitinib in extraskeletal myxoid chondrosarcoma. Eur J Cancer 50 (9): 1657-64, 2014.
- Leavey PJ, Laack NN, Krailo MD, et al.: Phase III Trial Adding Vincristine-Topotecan-Cyclophosphamide to the Initial Treatment of Patients With Nonmetastatic Ewing Sarcoma: A Children's Oncology Group Report. J Clin Oncol 39 (36): 4029-4038, 2021.
- Leuschner I, Radig K, Harms D: Desmoplastic small round cell tumor. Semin Diagn Pathol 13 (3): 204-12, 1996.
- Kushner BH, LaQuaglia MP, Wollner N, et al.: Desmoplastic small round-cell tumor: prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol 14 (5): 1526-31, 1996.
- Saab R, Khoury JD, Krasin M, et al.: Desmoplastic small round cell tumor in childhood: the St. Jude Children's Research Hospital experience. Pediatr Blood Cancer 49 (3): 274-9, 2007.
- Wang LL, Perlman EJ, Vujanic GM, et al.: Desmoplastic small round cell tumor of the kidney in childhood. Am J Surg Pathol 31 (4): 576-84, 2007.
- Hayes-Jordan A, LaQuaglia MP, Modak S: Management of desmoplastic small round cell tumor. Semin Pediatr Surg 25 (5): 299-304, 2016.
- Arora VC, Price AP, Fleming S, et al.: Characteristic imaging features of desmoplastic small round cell tumour. Pediatr Radiol 43 (1): 93-102, 2013.
- Gerald WL, Ladanyi M, de Alava E, et al.: Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round-cell tumor and its variants. J Clin Oncol 16 (9): 3028-36, 1998.
- Slotkin EK, Bowman AS, Levine MF, et al.: Comprehensive Molecular Profiling of Desmoplastic Small Round Cell Tumor. Mol Cancer Res 19 (7): 1146-1155, 2021.
- Chow WA, Yee JK, Tsark W, et al.: Recurrent secondary genomic alterations in desmoplastic small round cell tumors. BMC Med Genet 21 (1): 101, 2020.
- Lal DR, Su WT, Wolden SL, et al.: Results of multimodal treatment for desmoplastic small round cell tumors. J Pediatr Surg 40 (1): 251-5, 2005.
- Philippe-Chomette P, Kabbara N, Andre N, et al.: Desmoplastic small round cell tumors with EWS-WT1 fusion transcript in children and young adults. Pediatr Blood Cancer 58 (6): 891-7, 2012.
- Sedig L, Geiger J, Mody R, et al.: Paratesticular desmoplastic small round cell tumors: A case report and review of the literature. Pediatr Blood Cancer 64 (12): , 2017.
- Subbiah V, Lamhamedi-Cherradi SE, Cuglievan B, et al.: Multimodality Treatment of Desmoplastic Small Round Cell Tumor: Chemotherapy and Complete Cytoreductive Surgery Improve Patient Survival. Clin Cancer Res 24 (19): 4865-4873, 2018.
- Schwarz RE, Gerald WL, Kushner BH, et al.: Desmoplastic small round cell tumors: prognostic indicators and results of surgical management. Ann Surg Oncol 5 (5): 416-22, 1998 Jul-Aug.
- Goodman KA, Wolden SL, La Quaglia MP, et al.: Whole abdominopelvic radiotherapy for desmoplastic small round-cell tumor. Int J Radiat Oncol Biol Phys 54 (1): 170-6, 2002.
- Osborne EM, Briere TM, Hayes-Jordan A, et al.: Survival and toxicity following sequential multimodality treatment including whole abdominopelvic radiotherapy for patients with desmoplastic small round cell tumor. Radiother Oncol 119 (1): 40-4, 2016.
- Atallah V, Honore C, Orbach D, et al.: Role of Adjuvant Radiation Therapy After Surgery for Abdominal Desmoplastic Small Round Cell Tumors. Int J Radiat Oncol Biol Phys 95 (4): 1244-53, 2016.
- Hayes-Jordan AA, Coakley BA, Green HL, et al.: Desmoplastic Small Round Cell Tumor Treated with Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy: Results of a Phase 2 Trial. Ann Surg Oncol 25 (4): 872-877, 2018.
- Scalabre A, Philippe-Chomette P, Passot G, et al.: Cytoreductive surgery and hyperthermic intraperitoneal perfusion with chemotherapy in children with peritoneal tumor spread: A French nationwide study over 14 years. Pediatr Blood Cancer 65 (4): , 2018.
- Honoré C, Atallah V, Mir O, et al.: Abdominal desmoplastic small round cell tumor without extraperitoneal metastases: Is there a benefit for HIPEC after macroscopically complete cytoreductive surgery? PLoS One 12 (2): e0171639, 2017.
- Stiles ZE, Murphy AJ, Anghelescu DL, et al.: Desmoplastic Small Round Cell Tumor: Long-Term Complications After Cytoreduction and Hyperthermic Intraperitoneal Chemotherapy. Ann Surg Oncol 27 (1): 171-178, 2020.
- Cook RJ, Wang Z, Arora M, et al.: Clinical outcomes of patients with desmoplastic small round cell tumor of the peritoneum undergoing autologous HCT: a CIBMTR retrospective analysis. Bone Marrow Transplant 47 (11): 1455-8, 2012.
- Tarek N, Hayes-Jordan A, Salvador L, et al.: Recurrent desmoplastic small round cell tumor responding to an mTOR inhibitor containing regimen. Pediatr Blood Cancer 65 (1): , 2018.
- Kodet R, Newton WA, Sachs N, et al.: Rhabdoid tumors of soft tissues: a clinicopathologic study of 26 cases enrolled on the Intergroup Rhabdomyosarcoma Study. Hum Pathol 22 (7): 674-84, 1991.
- Biegel JA, Zhou JY, Rorke LB, et al.: Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59 (1): 74-9, 1999.
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011.
- Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012.
- Teplick A, Kowalski M, Biegel JA, et al.: Educational paper: screening in cancer predisposition syndromes: guidelines for the general pediatrician. Eur J Pediatr 170 (3): 285-94, 2011.
- Mitchell SG, Pencheva B, Porter CC: Germline Genetics and Childhood Cancer: Emerging Cancer Predisposition Syndromes and Psychosocial Impacts. Curr Oncol Rep 21 (10): 85, 2019.
- Foulkes WD, Kamihara J, Evans DGR, et al.: Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin Cancer Res 23 (12): e62-e67, 2017.
- Morgan KM, Siow VS, Strotmeyer S, et al.: Characteristics and Outcomes in Pediatric Non-Central Nervous System Malignant Rhabdoid Tumors: A Report from the National Cancer Database. Ann Surg Oncol 29 (1): 671-678, 2022.
- Sultan I, Qaddoumi I, Rodríguez-Galindo C, et al.: Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr Blood Cancer 54 (1): 35-40, 2010.
- Nemes K, Bens S, Kachanov D, et al.: Clinical and genetic risk factors define two risk groups of extracranial malignant rhabdoid tumours (eMRT/RTK). Eur J Cancer 142: 112-122, 2021.
- Puri DR, Meyers PA, Kraus DH, et al.: Radiotherapy in the multimodal treatment of extrarenal extracranial malignant rhabdoid tumors. Pediatr Blood Cancer 50 (1): 167-9, 2008.
- Madigan CE, Armenian SH, Malogolowkin MH, et al.: Extracranial malignant rhabdoid tumors in childhood: the Childrens Hospital Los Angeles experience. Cancer 110 (9): 2061-6, 2007.
- Bourdeaut F, Fréneaux P, Thuille B, et al.: Extra-renal non-cerebral rhabdoid tumours. Pediatr Blood Cancer 51 (3): 363-8, 2008.
- Wetmore C, Boyett J, Li S, et al.: Alisertib is active as single agent in recurrent atypical teratoid rhabdoid tumors in 4 children. Neuro Oncol 17 (6): 882-8, 2015.
- Folpe A, Inwards C, eds.: Bone and Soft Tissue Pathology: A Volume in the Foundations in Diagnostic Pathology. WB Saunders Co, 2010.
- Martignoni G, Pea M, Reghellin D, et al.: Molecular pathology of lymphangioleiomyomatosis and other perivascular epithelioid cell tumors. Arch Pathol Lab Med 134 (1): 33-40, 2010.
- Bissler JJ, McCormack FX, Young LR, et al.: Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 358 (2): 140-51, 2008.
- Davies DM, Johnson SR, Tattersfield AE, et al.: Sirolimus therapy in tuberous sclerosis or sporadic lymphangioleiomyomatosis. N Engl J Med 358 (2): 200-3, 2008.
- Agaram NP, Sung YS, Zhang L, et al.: Dichotomy of Genetic Abnormalities in PEComas With Therapeutic Implications. Am J Surg Pathol 39 (6): 813-25, 2015.
- Armah HB, Parwani AV: Perivascular epithelioid cell tumor. Arch Pathol Lab Med 133 (4): 648-54, 2009.
- Alaggio R, Cecchetto G, Martignoni G, et al.: Malignant perivascular epithelioid cell tumor in children: description of a case and review of the literature. J Pediatr Surg 47 (6): e31-40, 2012.
- Wagner AJ, Malinowska-Kolodziej I, Morgan JA, et al.: Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol 28 (5): 835-40, 2010.
- Wagner AJ, Ravi V, Riedel RF, et al.: nab-Sirolimus for Patients With Malignant Perivascular Epithelioid Cell Tumors. J Clin Oncol 39 (33): 3660-3670, 2021.
- Laetsch TW, Roy A, Xu L, et al.: Undifferentiated Sarcomas in Children Harbor Clinically Relevant Oncogenic Fusions and Gene Copy-Number Alterations: A Report from the Children's Oncology Group. Clin Cancer Res 24 (16): 3888-3897, 2018.
- Randall RL, Albritton KH, Ferney BJ, et al.: Malignant fibrous histiocytoma of soft tissue: an abandoned diagnosis. Am J Orthop 33 (12): 602-8, 2004.
- Fletcher CDM, Bridge JA, Hogendoorn P, et al., eds.: WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. IARC Press, 2013.
- Alaggio R, Collini P, Randall RL, et al.: Undifferentiated high-grade pleomorphic sarcomas in children: a clinicopathologic study of 10 cases and review of literature. Pediatr Dev Pathol 13 (3): 209-17, 2010 May-Jun.
- Daw NC, Billups CA, Pappo AS, et al.: Malignant fibrous histiocytoma and other fibrohistiocytic tumors in pediatric patients: the St. Jude Children's Research Hospital experience. Cancer 97 (11): 2839-47, 2003.
- Bjerkehagen B, Smeland S, Walberg L, et al.: Radiation-induced sarcoma: 25-year experience from the Norwegian Radium Hospital. Acta Oncol 47 (8): 1475-82, 2008.
- Le Guellec S, Chibon F, Ouali M, et al.: Are peripheral purely undifferentiated pleomorphic sarcomas with MDM2 amplification dedifferentiated liposarcomas? Am J Surg Pathol 38 (3): 293-304, 2014.
- Tawbi HA, Burgess M, Bolejack V, et al.: Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol 18 (11): 1493-1501, 2017.
- Sloan EA, Gupta R, Koelsche C, et al.: Intracranial mesenchymal tumors with FET-CREB fusion are composed of at least two epigenetic subgroups distinct from meningioma and extracranial sarcomas. Brain Pathol 32 (4): e13037, 2022.
- Sloan EA, Chiang J, Villanueva-Meyer JE, et al.: Intracranial mesenchymal tumor with FET-CREB fusion-A unifying diagnosis for the spectrum of intracranial myxoid mesenchymal tumors and angiomatoid fibrous histiocytoma-like neoplasms. Brain Pathol 31 (4): e12918, 2021.