Complications, Graft-Versus-Host Disease, and Late Effects After Pediatric Hematopoietic Stem Cell Transplant (PDQ®): Treatment - Health Professional Information [NCI]
Hematopoietic Stem Cell Transplant (HSCT)–Related Acute Complications
Infectious Risks and Immune Recovery After Transplant
Defective immune reconstitution is a major barrier to successful HSCT, regardless of graft source.[
Factors that can significantly slow immune recovery include the following:[
- Graft manipulation (removal of T cells).
- Stem cell source (slow recovery with cord blood).
- Chronic graft-versus-host disease (GVHD).
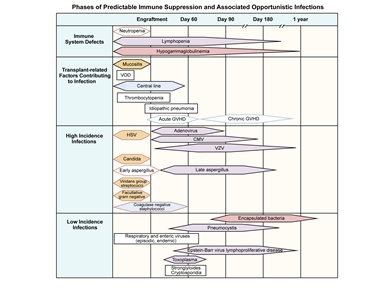
Figure 1. Phases of predictable immune suppression with their opportunistic infections among allogeneic hematopoietic stem cell transplant recipients. Adapted from Burik and Freifeld. This figure was published in Clinical Oncology, 3rd edition, Abeloff et al., Chapter: Infection in the severely immunocompromised patient, Pages 941–956, Copyright Elsevier (2004).
Bacterial infections tend to occur in the first few weeks after transplant during the neutropenic phase, when mucosal barriers are damaged from the conditioning regimen. There is significant ongoing research into the role of prophylactic antibacterial medications during the neutropenic phase.[
A joint effort between the Centers for Disease Control and Prevention, the Infectious Disease Society of America, and the American Society of Transplantation and Cellular Therapy established guidelines for the prevention of infections after HSCT.[
Prophylaxis against fungal infections is standard during the first several months after transplant and may be considered for patients with chronic GVHD who are at high risk of fungal infection. Antifungal prophylaxis must be tailored to the patient's underlying immune status. Pneumocystis infections can occur in all patients after bone marrow transplants, and prophylaxis is mandatory.[
After HSCT, viral infections can be a major source of mortality, especially after T-cell–depleted or cord blood procedures. Types of viral infections include the following:
- Cytomegalovirus (CMV). CMV infection has been a major cause of mortality in the past, but today, effective drugs to treat CMV are available. In addition, preventive strategies, including quantitative polymerase chain reaction (PCR) monitoring followed by preemptive therapy with ganciclovir, have been developed. In addition, the U.S. Food and Drug Administration (FDA) approved letermovir for CMV prophylaxis in adults. There is solid experience using letermovir in children aged 12 years and older and emerging experience in children younger than 12 years.[
9 ,10 ] - Epstein-Barr virus (EBV). EBV rarely causes lymphoproliferative disease and is generally associated with intensive, multidrug GVHD therapy or T-cell–depleted HSCT.
- Adenovirus. Adenovirus infection is a major issue in T-cell–depleted transplant, and monitoring by quantitative blood PCR followed by therapy with cidofovir or brincidofovir (available through a compassionate-use protocol) has led to a major decrease in morbidity.[
11 ] - Other. Other viruses have been implicated in hemorrhagic cystitis (BK virus), encephalitis and poor count recovery (human herpes virus 6), and other clinical issues.[
6 ] One study suggested that high BK viral loads early after transplant (4–7 weeks) may be associated with long-term decreases in glomerular filtration rate.[12 ]
Careful viral monitoring is essential during high-risk allogeneic procedures.
Late bacterial infections can occur in patients who have central lines or patients with significant chronic GVHD. These patients are susceptible to infection with encapsulated organisms, particularly pneumococcus. Despite reimmunization, these patients can sometimes develop significant infections, and continued prophylaxis is recommended until a serological response to immunizations has been documented. Occasionally, postallogeneic HSCT patients can become functionally asplenic, and antibiotic prophylaxis is recommended. Patients should remain on infection prophylaxis (e.g., Pneumocystis jirovecii pneumonia prophylaxis) until immune recovery. Time to immune recovery varies but ranges from 3 months to 9 months after autologous HSCT, and 9 months to 24 months after allogeneic HSCT without GVHD. Patients with active chronic GVHD may have persistent immunosuppression for years. Many centers monitor T-cell subset recovery after bone marrow transplants as a guide to infection risk.[
Vaccination after transplant
International transplant and infectious disease groups have developed specific guidelines for the administration of vaccines after autologous and allogeneic transplants.[
Vaccination recommendations should be reconsidered at times of local endemic or epidemic disease outbreaks. In those settings, earlier vaccination with killed vaccines may be implemented, acknowledging limited host responses. SARS-CoV-2 vaccination recommendations have been included in a recently updated consensus guideline on vaccines after HSCT.[
| Autologous HSCT | 6 Mob | 8 Mob | 12 Mob | 24 Mob |
|---|---|---|---|---|
| Allogeneic HSCT (if not immunized before 12 mo post-HSCT; start regardless of GVHD status or immunosuppression) | 12 mob(sooner if off immunosuppression) | 14 mob(or 2 mo after first dose) | 18 mob(or 6 mo after first dose) | 24 mob |
| GVHD = graft-versus-host disease; IM = intramuscular; PO = orally. | ||||
| a Adapted from Tomblyn et al.,[ |
||||
| b Times indicated are times posttransplant (day 0). | ||||
| c Use of Tdap is acceptable if DTap is not available. | ||||
| d Titers may be considered for pediatric patients and patients with GVHD who received immunizations while on immune suppression (minimum 6–8 weeks after last vaccination). | ||||
| e May start as soon as 4 months post-HSCT or sooner for patients with CD4 counts >200/mcL or at any time during an epidemic. If given <6 months after HSCT, may require second dose. Children younger than 9 years require second dose, separated by 1 month. | ||||
| f Consider pre- or postvaccine (at least 6–8 weeks after) titers. | ||||
| g PCV 7 at 24 months only for patients with GVHD; all other patients can get PPV 23. | ||||
| h Pediatric patients should receive two doses at least 1 month apart. | ||||
| Inactivated Vaccines | ||||
| Diphtheria, tetanus, acellular pertussis (DTap) | Xc | Xc | Xc,d | |
| Haemophilus influenzae (Hib) | X | X | Xd | |
| Hepatitis B (HepB) | X | X | Xd | |
| Inactive polio (IPV) | X | X | Xd | |
| Influenza—seasonal injection (IM) | Xe | |||
| Pneumococcal conjugate (PCV 7, PCV 13) | Xf | X | Xd,f,g | |
| Pneumococcal polysaccharide (PPV 23) | Xd,f,g | |||
| Live Attenuated Vaccines(contraindicated in patients with active GVHD or on immunosuppression) | ||||
| Measles, mumps, rubella | Xd,h | |||
| Optional Inactivated Vaccines | ||||
| Hepatitis A | Optional | |||
| Meningococcal | Xd(for high-risk patients) | |||
| Optional Live Vaccines(contraindicated in patients with active GVHD or on immunosuppression) | ||||
| Chicken pox (varicella vaccine) | Optional | |||
| Rabies | May be considered at 12–24 mo if exposed | |||
| Yellow fever, tick-borne encephalitis (TBE), Japanese B encephalitis | For travel in endemic areas | |||
| Contraindicated Vaccines | ||||
| Intranasal influenza (trivalent live-attenuated influenza vaccine) —household contacts and caregivers should not receive within 2 weeks before contact with HSCT recipient;shingles;bacillus Calmette-Guerin (BCG);oral polio vaccine (OPV);cholera;typhoid vaccine (PO, IM);rotavirus. | ||||
Sinusoidal Obstruction Syndrome/Veno-Occlusive Disease (SOS/VOD)
Pathologically, SOS/VOD of the liver is the result of damage to the hepatic sinusoids, resulting in biliary obstruction. This syndrome has been estimated to occur in 15% to 40% of pediatric patients who undergo myeloablative transplants.[
Risk factors for SOS/VOD include the following:[
- Use of busulfan (especially before therapeutic pharmacokinetic monitoring).
- Total-body irradiation.
- Serious infection.
- GVHD.
- Preexisting liver dysfunction caused by hepatitis or iron overload.
SOS/VOD is defined clinically by the following:
- Right upper quadrant pain with hepatomegaly.
- Fluid retention (weight gain and ascites).
- Hyperbilirubinemia.
Life-threatening SOS/VOD generally occurs soon after transplant and is characterized by multiorgan system failure.[
Diagnosis of SOS/VOD
Older definitions of SOS/VOD include the modified Seattle criteria or the Baltimore criteria.
- In the Seattle criteria, at least two of the following must be present by day 20 post HSCT:[
22 ]- Bilirubin level higher than 2 mg/dL.
- Hepatomegaly or right upper quadrant pain.
- Weight gain (>2%).
- In the Baltimore criteria, a bilirubin level of 2 mg/dL or higher and at least two of the following must be present by day 21 post HSCT:[
23 ]- Painful hepatomegaly.
- Weight gain (>5%).
- Ascites.
These definitions are inadequate, especially in pediatric practice, as they do not recognize late-onset SOS/VOD or VOD with normal bilirubin levels.
The European Society for Blood and Marrow Transplantation (EBMT) have published revised criteria that are now broadly in use.[
- Unexplained consumptive and transfusion-refractory thrombocytopenia.
- Otherwise unexplained weight gain on three consecutive days, despite the use of diuretics, or weight gain greater than 5% above baseline value.
- Hepatomegaly above baseline value (best if confirmed by imaging).
- Ascites above baseline value (best if confirmed by imaging).
- Rising bilirubin level from a baseline value on three consecutive days or bilirubin level of 2 mg/dL or higher within 72 hours.
An additional modification of the diagnostic algorithm (Cairo/Cooke criteria) has been proposed, which allows for flexibility with symptoms in unusual situations.[
Prevention and treatment of SOS/VOD
Approaches to both prevention and treatment with agents such as heparin, protein C, and antithrombin III have been studied, with mixed results.[
Another agent with demonstrated activity is defibrotide, a mixture of oligonucleotides with antithrombotic and fibrinolytic effects on microvascular endothelium. Studies of defibrotide have shown the following:
- Decreased mortality in patients who were treated with defibrotide for severe SOS/VOD, compared with historical controls.[
29 ,30 ,31 ,32 ]; [33 ][Level of evidence C1] - Decreased SOS/VOD mortality associated with the early initiation of defibrotide treatment soon after diagnostic criteria for SOS/VOD were met.[
34 ][Level of evidence B4] - Efficacy in decreasing SOS/VOD incidence when used prophylactically.[
35 ][Level of evidence A1] However, a second study was closed due to a lack of efficacy, questioning the validity of prophylactic defibrotide use.[36 ]
The FDA approved defibrotide for the treatment of patients who have hepatic SOS/VOD with renal or pulmonary dysfunction after HSCT.
The British Society for Blood and Marrow Transplantation (BSBMT) published evidence-guided recommendations for the diagnosis and management of SOS/VOD.[
Transplant-Associated Thrombotic Microangiopathy (TA-TMA)
Although TA-TMA clinically mirrors hemolytic uremic syndrome, its causes and clinical course differ from those of other hemolytic uremic syndrome–like diseases. Studies have linked this syndrome with dysregulation of complement pathways.[
Diagnostic criteria for this syndrome have been updated based on expert consensus opinion and are a modification of criteria published in 2014 (see
| Biopsy-proven disease (kidney or GI) OR | |
|---|---|
| Clinical criteria: Must meet ≥4 of the following 7 criteria within 14 days at 2 consecutive time points | |
| AIHA = autoimmune hemolytic anemia; BP = blood pressure; GI = gastrointestinal; LDH = lactate dehydrogenase; pRBCs = packed red blood cells; PRCA = pure red cell aplasia; rUPCR = random urine protein to creatinine ratio; ULN = upper limit of normal. | |
| a Reprinted with permission from Schoettler et al., which is available under the Creative Commons CC-BY-NC-ND license.[ |
|
| b Indicates clarification from published Jodele et al. criteria.[ |
|
| Anemiab | Defined as one of the following: |
| 1. Failure to achieve transfusion independence for pRBCs despite evidence of neutrophil engraftment | |
| 2. Hemoglobin decline from patient's baseline by 1 g/dL | |
| 3. New onset of transfusion dependence | |
| Rule out other causes of anemia, such as AIHA and PRCA | |
| Thrombocytopeniab | Defined as one of the following: |
| 1. Failure to achieve platelet engraftment | |
| 2. Higher than expected platelet transfusion needs | |
| 3. Refractoriness to platelet transfusion | |
| 4. 50% reduction or greater in baseline platelet count after full platelet engraftment | |
| Elevated LDH | >ULN for age |
| Schistocytes | Present |
| Hypertension | >99th percentile for age (<18 y), or systolic BP ≥140 mm Hg or diastolic BP ≥90 mm Hg (≥18 y) |
| Elevated sC5b-9 | ≥ULN |
| Proteinuria | ≥1 mg/mg rUPCR |
Evidence (impact of TA-TMA on HSCT outcomes):
- A multicenter study of TA-TMA in pediatric patients used the following definition of TA-TMA:[
46 ]- Histological evidence of TA-TMA, or
- Presence of at least four of the following laboratory and clinical markers diagnostic for TA-TMA:
- Lactate dehydrogenase (LDH) levels above reference value for age.
- Schistocytes on peripheral blood smear.
- De novo thrombocytopenia or requirement for platelet transfusions.
- De novo anemia or requirement for red blood cell transfusions.
- Hypertension greater than 99% for age (aged <18 years) or 140/90 mm Hg (aged ≥18 years) requiring ≥2 antihypertensive agents.
- Proteinuria ≥30 mg/dL on random urine analysis twice or random urine protein to creatinine ratio >1 mg/mg.
- Terminal complement activation: Elevated plasma sC5b-9 above normal limit (≥244 ng/mL).
- This study demonstrated the following results:
- In 614 sequential patients who underwent allogeneic or autologous HSCT, 19% of allogeneic recipients and 10% of autologous recipients developed TA-TMA.
- Patients who developed TA-TMA had increased rates of acute GVHD and steroid-refractory GVHD, intensive care unit admission, invasive ventilation, pericardial effusions, pulmonary hypertension, dialysis or continuous renal replacement therapy, acute kidney injury, and VOD.
- In patients who underwent allogeneic HSCT, treatment-related mortality during the first 6 months was significantly higher in patients with TA-TMA than in those without TA-TMA (20% vs. 3%; P ≤ .0001).
- In patients who underwent autologous HSCT, the overall survival (OS) rate during the first 6 months was significantly lower in patients with TA-TMA than in those without TA-TMA (79% vs. 98%; P = .001).
Treatment of TA-TMA
Treatment for TA-TMA includes the following:
- Cessation of the calcineurin inhibitor and substitution with other immune suppressants, if necessary.
- Careful management of hypertension and renal damage by dialysis, if necessary.
Prognosis for normal kidney function when disease is caused by calcineurin inhibitors alone is generally poor. However, most TA-TMA that is associated with the combination of a calcineurin inhibitor and sirolimus has been reversed after sirolimus is discontinued, and in some cases, after both medications are stopped.[
Some evidence suggests a role for complement modulation (c5, eculizumab therapy) in preserving renal function. Further assessment of the role of this medication in treating this complication is ongoing.[
Evidence (treatment of high-risk TA-TMA with eculizumab):
A prospective multicenter trial enrolled 21 patients with high-risk TA-TMA and multisystem organ dysfunction. The eculizumab dosing regimen included intensive loading, induction, and maintenance phases for up to 24 weeks of therapy.[
- The primary outcome was met, with an OS rate of 71% at 6 months after HSCT (vs. 18% for untreated historical controls; P < .0001).
- Eleven of fifteen survivors (73%) had fully recovered organ function at the time of reporting.
Idiopathic Pneumonia Syndrome (IPS)
IPS is characterized by diffuse, noninfectious lung injury that occurs between 14 and 90 days after the infusion of donor cells. Possible etiologies include direct toxic effects of conditioning regimens and occult infection leading to secretion of high levels of inflammatory cytokines into the alveoli.[
The incidence of IPS appears to be decreasing, possibly because of less intensive preparative regimens, better HLA matching, and better definition of occult infections through PCR testing of blood and bronchioalveolar specimens. Mortality rates of 50% to 70% have been reported.[
Diagnostic criteria include the following signs and symptoms in the absence of documented infectious organisms:[
- Pneumonia.
- Evidence of nonlobar radiographic infiltrates.
- Abnormal pulmonary function.
Early assessment by bronchioalveolar lavage to rule out infection is important.
Treatment of IPS
The traditional therapy for IPS has been high-dose methylprednisolone and pulmonary support.
Etanercept is a soluble fusion protein that joins the extracellular ligand-binding domain of the tumor necrosis factor (TNF)–alpha receptor to the Fc region of the immunoglobulin G1 antibody. It acts by blocking TNF-alpha signaling. The addition of etanercept to steroid therapies has shown promising short-term outcomes (extubation, improved short-term survival) in single-center studies.[
Autoimmune Cytopenias (AIC)
AIC after allogeneic HSCT can be restricted to one cell lineage (e.g., autoimmune hemolytic anemia), two cell lineages, or three cell lineages. Most data about AIC in pediatric patients after HSCT are reported from single-center experiences, with the number of cases ranging from 20 to 30 over a 10- to 20-year period.[
The National Institutes of Health task force on chronic GVHD has recognized AIC as a possible atypical feature of chronic GVHD (although they may be distinct pathologically).[
Treatment of AIC
The most common first-line therapy for AIC has been corticosteroids.[
Epstein-Barr Virus (EBV)–Associated Lymphoproliferative Disorder
After HSCT, EBV infection incidence increases through childhood, from approximately 40% in children aged 4 years to more than 80% in teenagers. Patients with a history of previous EBV infection are at risk of EBV reactivation when undergoing HSCT procedures that result in intense, prolonged lymphopenia (T-cell–depleted procedures, use of antithymocyte globulin [ATG] or alemtuzumab, and, to a lesser degree, use of cord blood).[
Features of EBV reactivation can vary, from an isolated increase in EBV titers in the bloodstream as measured by PCR to an aggressive monoclonal disease with marked lymphadenopathy presenting as lymphoma (lymphoproliferative disorder).
Treatment of EBV-associated lymphoproliferative disorder
Isolated bloodstream reactivation of EBV can improve in some cases without therapy as immune function improves. However, lymphoproliferative disorder requires more aggressive therapy.
Treatment of EBV-associated lymphoproliferative disorder involves decreasing immune suppression and treatment with chemotherapy agents such as cyclophosphamide. CD20-positive EBV-associated lymphoproliferative disorder and EBV reactivation have been shown to respond to therapy with the CD20 monoclonal antibody therapy rituximab.[
Improved understanding of the risk of EBV reactivation, early monitoring, and aggressive therapy have significantly decreased the risk of mortality from this challenging complication.
Acute GVHD
GVHD is the result of immunologic activation of donor lymphocytes targeting major or minor HLA disparities in the tissues of a recipient.[
Typically, acute GVHD presents with at least one of the following:
- Skin rash.
- Hyperbilirubinemia.
- Secretory diarrhea.
Acute GVHD is classified by staging the severity of skin, liver, and gastrointestinal involvement, and further combining the individual staging of these three areas into an overall grade that is prognostically significant (see Tables
| Stage | Skin | Liver (bilirubin)b | GI/Gut (stool output per day)c | |
|---|---|---|---|---|
| | | | Adult | Child |
| BSA = body surface area; GI = gastrointestinal. | ||||
| a Adapted from Harris et al.[ |
||||
| b There is no modification of liver staging for other causes of hyperbilirubinemia. | ||||
| c For GI staging: Theadult stool output values should be used for patients weighing >50 kg. Use 3-day averages for GI staging based on stool output. If stool and urine are mixed, stool output is presumed to be 50% of total stool/urine mix. | ||||
| d If results of colon or rectal biopsy are positive but stool output is <500 mL/day (<10 mL/kg/day), then consider as GI stage 0. | ||||
| e For stage 4 GI: the termsevere abdominal pain will be defined as having both (a) pain control requiring treatment with opioids or an increased dose in ongoing opioid use and (b) pain that significantly impacts performance status, as determined by the treating physician. | ||||
| 0 | No GVHD rash | <2 mg/dL | <500 mL or <3 episodes/day | <10 mL/kg or <4 episodes/day |
| 1 | Maculopapular rash <25% BSA | 2–3 mg/dL | 500–999 mLd or 3–4 episodes/day | 10–19.9 mL/kg or 4–6 episodes/day; persistent nausea, vomiting, or anorexia, with a positive result from upper GI biopsy |
| 2 | Maculopapular rash 25%–50% BSA | 3.1–6 mg/dL | 1,000–1,500 mL or 5–7 episodes/day | 20–30 mL/kg or 7–10 episodes/day |
| 3 | Maculopapular rash >50% BSA | 6.1–15 mg/dL | >1,500 mL or >7 episodes/day | >30 mL/kg or >10 episodes/day |
| 4 | Generalized erythroderma plus bullous formation and desquamation >5% BSA | >15 mg/dL | Severe abdominal paine with or without ileus, or grossly bloody stool (regardless of stool volume) | Severe abdominal paine with or without ileus, or grossly bloody stool (regardless of stool volume) |
| GI = gastrointestinal. | |
| Grade 0: | No stage 1–4 of any organ |
| Grade I: | Stage 1–2 skin and no liver or gut involvement |
| Grade II: | Stage 3 skin and/or stage 1 liver involvement and/or stage 1 GI |
| Grade III: | Stage 0–3 skin, with stage 2–3 liver and/or stage 2–3 GI |
| Grade IV: | Stage 4 skin, liver, or GI involvement |
Because the outcomes of patients with different grades of acute GVHD vary, investigators have sought to more precisely define acute GVHD risk based on serum biomarkers. A study that included both adults and children used a score calculated based on the levels of a combination of three biomarkers (tumor necrosis factor receptor 1 [TNFR1], suppression of tumorigenicity 2 [ST2], and regenerating islet-derived 3-alpha [REG3-alpha]), measured at the onset of acute GVHD. Investigators were able to define patients with low (8%), intermediate (27%), and high (46%, P < .0001) risk of 6-month mortality. The biomarker score was more sensitive and specific for predicting survival than clinical staging.[
Prevention and treatment of acute GVHD
Morbidity and mortality from acute GVHD can be reduced through immune suppressive medications given prophylactically or T-cell depletion of grafts, either ex vivo by actual removal of cells from a graft or in vivo with anti–T-lymphocyte antibodies (ATG or anti-CD52 [alemtuzumab]). A newer approach includes administering posttransplant cyclophosphamide on days 3 and 4 after HSCT.[
Complete elimination of acute GVHD with intense T-cell depletion has generally resulted in increased relapse, more infectious morbidity, and increased EBV-associated lymphoproliferative disorder. Because of this issue, most HSCT GVHD prophylaxis approaches try to balance risk by giving sufficient immune suppression to prevent severe acute and/or chronic GVHD but not completely remove GVHD risk.
GVHD prophylaxis approaches
GVHD prophylaxis has evolved, from one approach for all donor types to specific and varied approaches tailored to the following factors:
- Stem cell sources. For example, using higher intensity prophylaxis for mismatched bone marrow/peripheral blood stem cell (PBSC) HSCT, compared with low intensity/early immune tapering for matched-sibling bone marrow HSCT.[
79 ] - Clinical situations. For example, using planned early tapering of prophylaxis for high-risk disease to stimulate the graft-versus-leukemia (GVL) effect.[
79 ] - Intensity of the HSCT procedure. For example, using less intense prophylaxis for reduced-intensity regimens to increase the GVL effect because of the absence of myeloablation for disease control.
Because of these factors, it is best to consider the combination of preparative regimen, GVHD prophylaxis, and stem cell source as a unit because survival and toxicity outcomes vary if any of these three elements change.
GVHD prophylaxis for matched-sibling HSCT
The most commonly used GVHD prophylaxis approaches in pediatrics for matched-sibling HSCT consist of a calcineurin inhibitor (cyclosporine or tacrolimus), either as a single agent or in combination with methotrexate.[
A calcineurin inhibitor in combination with mycophenolate mofetil has also been used with matched-sibling HSCT, especially when reduced-intensity conditioning approaches are used.[
GVHD prophylaxis for matched unrelated-donor HSCT
A calcineurin inhibitor in combination with methotrexate (10 mg/m2 for four doses) has been a standard approach that leads to excellent outcomes.[
A number of studies have assessed the role of ATG or alemtuzumab (both considered serotherapy, antibodies that deplete T cells) in improving outcomes after unrelated-donor bone marrow transplant.[
A combined adult and pediatric study compared abatacept (T-cell costimulatory blocker) plus a calcineurin inhibitor/methotrexate with placebo plus a calcineurin inhibitor/methotrexate. Patients who received abatacept had improved rates of grades 2 to 4 acute GVHD and severe GVHD-free survival.[
GVHD prophylaxis for mismatched unrelated-donor HSCT
Use of a calcineurin inhibitor/methotrexate for mismatched unrelated-donor HSCT has led to higher rates of severe GVHD and lower rates of survival. This outcome is partially mitigated by the use of serotherapy in combination with a calcineurin inhibitor/methotrexate. Using this combined approach, the International BFM Study Group has considered the use of a single-antigen mismatched donor (7/8 or 9/10) to be a well-matched donor, with outcomes similar to those with matched unrelated donors.[
A prospective trial in mismatched unrelated-donor recipients added abatacept to a calcineurin inhibitor/methotrexate regimen. The study showed a marked improvement in severe acute GVHD and OS, compared with a Center for International Blood and Marrow Transplant Research (CIBMTR) control trial that used a calcineurin inhibitor/methotrexate alone.[
GVHD prophylaxis for unrelated-donor cord blood HSCT
A calcineurin inhibitor/methotrexate and cyclosporine/prednisone regimen has been used as GVHD prophylaxis in cord blood HSCT. However, a number of studies in pediatric patients have documented better survival and GVHD outcomes using a calcineurin inhibitor/mycophenolate mofetil combination.[
GVHD prophylaxis for haploidentical-donor HSCT
Early approaches using various intensities of GVHD prophylaxis and different types of T-cell depletion led to relatively poor rates of survival and high rates of GVHD when haploidentical donors were used.[
The other widely used approach to haploidentical HSCT in pediatrics is T-cell receptor (TCR) alpha beta/CD19 depletion. Using this process, several pediatric groups have demonstrated outcomes similar to those for fully matched stem cell sources, with low rates of GVHD.[
Similar to unrelated-donor bone marrow and cord blood HSCT, outcomes improved after TCR alpha beta/CD19 depletion when rATG was targeted to specific pre- and post-HSCT exposures.[
Nutritional approaches to prevent GVHD
Other nonimmune approaches to prevent GVHD are emerging. In a double-blind randomized study, patients with low vitamin A levels received either one pretransplant dose of vitamin A or a placebo. Patients who received vitamin A had statistically less acute GVHD (grades II to IV), acute gastrointestinal GVHD, and chronic GVHD.[
Steroid-refractory acute GVHD
When significant acute GVHD occurs, first-line therapy is generally methylprednisolone.[
References:
- Antin JH: Immune reconstitution: the major barrier to successful stem cell transplantation. Biol Blood Marrow Transplant 11 (2 Suppl 2): 43-5, 2005.
- Fry TJ, Mackall CL: Immune reconstitution following hematopoietic progenitor cell transplantation: challenges for the future. Bone Marrow Transplant 35 (Suppl 1): S53-7, 2005.
- Wingard JR, Majhail NS, Brazauskas R, et al.: Long-term survival and late deaths after allogeneic hematopoietic cell transplantation. J Clin Oncol 29 (16): 2230-9, 2011.
- Bunin N, Small T, Szabolcs P, et al.: NCI, NHLBI/PBMTC first international conference on late effects after pediatric hematopoietic cell transplantation: persistent immune deficiency in pediatric transplant survivors. Biol Blood Marrow Transplant 18 (1): 6-15, 2012.
- Burik JH, Freifeld AG: Infection in the severely immunocompromised patient. In: Abeloff MD, Armitage JO, Niederhuber JE, et al.: Clinical Oncology. 3rd ed. Elsevier, Churchill Livingstone, 2004, pp 941-56.
- Tomblyn M, Chiller T, Einsele H, et al.: Guidelines for preventing infectious complications among hematopoietic cell transplantation recipients: a global perspective. Biol Blood Marrow Transplant 15 (10): 1143-238, 2009.
- Centers for Disease Control and Prevention, Infectious Disease Society of America, American Society of Blood and Marrow Transplantation: Guidelines for preventing opportunistic infections among hematopoietic stem cell transplant recipients. MMWR Recomm Rep 49 (RR-10): 1-125, CE1-7, 2000.
- Levy ER, Musick L, Zinter MS, et al.: Safe and Effective Prophylaxis with Bimonthly Intravenous Pentamidine in the Pediatric Hematopoietic Stem Cell Transplant Population. Pediatr Infect Dis J 35 (2): 135-41, 2016.
- Russo D, Schmitt M, Pilorge S, et al.: Efficacy and safety of extended duration letermovir prophylaxis in recipients of haematopoietic stem-cell transplantation at risk of cytomegalovirus infection: a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Haematol 11 (2): e127-e135, 2024.
- Galaverna F, Baccelli F, Zama D, et al.: Letermovir for Cytomegalovirus infection in pediatric patients undergoing allogenic hematopoietic stem cell transplantation: a real-life study by the Infectious Diseases Working Group of Italian Association of Pediatric Hematology-Oncology (AIEOP). Bone Marrow Transplant 59 (4): 505-512, 2024.
- Hiwarkar P, Amrolia P, Sivaprakasam P, et al.: Brincidofovir is highly efficacious in controlling adenoviremia in pediatric recipients of hematopoietic cell transplant. Blood 129 (14): 2033-2037, 2017.
- Wychera C, Imlay HN, Duke ER, et al.: BK Viremia and Changes in Estimated Glomerular Filtration Rate in Children and Young Adults after Hematopoietic Cell Transplantation. Transplant Cell Ther 29 (3): 187.e1-187.e8, 2023.
- Rubin LG, Levin MJ, Ljungman P, et al.: 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clin Infect Dis 58 (3): e44-100, 2014.
- Cordonnier C, Einarsdottir S, Cesaro S, et al.: Vaccination of haemopoietic stem cell transplant recipients: guidelines of the 2017 European Conference on Infections in Leukaemia (ECIL 7). Lancet Infect Dis 19 (6): e200-e212, 2019.
- Miller P, Patel SR, Skinner R, et al.: Joint consensus statement on the vaccination of adult and paediatric haematopoietic stem cell transplant recipients: Prepared on behalf of the British society of blood and marrow transplantation and cellular therapy (BSBMTCT), the Children's cancer and Leukaemia Group (CCLG), and British Infection Association (BIA). J Infect 86 (1): 1-8, 2023.
- Hill JA, Martens MJ, Young JH, et al.: SARS-CoV-2 vaccination in the first year after allogeneic hematopoietic cell transplant: a prospective, multicentre, observational study. EClinicalMedicine 59: 101983, 2023.
- Kumar D, Chen MH, Welsh B, et al.: A randomized, double-blind trial of pneumococcal vaccination in adult allogeneic stem cell transplant donors and recipients. Clin Infect Dis 45 (12): 1576-82, 2007.
- Reiss U, Cowan M, McMillan A, et al.: Hepatic venoocclusive disease in blood and bone marrow transplantation in children and young adults: incidence, risk factors, and outcome in a cohort of 241 patients. J Pediatr Hematol Oncol 24 (9): 746-50, 2002.
- Cesaro S, Pillon M, Talenti E, et al.: A prospective survey on incidence, risk factors and therapy of hepatic veno-occlusive disease in children after hematopoietic stem cell transplantation. Haematologica 90 (10): 1396-404, 2005.
- Bearman SI: The syndrome of hepatic veno-occlusive disease after marrow transplantation. Blood 85 (11): 3005-20, 1995.
- Myers KC, Dandoy C, El-Bietar J, et al.: Veno-occlusive disease of the liver in the absence of elevation in bilirubin in pediatric patients after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 21 (2): 379-81, 2015.
- Carreras E, Dufour C, Mohty M, et al., eds.: The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies. 7th edition. Cham (CH): Springer, 2019.
Available online . Last accessed May 7, 2024. - Jones RJ, Lee KS, Beschorner WE, et al.: Venoocclusive disease of the liver following bone marrow transplantation. Transplantation 44 (6): 778-83, 1987.
- Mohty M, Malard F, Abecassis M, et al.: Revised diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in adult patients: a new classification from the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant 51 (7): 906-12, 2016.
- Corbacioglu S, Carreras E, Ansari M, et al.: Diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in pediatric patients: a new classification from the European society for blood and marrow transplantation. Bone Marrow Transplant 53 (2): 138-145, 2018.
- Cairo MS, Cooke KR, Lazarus HM, et al.: Modified diagnostic criteria, grading classification and newly elucidated pathophysiology of hepatic SOS/VOD after haematopoietic cell transplantation. Br J Haematol 190 (6): 822-836, 2020.
- Ruutu T, Eriksson B, Remes K, et al.: Ursodeoxycholic acid for the prevention of hepatic complications in allogeneic stem cell transplantation. Blood 100 (6): 1977-83, 2002.
- Myers KC, Lawrence J, Marsh RA, et al.: High-dose methylprednisolone for veno-occlusive disease of the liver in pediatric hematopoietic stem cell transplantation recipients. Biol Blood Marrow Transplant 19 (3): 500-3, 2013.
- Richardson PG, Murakami C, Jin Z, et al.: Multi-institutional use of defibrotide in 88 patients after stem cell transplantation with severe veno-occlusive disease and multisystem organ failure: response without significant toxicity in a high-risk population and factors predictive of outcome. Blood 100 (13): 4337-43, 2002.
- Corbacioglu S, Kernan N, Lehmann L, et al.: Defibrotide for the treatment of hepatic veno-occlusive disease in children after hematopoietic stem cell transplantation. Expert Rev Hematol 5 (3): 291-302, 2012.
- Richardson PG, Soiffer RJ, Antin JH, et al.: Defibrotide for the treatment of severe hepatic veno-occlusive disease and multiorgan failure after stem cell transplantation: a multicenter, randomized, dose-finding trial. Biol Blood Marrow Transplant 16 (7): 1005-17, 2010.
- Dignan FL, Wynn RF, Hadzic N, et al.: BCSH/BSBMT guideline: diagnosis and management of veno-occlusive disease (sinusoidal obstruction syndrome) following haematopoietic stem cell transplantation. Br J Haematol 163 (4): 444-57, 2013.
- Strouse C, Richardson P, Prentice G, et al.: Defibrotide for Treatment of Severe Veno-Occlusive Disease in Pediatrics and Adults: An Exploratory Analysis Using Data from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant 22 (7): 1306-1312, 2016.
- Richardson PG, Smith AR, Triplett BM, et al.: Earlier defibrotide initiation post-diagnosis of veno-occlusive disease/sinusoidal obstruction syndrome improves Day +100 survival following haematopoietic stem cell transplantation. Br J Haematol 178 (1): 112-118, 2017.
- Corbacioglu S, Cesaro S, Faraci M, et al.: Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial. Lancet 379 (9823): 1301-9, 2012.
- Grupp SA, Corbacioglu S, Kang HJ, et al.: Defibrotide plus best standard of care compared with best standard of care alone for the prevention of sinusoidal obstruction syndrome (HARMONY): a randomised, multicentre, phase 3 trial. Lancet Haematol 10 (5): e333-e345, 2023.
- Ruutu T, Juvonen E, Remberger M, et al.: Improved survival with ursodeoxycholic acid prophylaxis in allogeneic stem cell transplantation: long-term follow-up of a randomized study. Biol Blood Marrow Transplant 20 (1): 135-8, 2014.
- Bajwa RPS, Mahadeo KM, Taragin BH, et al.: Consensus Report by Pediatric Acute Lung Injury and Sepsis Investigators and Pediatric Blood and Marrow Transplantation Consortium Joint Working Committees: Supportive Care Guidelines for Management of Veno-Occlusive Disease in Children and Adolescents, Part 1: Focus on Investigations, Prophylaxis, and Specific Treatment. Biol Blood Marrow Transplant 23 (11): 1817-1825, 2017.
- Mahadeo KM, McArthur J, Adams RH, et al.: Consensus Report by the Pediatric Acute Lung Injury and Sepsis Investigators and Pediatric Blood and Marrow Transplant Consortium Joint Working Committees on Supportive Care Guidelines for Management of Veno-Occlusive Disease in Children and Adolescents: Part 2-Focus on Ascites, Fluid and Electrolytes, Renal, and Transfusion Issues. Biol Blood Marrow Transplant 23 (12): 2023-2033, 2017.
- Ovchinsky N, Frazier W, Auletta JJ, et al.: Consensus Report by the Pediatric Acute Lung Injury and Sepsis Investigators and Pediatric Blood and Marrow Transplantation Consortium Joint Working Committees on Supportive Care Guidelines for Management of Veno-Occlusive Disease in Children and Adolescents, Part 3: Focus on Cardiorespiratory Dysfunction, Infections, Liver Dysfunction, and Delirium. Biol Blood Marrow Transplant 24 (2): 207-218, 2018.
- Jodele S, Licht C, Goebel J, et al.: Abnormalities in the alternative pathway of complement in children with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Blood 122 (12): 2003-7, 2013.
- Cutler C, Henry NL, Magee C, et al.: Sirolimus and thrombotic microangiopathy after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 11 (7): 551-7, 2005.
- Schoettler ML, Carreras E, Cho B, et al.: Harmonizing Definitions for Diagnostic Criteria and Prognostic Assessment of Transplantation-Associated Thrombotic Microangiopathy: A Report on Behalf of the European Society for Blood and Marrow Transplantation, American Society for Transplantation and Cellular Therapy, Asia-Pacific Blood and Marrow Transplantation Group, and Center for International Blood and Marrow Transplant Research. Transplant Cell Ther 29 (3): 151-163, 2023.
- Jodele S, Dandoy CE, Aguayo-Hiraldo P, et al.: A prospective multi-institutional study of eculizumab to treat high-risk stem cell transplantation-associated TMA. Blood 143 (12): 1112-1123, 2024.
- Jodele S, Davies SM, Lane A, et al.: Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood 124 (4): 645-53, 2014.
- Dandoy CE, Rotz S, Alonso PB, et al.: A pragmatic multi-institutional approach to understanding transplant-associated thrombotic microangiopathy after stem cell transplant. Blood Adv 5 (1): 1-11, 2021.
- Jodele S, Fukuda T, Vinks A, et al.: Eculizumab therapy in children with severe hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Biol Blood Marrow Transplant 20 (4): 518-25, 2014.
- Jodele S, Fukuda T, Mizuno K, et al.: Variable Eculizumab Clearance Requires Pharmacodynamic Monitoring to Optimize Therapy for Thrombotic Microangiopathy after Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant 22 (2): 307-315, 2016.
- Schoettler M, Lehmann L, Li A, et al.: Thrombotic Microangiopathy Following Pediatric Autologous Hematopoietic Cell Transplantation: A Report of Significant End-Organ Dysfunction in Eculizumab-Treated Survivors. Biol Blood Marrow Transplant 25 (5): e163-e168, 2019.
- Jodele S, Dandoy CE, Lane A, et al.: Complement blockade for TA-TMA: lessons learned from a large pediatric cohort treated with eculizumab. Blood 135 (13): 1049-1057, 2020.
- Svec P, Elfeky R, Galimard JE, et al.: Use of eculizumab in children with allogeneic haematopoietic stem cell transplantation associated thrombotic microangiopathy - a multicentre retrospective PDWP and IEWP EBMT study. Bone Marrow Transplant 58 (2): 129-141, 2023.
- Benítez Carabante MI, Bueno D, Alonso García L, et al.: Use of Eculizumab in Pediatric Patients with High-Risk Transplantation-Associated Thrombotic Microangiopathy: Outcomes and Risk Factors Associated with Response and Survival. A Retrospective Study on Behalf of the Spanish Group for Hematopoietic Transplantation and Cellular Therapy (GETH-TC). Transplant Cell Ther 30 (6): 601.e1-601.e13, 2024.
- Kantrow SP, Hackman RC, Boeckh M, et al.: Idiopathic pneumonia syndrome: changing spectrum of lung injury after marrow transplantation. Transplantation 63 (8): 1079-86, 1997.
- Clark JG, Hansen JA, Hertz MI, et al.: NHLBI workshop summary. Idiopathic pneumonia syndrome after bone marrow transplantation. Am Rev Respir Dis 147 (6 Pt 1): 1601-6, 1993.
- Yanik GA, Ho VT, Levine JE, et al.: The impact of soluble tumor necrosis factor receptor etanercept on the treatment of idiopathic pneumonia syndrome after allogeneic hematopoietic stem cell transplantation. Blood 112 (8): 3073-81, 2008.
- Yanik GA, Grupp SA, Pulsipher MA, et al.: TNF-receptor inhibitor therapy for the treatment of children with idiopathic pneumonia syndrome. A joint Pediatric Blood and Marrow Transplant Consortium and Children's Oncology Group Study (ASCT0521). Biol Blood Marrow Transplant 21 (1): 67-73, 2015.
- Szanto CL, Langenhorst J, de Koning C, et al.: Predictors for Autoimmune Cytopenias after Allogeneic Hematopoietic Cell Transplantation in Children. Biol Blood Marrow Transplant 26 (1): 114-122, 2020.
- Koo J, Giller RH, Quinones R, et al.: Autoimmune cytopenias following allogeneic hematopoietic stem cell transplant in pediatric patients: Response to therapy and late effects. Pediatr Blood Cancer 67 (9): e28591, 2020.
- O'Brien TA, Eastlund T, Peters C, et al.: Autoimmune haemolytic anaemia complicating haematopoietic cell transplantation in paediatric patients: high incidence and significant mortality in unrelated donor transplants for non-malignant diseases. Br J Haematol 127 (1): 67-75, 2004.
- Cuvelier GDE, Schoettler M, Buxbaum NP, et al.: Toward a Better Understanding of the Atypical Features of Chronic Graft-Versus-Host Disease: A Report from the 2020 National Institutes of Health Consensus Project Task Force. Transplant Cell Ther 28 (8): 426-445, 2022.
- Hillier K, Harris EM, Berbert L, et al.: Characteristics and outcomes of autoimmune hemolytic anemia after pediatric allogeneic stem cell transplant. Pediatr Blood Cancer 69 (1): e29410, 2022.
- Even-Or E, Schejter YD, NaserEddin A, et al.: Autoimmune Cytopenias Post Hematopoietic Stem Cell Transplantation in Pediatric Patients With Osteopetrosis and Other Nonmalignant Diseases. Front Immunol 13: 879994, 2022.
- Gerritsen EJ, Stam ED, Hermans J, et al.: Risk factors for developing EBV-related B cell lymphoproliferative disorders (BLPD) after non-HLA-identical BMT in children. Bone Marrow Transplant 18 (2): 377-82, 1996.
- Shapiro RS, McClain K, Frizzera G, et al.: Epstein-Barr virus associated B cell lymphoproliferative disorders following bone marrow transplantation. Blood 71 (5): 1234-43, 1988.
- Brunstein CG, Weisdorf DJ, DeFor T, et al.: Marked increased risk of Epstein-Barr virus-related complications with the addition of antithymocyte globulin to a nonmyeloablative conditioning prior to unrelated umbilical cord blood transplantation. Blood 108 (8): 2874-80, 2006.
- Blaes AH, Cao Q, Wagner JE, et al.: Monitoring and preemptive rituximab therapy for Epstein-Barr virus reactivation after antithymocyte globulin containing nonmyeloablative conditioning for umbilical cord blood transplantation. Biol Blood Marrow Transplant 16 (2): 287-91, 2010.
- Kuehnle I, Huls MH, Liu Z, et al.: CD20 monoclonal antibody (rituximab) for therapy of Epstein-Barr virus lymphoma after hemopoietic stem-cell transplantation. Blood 95 (4): 1502-5, 2000.
- Styczynski J, Gil L, Tridello G, et al.: Response to rituximab-based therapy and risk factor analysis in Epstein Barr Virus-related lymphoproliferative disorder after hematopoietic stem cell transplant in children and adults: a study from the Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Clin Infect Dis 57 (6): 794-802, 2013.
- Liu Z, Savoldo B, Huls H, et al.: Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes for the prevention and treatment of EBV-associated post-transplant lymphomas. Recent Results Cancer Res 159: 123-33, 2002.
- Bollard CM, Heslop HE: T cells for viral infections after allogeneic hematopoietic stem cell transplant. Blood 127 (26): 3331-40, 2016.
- Keller MD, Hanley PJ, Chi YY, et al.: Antiviral cellular therapy for enhancing T-cell reconstitution before or after hematopoietic stem cell transplantation (ACES): a two-arm, open label phase II interventional trial of pediatric patients with risk factor assessment. Nat Commun 15 (1): 3258, 2024.
- Ferrara JL, Levine JE, Reddy P, et al.: Graft-versus-host disease. Lancet 373 (9674): 1550-61, 2009.
- Przepiorka D, Weisdorf D, Martin P, et al.: 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant 15 (6): 825-8, 1995.
- Harris AC, Young R, Devine S, et al.: International, Multicenter Standardization of Acute Graft-versus-Host Disease Clinical Data Collection: A Report from the Mount Sinai Acute GVHD International Consortium. Biol Blood Marrow Transplant 22 (1): 4-10, 2016.
- Levine JE, Braun TM, Harris AC, et al.: A prognostic score for acute graft-versus-host disease based on biomarkers: a multicentre study. Lancet Haematol 2 (1): e21-9, 2015.
- Srinagesh HK, Özbek U, Kapoor U, et al.: The MAGIC algorithm probability is a validated response biomarker of treatment of acute graft-versus-host disease. Blood Adv 3 (23): 4034-4042, 2019.
- Luznik L, O'Donnell PV, Fuchs EJ: Post-transplantation cyclophosphamide for tolerance induction in HLA-haploidentical bone marrow transplantation. Semin Oncol 39 (6): 683-93, 2012.
- Nunes NS, Kanakry CG: Mechanisms of Graft-versus-Host Disease Prevention by Post-transplantation Cyclophosphamide: An Evolving Understanding. Front Immunol 10: 2668, 2019.
- Watkins B, Williams KM: Controversies and expectations for the prevention of GVHD: A biological and clinical perspective. Front Immunol 13: 1057694, 2022.
- Locatelli F, Zecca M, Rondelli R, et al.: Graft versus host disease prophylaxis with low-dose cyclosporine-A reduces the risk of relapse in children with acute leukemia given HLA-identical sibling bone marrow transplantation: results of a randomized trial. Blood 95 (5): 1572-9, 2000.
- Storb R, Deeg HJ, Pepe M, et al.: Methotrexate and cyclosporine versus cyclosporine alone for prophylaxis of graft-versus-host disease in patients given HLA-identical marrow grafts for leukemia: long-term follow-up of a controlled trial. Blood 73 (6): 1729-34, 1989.
- Pulsipher MA, Langholz B, Wall DA, et al.: The addition of sirolimus to tacrolimus/methotrexate GVHD prophylaxis in children with ALL: a phase 3 Children's Oncology Group/Pediatric Blood and Marrow Transplant Consortium trial. Blood 123 (13): 2017-25, 2014.
- Ueda Oshima M, Storer BE, Qiu H, et al.: Long-term Outcomes with Nonmyeloablative HLA-Identical Related Hematopoietic Cell Transplantation Using Tacrolimus and Mycophenolate Mofetil for Graft-versus-Host Disease Prophylaxis. Transplant Cell Ther 27 (2): 163.e1-163.e7, 2021.
- Mehta RS, Saliba RM, Rondon G, et al.: Post-Transplantation Cyclophosphamide Versus Tacrolimus and Methotrexate Graft-Versus-Host Disease Prophylaxis for HLA-Matched Donor Transplantation. Transplant Cell Ther 28 (10): 695.e1-695.e10, 2022.
- Borovkova AS, Paina OV, Semenova EV, et al.: Post-transplant сyclophosphamide after matched donor hematopoietic stem cell transplantation in children with acute leukemia. Clin Transplant 38 (1): e15181, 2024.
- Fierro-Pineda JC, Tsai HL, Blackford A, et al.: Prospective PTCTC trial of myeloablative haplo-BMT with posttransplant cyclophosphamide for pediatric acute leukemias. Blood Adv 7 (18): 5639-5648, 2023.
- McCurdy SR, Kanakry CG, Tsai HL, et al.: Development of Grade II Acute Graft-versus-Host Disease Is Associated with Improved Survival after Myeloablative HLA-Matched Bone Marrow Transplantation using Single-Agent Post-Transplant Cyclophosphamide. Biol Blood Marrow Transplant 25 (6): 1128-1135, 2019.
- Kanakry CG, O'Donnell PV, Furlong T, et al.: Multi-institutional study of post-transplantation cyclophosphamide as single-agent graft-versus-host disease prophylaxis after allogeneic bone marrow transplantation using myeloablative busulfan and fludarabine conditioning. J Clin Oncol 32 (31): 3497-505, 2014.
- Soiffer RJ, Kim HT, McGuirk J, et al.: Prospective, Randomized, Double-Blind, Phase III Clinical Trial of Anti-T-Lymphocyte Globulin to Assess Impact on Chronic Graft-Versus-Host Disease-Free Survival in Patients Undergoing HLA-Matched Unrelated Myeloablative Hematopoietic Cell Transplantation. J Clin Oncol 35 (36): 4003-4011, 2017.
- Socié G, Schmoor C, Bethge WA, et al.: Chronic graft-versus-host disease: long-term results from a randomized trial on graft-versus-host disease prophylaxis with or without anti-T-cell globulin ATG-Fresenius. Blood 117 (23): 6375-82, 2011.
- Kröger N, Solano C, Wolschke C, et al.: Antilymphocyte Globulin for Prevention of Chronic Graft-versus-Host Disease. N Engl J Med 374 (1): 43-53, 2016.
- Admiraal R, Nierkens S, Bierings MB, et al.: Individualised dosing of anti-thymocyte globulin in paediatric unrelated allogeneic haematopoietic stem-cell transplantation (PARACHUTE): a single-arm, phase 2 clinical trial. Lancet Haematol 9 (2): e111-e120, 2022.
- Admiraal R, van Kesteren C, Jol-van der Zijde CM, et al.: Association between anti-thymocyte globulin exposure and CD4+ immune reconstitution in paediatric haemopoietic cell transplantation: a multicentre, retrospective pharmacodynamic cohort analysis. Lancet Haematol 2 (5): e194-203, 2015.
- Watkins B, Qayed M, McCracken C, et al.: Phase II Trial of Costimulation Blockade With Abatacept for Prevention of Acute GVHD. J Clin Oncol 39 (17): 1865-1877, 2021.
- Peters C, Schrappe M, von Stackelberg A, et al.: Stem-cell transplantation in children with acute lymphoblastic leukemia: A prospective international multicenter trial comparing sibling donors with matched unrelated donors-The ALL-SCT-BFM-2003 trial. J Clin Oncol 33 (11): 1265-74, 2015.
- Kean LS, Burns LJ, Kou TD, et al.: Abatacept for acute graft-versus-host disease prophylaxis after unrelated donor hematopoietic cell transplantation. Blood 144 (17): 1834-1845, 2024.
- Shaw BE, Jimenez-Jimenez AM, Burns LJ, et al.: National Marrow Donor Program-Sponsored Multicenter, Phase II Trial of HLA-Mismatched Unrelated Donor Bone Marrow Transplantation Using Post-Transplant Cyclophosphamide. J Clin Oncol 39 (18): 1971-1982, 2021.
- Wagner JE, Eapen M, Carter S, et al.: One-unit versus two-unit cord-blood transplantation for hematologic cancers. N Engl J Med 371 (18): 1685-94, 2014.
- Eapen M, Kurtzberg J, Zhang MJ, et al.: Umbilical Cord Blood Transplantation in Children with Acute Leukemia: Impact of Conditioning on Transplantation Outcomes. Biol Blood Marrow Transplant 23 (10): 1714-1721, 2017.
- Lindemans CA, Chiesa R, Amrolia PJ, et al.: Impact of thymoglobulin prior to pediatric unrelated umbilical cord blood transplantation on immune reconstitution and clinical outcome. Blood 123 (1): 126-32, 2014.
- Admiraal R, Lindemans CA, van Kesteren C, et al.: Excellent T-cell reconstitution and survival depend on low ATG exposure after pediatric cord blood transplantation. Blood 128 (23): 2734-2741, 2016.
- Admiraal R, Versluijs AB, Huitema ADR, et al.: High-dose individualized antithymocyte globulin with therapeutic drug monitoring in high-risk cord blood transplant. Cytotherapy 26 (6): 599-605, 2024.
- Pulsipher MA: Haplo is the new black. Blood 124 (5): 675-6, 2014.
- Pulsipher MA, Ahn KW, Bunin NJ, et al.: KIR-favorable TCR-αβ/CD19-depleted haploidentical HCT in children with ALL/AML/MDS: primary analysis of the PTCTC ONC1401 trial. Blood 140 (24): 2556-2572, 2022.
- Bertaina A, Zecca M, Buldini B, et al.: Unrelated donor vs HLA-haploidentical α/β T-cell- and B-cell-depleted HSCT in children with acute leukemia. Blood 132 (24): 2594-2607, 2018.
- Merli P, Algeri M, Galaverna F, et al.: TCRαβ/CD19 cell-depleted HLA-haploidentical transplantation to treat pediatric acute leukemia: updated final analysis. Blood 143 (3): 279-289, 2024.
- Mehta PA, Davies SM, Leemhuis T, et al.: Radiation-free, alternative-donor HCT for Fanconi anemia patients: results from a prospective multi-institutional study. Blood 129 (16): 2308-2315, 2017.
- Bleakley M, Heimfeld S, Loeb KR, et al.: Outcomes of acute leukemia patients transplanted with naive T cell-depleted stem cell grafts. J Clin Invest 125 (7): 2677-89, 2015.
- Dvorak CC, Long-Boyle JR, Holbrook-Brown L, et al.: Effect of rabbit ATG PK on outcomes after TCR-αβ/CD19-depleted pediatric haploidentical HCT for hematologic malignancy. Blood Adv 8 (23): 6003-6014, 2024.
- Khandelwal P, Langenberg L, Luebbering N, et al.: A randomized phase 2 trial of oral vitamin A for graft-versus-host disease in children and young adults. Blood 143 (12): 1181-1192, 2024.
- Jacobsohn DA: Acute graft-versus-host disease in children. Bone Marrow Transplant 41 (2): 215-21, 2008.
- Deeg HJ: How I treat refractory acute GVHD. Blood 109 (10): 4119-26, 2007.
- Jagasia M, Perales MA, Schroeder MA, et al.: Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): a multicenter, open-label phase 2 trial. Blood 135 (20): 1739-1749, 2020.
- Laisne L, Neven B, Dalle JH, et al.: Ruxolitinib in children with steroid-refractory acute graft-versus-host disease: A retrospective multicenter study of the pediatric group of SFGM-TC. Pediatr Blood Cancer 67 (9): e28233, 2020.
- Locatelli F, Kang HJ, Bruno B, et al.: Ruxolitinib for pediatric patients with treatment-naïve and steroid-refractory acute graft-versus-host disease: the REACH4 study. Blood 144 (20): 2095-2106, 2024.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
Page Footer
I want to...
Audiences
Secure Member Sites
The Cigna Group Information
Disclaimer
Individual and family medical and dental insurance plans are insured by Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc., and Cigna HealthCare of Texas, Inc. Group health insurance and health benefit plans are insured or administered by CHLIC, Connecticut General Life Insurance Company (CGLIC), or their affiliates (see
All insurance policies and group benefit plans contain exclusions and limitations. For availability, costs and complete details of coverage, contact a licensed agent or Cigna sales representative. This website is not intended for residents of New Mexico.