Ewing Sarcoma and Undifferentiated Small Round Cell Sarcomas of Bone and Soft Tissue Treatment (PDQ®): Treatment - Health Professional Information [NCI]
General Information About Ewing Sarcoma and Undifferentiated Small Round Cell Sarcomas of Bone and Soft Tissue
Dramatic improvements in survival have been achieved for children and adolescents with cancer.[
Studies using immunohistochemical markers,[
The World Health Organization (WHO) classification of tumors of soft tissue and bone was modified in 2020 to introduce a new chapter on undifferentiated small round cell sarcomas of bone and soft tissue. This WHO chapter consists of Ewing sarcoma and three main categories, including round cell sarcomas with EWSR1::non-ETS fusions, CIC-rearranged sarcoma, and sarcomas with BCOR genetic alterations.[
Before the widespread availability of genomic testing, Ewing sarcoma was identified by the appearance of small, round, blue cells on light microscopic examination, along with positive staining for CD99 by immunohistochemistry. The identification of the recurring t(11;22) translocation in most Ewing sarcoma tumors led to the discovery that most tumors classified as Ewing sarcoma had a translocation that juxtaposed a portion of the EWSR1 gene to a portion of a gene in the ETS family, resulting in a transforming transcript. Not all undifferentiated small round cell sarcomas of bone and soft tissue have such a translocation. Further research identified additional genetic changes, including tumors with translocations of the CIC gene or the BCOR gene. These groups of tumors occur much less frequently than Ewing sarcoma, and data on these patients are based on smaller sample sizes and less homogeneous treatment; therefore, patient outcomes are harder to quantify with precision. Most of these tumors have been treated with regimens designed for Ewing sarcoma, and the consensus was that they were often included in clinical trials for the treatment of Ewing sarcoma, sometimes referred to as translocation-negative Ewing sarcoma. It is now agreed that these tumors are sufficiently different from Ewing sarcoma and that they should be stratified and analyzed separately from Ewing sarcoma, even if they are treated with similar therapy. In this summary, these tumors are described separately. For more information about these smaller groups of tumors, see the following sections:
-
Undifferentiated Small Round Cell Sarcomas With BCOR Genetic Alterations . -
Undifferentiated Small Round Cell Sarcomas With CIC Genetic Alterations . -
Undifferentiated Small Round Cell Sarcomas With EWSR1::non-ETS Fusions .
Incidence
In the United States between 2016 and 2020, the National Childhood Cancer Registry (NCCR) reported an incidence rate of Ewing sarcoma and related sarcomas of bone of 3.0 cases per 1 million in children and adolescents younger than 20 years.[
| Age (years) | Rate per 1,000,000 | 95% Confidence Interval |
|---|---|---|
| a Source: National Childhood Cancer Registry (NCCR) Explorer.[ |
||
| <1 | 0.5 | 0.2–1.1 |
| 1–4 | 1 | 0.7–1.3 |
| 5–9 | 2.3 | 1.9–2.6 |
| 10–14 | 4.3 | 3.9–4.9 |
| 15–19 | 4.5 | 4.0–5.0 |
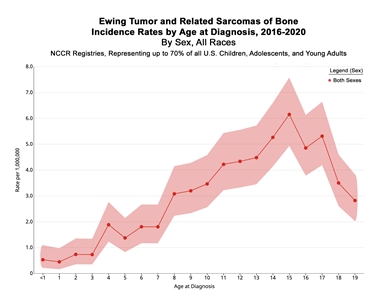
Figure 1. Incidence rates of Ewing tumor and related sarcomas of bone by age at diagnosis in the National Childhood Cancer Registry (NCCR) from 2016 to 2020. Credit: NCCR*Explorer: An interactive website for NCCR cancer statistics [Internet]. National Cancer Institute; 2023 Sep 7. [updated: 2023 Sep 8; cited 2024 Sep 4]. Available from: https://nccrexplorer.ccdi.cancer.gov.
The incidence of Ewing sarcoma in the United States is nine times greater in White people than in Black people, with an intermediate incidence in Asian people.[
Based on data from 1,426 patients entered on European Intergroup Cooperative Ewing Sarcoma Studies, 59% of patients are male and 41% are female.[
Genetic Predisposition to Ewing Sarcoma
Conventional understanding of translocation-driven sarcoma such as Ewing sarcoma suggests that these patients do not have a genetic predisposition.[
Genome-wide association studies have identified susceptibility loci for Ewing sarcoma at 1p36.22, 10q21, and 15q15.[
Clinical Presentation
Clinical presentation of Ewing sarcoma varies and depends on the tumor's size and location.
Primary sites of bone disease are listed in
| Primary Site | Incidence Rate |
|---|---|
| Skull | 5% |
| Spine | 7% |
| Rib | 11% |
| Sternum, scapula, and clavicle | 5% |
| Humerus | 7% |
| Radius, ulna, hand | 2% |
| Pelvis | 18% |
| Femur | 11% |
| Tibia, fibula, patella, foot | 14% |
| Soft tissue | 19% |
The time from the first symptom to diagnosis of Ewing sarcoma is often long, with a median interval reported from 2 to 5 months. Longer times are associated with older age and pelvic primary sites. Time from the first symptom to diagnosis has not been associated with metastasis, surgical outcome, or survival.[
Approximately 25% of patients with Ewing sarcoma have metastatic disease at the time of diagnosis, with lung, bone, and bone marrow being the most common metastatic sites.[
A retrospective analysis examined patients treated on two Children's Oncology Group (COG) studies, INT-0154 and AEWS0031 (NCT00006734). This study compared the clinical characteristics of 213 patients with extraskeletal primary Ewing sarcoma with those of 826 patients with primary Ewing sarcoma of bone.[
The Surveillance, Epidemiology, and End Results (SEER) Program database was used to compare patients younger than 40 years with Ewing sarcoma who presented with skeletal and extraosseous primary sites (see
| Characteristic | Extraosseous Ewing Sarcoma | Skeletal Ewing Sarcoma | P Value |
|---|---|---|---|
| a Adapted from Applebaum et al.[ |
|||
| Mean age (range), years | 20 (0–39) | 16 (0–39) | <.001 |
| Male | 53% | 63% | <.001 |
| White race | 85% | 93% | <.001 |
| Axial primary sites | 73% | 54% | <.001 |
| Pelvic primary sites | 20% | 27% | .001 |
Diagnostic Evaluation
The following tests and procedures may be used to diagnose or stage Ewing sarcoma:
- Physical examination and history.
- Magnetic resonance imaging (MRI) of primary tumor site.
- Computed tomography (CT) scan of chest.
- Positron emission tomography (PET) scan.
- Bone scan. Bone scan was traditionally routinely performed on all patients with Ewing sarcoma for staging. However, many investigators believe that the PET scan can replace the bone scan.[
25 ,26 ] - Bone marrow aspiration and biopsy.
- X-ray of primary bone sites.
- Complete blood count.
- Blood chemistry studies, such as lactate dehydrogenase (LDH).
Skip metastasis evaluation is important for primary appendicular bone tumors. Thus, imaging of the entire involved bone is standardly performed. In one retrospective study, skip metastasis was seen in 15.8% of patients. The presence of skip metastasis was associated with an increased risk of distant metastatic disease.[
Omission of bone marrow biopsy and aspiration may be considered, when fluorine F 18-fludeoxyglucose (18F-FDG) PET imaging is used, in patients with otherwise localized disease after initial staging studies. A systematic review of Ewing sarcoma studies was performed to assess the incidence of bone marrow metastasis and the role of 18F-FDG PET imaging to detect bone marrow metastasis.[
Prognostic Factors
The two major types of prognostic factors for patients with Ewing sarcoma are grouped as follows:
-
Pretreatment factors . -
Response to initial therapy factors .
Pretreatment factors
- Metastases: The presence or absence of metastatic disease is the single most powerful predictor of outcome. Any metastatic disease defined by standard imaging techniques or bone marrow aspirate/biopsy by morphology is an adverse prognostic factor. Metastases at diagnosis are detected in about 25% of patients.[
11 ]Patients with metastatic disease confined to the lung have a better prognosis than patients with extrapulmonary metastatic sites.[
29 ,30 ,31 ,32 ] The number of pulmonary lesions does not seem to correlate with outcome, but patients with unilateral lung involvement have a better prognosis than patients with bilateral lung involvement.[33 ]Patients with metastasis to only bone seem to have a better outcome than patients with metastases to both bone and lung.[
34 ,35 ]Based on an analysis from the SEER database, regional lymph node involvement in patients is associated with an inferior overall outcome when compared with patients without regional lymph node involvement.[
36 ] - Site of tumor: Patients with Ewing sarcoma in the distal extremities have more favorable outcomes. Patients with Ewing sarcoma in the proximal extremities have an intermediate prognosis, followed by patients with central or pelvic sites.[
29 ,31 ,32 ,37 ] However, a trial from the COG showed similar outcomes for patients with pelvic primary tumors compared with other sites.[21 ]One study retrospectively analyzed a single-institution's experience with visceral Ewing sarcoma. The study focused on surgical management and compared the outcomes of patients with visceral Ewing sarcoma with those of patients with osseous and soft tissue Ewing sarcoma.[
38 ] There were 156 patients with Ewing sarcoma identified: 117 osseous Ewing sarcomas, 20 soft tissue Ewing sarcomas, and 19 visceral Ewing sarcomas. Visceral Ewing sarcomas arose in the kidneys (n = 5), lungs (n = 5), intestines (n = 2), esophagus (n = 1), liver (n = 1), pancreas (n = 1), adrenal gland (n = 1), vagina (n = 1), brain (n = 1), and spinal cord (n = 1). Visceral Ewing sarcoma was more frequently metastatic at presentation (63.2%; P = .005). However, there was no significant difference in overall survival (OS) or relapse-free survival among the Ewing sarcoma groups, with similar follow-up intervals. - Extraskeletal versus skeletal primary tumors: The COG performed a retrospective analysis from two large cooperative trials that used similar treatment regimens.[
23 ] They identified 213 patients with extraskeletal primary tumors and 826 patients with skeletal primary tumors. Patients with extraskeletal primary tumors were more likely to have an axial primary site, less likely to have large primary tumors, and had a statistically significant better prognosis than did patients with skeletal primary tumors. - Tumor size or volume: Most studies have shown that tumor size or volume is an important prognostic factor. Cutoffs of a volume of 100 mL or 200 mL and/or single dimension greater than 8 cm are used to define larger tumors. Larger tumors tend to occur in unfavorable sites.[
31 ,32 ,39 ] - Age: Younger patients generally have a better prognosis than older patients, as noted in the following studies:[
13 ,29 ,32 ,37 ,40 ,41 ,42 ]- In North American studies, patients younger than 10 years had a better outcome than those aged 10 to 17 years at diagnosis (relative risk [RR], 1.4). Patients older than 18 years had an inferior outcome (RR, 2.5).[
43 ,44 ,45 ] - A retrospective review of two consecutive German trials for Ewing sarcoma identified 47 patients older than 40 years.[
46 ] With adequate multimodal therapy, survival was comparable to the survival observed in adolescents treated on the same trials. - Review of the SEER database from 1973 to 2011 identified 1,957 patients with Ewing sarcoma.[
47 ] Thirty-nine of these patients (2.0%) were younger than 12 months at diagnosis. Infants were less likely to receive radiation therapy and more likely to have soft tissue primary sites. Early death was more common in infants, but the OS did not differ significantly from that of older patients. - A European retrospective review identified 2,635 patients with Ewing sarcoma of bone.[
48 ] Sites of primary and metastatic tumors differed according to the age groups of young children (0–9 years), early adolescence (10–14 years), late adolescence (15–19 years), young adults (20–24 years), and adults (older than 24 years). Young children had the most striking differences in site of disease, with a lower proportion of pelvic primary and axial tumors. Young children also presented less often with metastatic disease at diagnosis.
- In North American studies, patients younger than 10 years had a better outcome than those aged 10 to 17 years at diagnosis (relative risk [RR], 1.4). Patients older than 18 years had an inferior outcome (RR, 2.5).[
- Sex: Females with Ewing sarcoma have a better prognosis than males with Ewing sarcoma.[
14 ,32 ,37 ] - Serum LDH: Increased serum LDH levels before treatment are associated with inferior prognosis. Increased LDH levels are also associated with large primary tumors and metastatic disease.[
37 ] - Pathological fracture: A single-institution retrospective analysis of 78 patients with Ewing sarcoma suggested that pathological fracture at initial presentation was associated with inferior event-free survival (EFS) and OS.[
49 ][Level of evidence C1] Another study found that pathological fracture at the time of diagnosis did not preclude surgical resection and was not associated with an adverse outcome.[50 ] - Previous treatment for cancer: In the SEER database, 58 patients with Ewing sarcoma were diagnosed after treatment for a previous malignancy (2.1% of patients with Ewing sarcoma). These patients were compared with 2,756 patients with Ewing sarcoma as a first cancer over the same period. Patients with Ewing sarcoma as a second malignant neoplasm were older (secondary Ewing sarcoma, mean age of 47.8 years; primary Ewing sarcoma, mean age of 22.5 years), more likely to have a primary tumor in an axial or extraskeletal site, and had a worse prognosis (5-year OS rates of 43.5% for patients with secondary Ewing sarcoma and 64.2% for patients with primary Ewing sarcoma).[
51 ] - Chromosomal alterations:
- Complex karyotype (defined as the presence of five or more independent chromosome abnormalities at diagnosis) and modal chromosome numbers lower than 50 appear to have adverse prognostic significance.[
52 ] - Gain of chromosome 1q and/or deletion of chromosome 16q has been associated with inferior prognosis for patients with Ewing sarcoma in several cohorts.[
53 ,54 ,55 ] These two chromosomal alterations commonly occur together across a range of cancer types, including Ewing sarcoma.[56 ] Their co-occurrence is likely a result of their derivation from an unbalanced t(1;16) translocation resulting in gain of chromosome 1q together with loss of chromosomal material from 16q.[57 ,58 ]
- Complex karyotype (defined as the presence of five or more independent chromosome abnormalities at diagnosis) and modal chromosome numbers lower than 50 appear to have adverse prognostic significance.[
- Detectable Ewing sarcoma cells, fusion transcripts, or circulating tumor DNA (ctDNA) in peripheral blood: Several techniques to evaluate the presence of Ewing sarcoma in the peripheral blood have been proposed. Flow cytometry for cells that express the CD99 antigen was not sufficiently sensitive to serve as a reliable biomarker.[
59 ,60 ] Reverse transcriptase–polymerase chain reaction (RT-PCR) for the EWSR1::FLI1 translocation was also not considered a reliable biomarker.[61 ]A more sensitive technique used patient-specific primers designed after identification of the specific translocation breakpoint in combination with droplet digital PCR to detect the EWSR1 fusion. This technique reported a sensitivity threshold of 0.009% to 0.018%.[
62 ] Levels of circulating cell-free DNA were higher in patients with metastatic disease than in patients with localized disease.A next-generation sequencing hybrid capture assay and an ultra-low-pass whole-genome sequencing assay were used to detect the EWSR1 fusion in ctDNA in banked plasma from patients with Ewing sarcoma. Among patients with newly diagnosed localized Ewing sarcoma, detectable ctDNA was associated with inferior 3-year EFS rates (48.6% vs. 82.1%; P = .006) and OS rates (79.8% vs. 92.6%; P = .01).[
63 ]ctDNA was separately assayed by digital-droplet PCR in 102 patients who were treated in the EWING2008 (NCT00987636) trial.[
64 ] Pretreatment ctDNA copy numbers correlated with EFS and OS. A reduction in ctDNA levels below the detection limit was observed in most patients after only two blocks of vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) induction chemotherapy. The persistence of ctDNA after two blocks of VIDE was a strong predictor of poor outcomes. - Detectable fusion transcripts in morphologically normal marrow: RT-PCR can be used to detect fusion transcripts in bone marrow. In a single retrospective study using patients with normal marrow morphology and no other metastatic site, fusion transcript detection in marrow or peripheral blood was associated with an increased risk of relapse.[
60 ] However, a larger cohort (n = 225) of patients with localized Ewing sarcoma did not show a difference in EFS or OS based on the detection of fusion transcripts in blood or bone marrow.[65 ] - Gene alterations: A prospective analysis of TP53 variants and/or CDKN2A deletions was done in patients with Ewing sarcoma enrolled on COG clinical trials. The analysis found no association of these alterations with EFS.[
66 ]In a study of 299 patients with Ewing sarcoma, 41 patients (14%) had STAG2 variants and 16 patients (5%) had TP53 variants.[
55 ] There was no association with OS for patients with either the STAG2 or TP53 variant alone. However, the nine patients (3%) with tumors that had both STAG2 and TP53 variants had a significantly decreased OS rate (<20% at 4 years).The COG analyzed STAG2 expression by immunohistochemistry in children with Ewing sarcoma who participated in frontline treatment trials.[
67 ] STAG2 was lost in 29 of 108 patients with localized disease and in 6 of 27 patients with metastatic disease. Among patients who had immunohistochemistry and sequencing performed, no cases (0 of 17) with STAG2 expression had STAG2 variants, and 2 of 7 cases with STAG2 loss had STAG2 variants. Among patients with localized disease, the 5-year EFS rate was 54% (95% CI, 34%–70%) for those with STAG2 loss, compared with 75% (95% CI, 63%–84%) for those with STAG2 expression (P = .0034).
The following are not considered to be adverse prognostic factors for Ewing sarcoma:
- Histopathology: The degree of neural differentiation is not a prognostic factor in Ewing sarcoma.[
68 ,69 ] - Fusion subtype: The EWSR1::ETS translocation associated with Ewing sarcoma can occur at several potential breakpoints in each of the genes that join to form the novel segment of DNA. Once thought to be significant,[
70 ] two large series have shown that the EWSR1::ETS translocation breakpoint site is not an adverse prognostic factor.[71 ,72 ]
Response to initial therapy factors
Multiple studies have shown that patients with minimal or no residual viable tumor after presurgical chemotherapy have a significantly better EFS than do patients with larger amounts of viable tumor.[
Patients with poor response to presurgical chemotherapy have an increased risk of local recurrence.[
A retrospective analysis of risk factors for recurrence was performed in patients who received initial chemotherapy and underwent surgical resection of the primary tumor.[
- Adverse risk factors for local recurrence were pelvic primary tumors (hazard ratio [HR], 2.04; 95% CI, 1.10–3.80) and marginal/intralesional resection (HR, 2.28; 95% CI, 1.25–4.16). The addition of radiation therapy was associated with improved outcome (HR, 0.52; 95% CI, 0.28–0.95).
- Adverse risk factors for developing new pulmonary metastasis were less than 90% necrosis (HR, 2.13; 95% CI, 1.13–4.00) and previous pulmonary metastasis (HR, 4.90; 95% CI, 2.28–8.52).
- Adverse risk factors for death included pulmonary metastasis (HR, 8.08; 95% CI, 4.01–16.29), bone or other metastasis (HR, 10.23; 95% CI, 4.90–21.36), and less than 90% necrosis (HR, 6.35; 95% CI, 3.18–12.69).
- Early local recurrence (0–24 months) negatively influenced survival (HR, 3.79; 95% CI, 1.34–10.76).
In a retrospective cohort of 148 patients with pulmonary metastatic Ewing sarcoma, 41.2% had radiographic resolution of lung nodules after initial induction chemotherapy.[
References:
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute.
Available online . Last accessed August 23, 2024. - Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute.
Available online . Last accessed September 5, 2024. - Olsen SH, Thomas DG, Lucas DR: Cluster analysis of immunohistochemical profiles in synovial sarcoma, malignant peripheral nerve sheath tumor, and Ewing sarcoma. Mod Pathol 19 (5): 659-68, 2006.
- Delattre O, Zucman J, Melot T, et al.: The Ewing family of tumors--a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 331 (5): 294-9, 1994.
- Dagher R, Pham TA, Sorbara L, et al.: Molecular confirmation of Ewing sarcoma. J Pediatr Hematol Oncol 23 (4): 221-4, 2001.
- Llombart-Bosch A, Carda C, Peydro-Olaya A, et al.: Soft tissue Ewing's sarcoma. Characterization in established cultures and xenografts with evidence of a neuroectodermic phenotype. Cancer 66 (12): 2589-601, 1990.
- Suvà ML, Riggi N, Stehle JC, et al.: Identification of cancer stem cells in Ewing's sarcoma. Cancer Res 69 (5): 1776-81, 2009.
- Tirode F, Laud-Duval K, Prieur A, et al.: Mesenchymal stem cell features of Ewing tumors. Cancer Cell 11 (5): 421-9, 2007.
- Choi JH, Ro JY: The 2020 WHO Classification of Tumors of Soft Tissue: Selected Changes and New Entities. Adv Anat Pathol 28 (1): 44-58, 2021.
- Esiashvili N, Goodman M, Marcus RB: Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol 30 (6): 425-30, 2008.
- Kim SY, Tsokos M, Helman LJ: Dilemmas associated with congenital ewing sarcoma family tumors. J Pediatr Hematol Oncol 30 (1): 4-7, 2008.
- van den Berg H, Dirksen U, Ranft A, et al.: Ewing tumors in infants. Pediatr Blood Cancer 50 (4): 761-4, 2008.
- Jawad MU, Cheung MC, Min ES, et al.: Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: an analysis of 1631 cases from the SEER database, 1973-2005. Cancer 115 (15): 3526-36, 2009.
- Beck R, Monument MJ, Watkins WS, et al.: EWS/FLI-responsive GGAA microsatellites exhibit polymorphic differences between European and African populations. Cancer Genet 205 (6): 304-12, 2012.
- Grünewald TG, Bernard V, Gilardi-Hebenstreit P, et al.: Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet 47 (9): 1073-8, 2015.
- Paulussen M, Craft AW, Lewis I, et al.: Results of the EICESS-92 Study: two randomized trials of Ewing's sarcoma treatment--cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol 26 (27): 4385-93, 2008.
- Gillani R, Camp SY, Han S, et al.: Germline predisposition to pediatric Ewing sarcoma is characterized by inherited pathogenic variants in DNA damage repair genes. Am J Hum Genet 109 (6): 1026-1037, 2022.
- Postel-Vinay S, Véron AS, Tirode F, et al.: Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet 44 (3): 323-7, 2012.
- Machiela MJ, Grünewald TGP, Surdez D, et al.: Genome-wide association study identifies multiple new loci associated with Ewing sarcoma susceptibility. Nat Commun 9 (1): 3184, 2018.
- Leavey PJ, Laack NN, Krailo MD, et al.: Phase III Trial Adding Vincristine-Topotecan-Cyclophosphamide to the Initial Treatment of Patients With Nonmetastatic Ewing Sarcoma: A Children's Oncology Group Report. J Clin Oncol 39 (36): 4029-4038, 2021.
- Brasme JF, Chalumeau M, Oberlin O, et al.: Time to diagnosis of Ewing tumors in children and adolescents is not associated with metastasis or survival: a prospective multicenter study of 436 patients. J Clin Oncol 32 (18): 1935-40, 2014.
- Cash T, McIlvaine E, Krailo MD, et al.: Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 63 (10): 1771-9, 2016.
- Applebaum MA, Worch J, Matthay KK, et al.: Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer 117 (13): 3027-32, 2011.
- Tal AL, Doshi H, Parkar F, et al.: The Utility of 18FDG PET/CT Versus Bone Scan for Identification of Bone Metastases in a Pediatric Sarcoma Population and a Review of the Literature. J Pediatr Hematol Oncol 43 (2): 52-58, 2021.
- Costelloe CM, Chuang HH, Daw NC: PET/CT of Osteosarcoma and Ewing Sarcoma. Semin Roentgenol 52 (4): 255-268, 2017.
- Saifuddin A, Michelagnoli M, Pressney I: Skip metastases in appendicular Ewing sarcoma: relationship to distant metastases at diagnosis, chemotherapy response and overall survival. Skeletal Radiol 52 (3): 585-591, 2023.
- Campbell KM, Shulman DS, Grier HE, et al.: Role of bone marrow biopsy for staging new patients with Ewing sarcoma: A systematic review. Pediatr Blood Cancer 68 (2): e28807, 2021.
- Cotterill SJ, Ahrens S, Paulussen M, et al.: Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol 18 (17): 3108-14, 2000.
- Miser JS, Krailo MD, Tarbell NJ, et al.: Treatment of metastatic Ewing's sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide--a Children's Cancer Group and Pediatric Oncology Group study. J Clin Oncol 22 (14): 2873-6, 2004.
- Rodríguez-Galindo C, Liu T, Krasin MJ, et al.: Analysis of prognostic factors in ewing sarcoma family of tumors: review of St. Jude Children's Research Hospital studies. Cancer 110 (2): 375-84, 2007.
- Karski EE, McIlvaine E, Segal MR, et al.: Identification of Discrete Prognostic Groups in Ewing Sarcoma. Pediatr Blood Cancer 63 (1): 47-53, 2016.
- Paulussen M, Ahrens S, Craft AW, et al.: Ewing's tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing's Sarcoma Studies patients. J Clin Oncol 16 (9): 3044-52, 1998.
- Paulussen M, Ahrens S, Burdach S, et al.: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9 (3): 275-81, 1998.
- Ladenstein R, Pötschger U, Le Deley MC, et al.: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28 (20): 3284-91, 2010.
- Applebaum MA, Goldsby R, Neuhaus J, et al.: Clinical features and outcomes in patients with Ewing sarcoma and regional lymph node involvement. Pediatr Blood Cancer 59 (4): 617-20, 2012.
- Bacci G, Longhi A, Ferrari S, et al.: Prognostic factors in non-metastatic Ewing's sarcoma tumor of bone: an analysis of 579 patients treated at a single institution with adjuvant or neoadjuvant chemotherapy between 1972 and 1998. Acta Oncol 45 (4): 469-75, 2006.
- Wallace MW, Niec JA, Ghani MOA, et al.: Distribution and Surgical Management of Visceral Ewing Sarcoma Among Children and Adolescents. J Pediatr Surg 58 (9): 1727-1735, 2023.
- Ahrens S, Hoffmann C, Jabar S, et al.: Evaluation of prognostic factors in a tumor volume-adapted treatment strategy for localized Ewing sarcoma of bone: the CESS 86 experience. Cooperative Ewing Sarcoma Study. Med Pediatr Oncol 32 (3): 186-95, 1999.
- De Ioris MA, Prete A, Cozza R, et al.: Ewing sarcoma of the bone in children under 6 years of age. PLoS One 8 (1): e53223, 2013.
- Huh WW, Daw NC, Herzog CE, et al.: Ewing sarcoma family of tumors in children younger than 10 years of age. Pediatr Blood Cancer 64 (4): , 2017.
- Ahmed SK, Randall RL, DuBois SG, et al.: Identification of Patients With Localized Ewing Sarcoma at Higher Risk for Local Failure: A Report From the Children's Oncology Group. Int J Radiat Oncol Biol Phys 99 (5): 1286-1294, 2017.
- Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348 (8): 694-701, 2003.
- Granowetter L, Womer R, Devidas M, et al.: Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol 27 (15): 2536-41, 2009.
- Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (33): 4148-54, 2012.
- Pieper S, Ranft A, Braun-Munzinger G, et al.: Ewing's tumors over the age of 40: a retrospective analysis of 47 patients treated according to the International Clinical Trials EICESS 92 and EURO-E.W.I.N.G. 99. Onkologie 31 (12): 657-63, 2008.
- Wong T, Goldsby RE, Wustrack R, et al.: Clinical features and outcomes of infants with Ewing sarcoma under 12 months of age. Pediatr Blood Cancer 62 (11): 1947-51, 2015.
- Worch J, Ranft A, DuBois SG, et al.: Age dependency of primary tumor sites and metastases in patients with Ewing sarcoma. Pediatr Blood Cancer 65 (9): e27251, 2018.
- Schlegel M, Zeumer M, Prodinger PM, et al.: Impact of Pathological Fractures on the Prognosis of Primary Malignant Bone Sarcoma in Children and Adults: A Single-Center Retrospective Study of 205 Patients. Oncology 94 (6): 354-362, 2018.
- Bramer JA, Abudu AA, Grimer RJ, et al.: Do pathological fractures influence survival and local recurrence rate in bony sarcomas? Eur J Cancer 43 (13): 1944-51, 2007.
- Applebaum MA, Goldsby R, Neuhaus J, et al.: Clinical features and outcomes in patients with secondary Ewing sarcoma. Pediatr Blood Cancer 60 (4): 611-5, 2013.
- Roberts P, Burchill SA, Brownhill S, et al.: Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing's sarcoma family of tumors: a study by the United Kingdom Cancer Cytogenetics and the Children's Cancer and Leukaemia Group. Genes Chromosomes Cancer 47 (3): 207-20, 2008.
- Hattinger CM, Pötschger U, Tarkkanen M, et al.: Prognostic impact of chromosomal aberrations in Ewing tumours. Br J Cancer 86 (11): 1763-9, 2002.
- Mackintosh C, Ordóñez JL, García-Domínguez DJ, et al.: 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene 31 (10): 1287-98, 2012.
- Tirode F, Surdez D, Ma X, et al.: Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 4 (11): 1342-53, 2014.
- Mrózek K, Bloomfield CD: Der(16)t(1;16) is a secondary chromosome aberration in at least eighteen different types of human cancer. Genes Chromosomes Cancer 23 (1): 78-80, 1998.
- Mugneret F, Lizard S, Aurias A, et al.: Chromosomes in Ewing's sarcoma. II. Nonrandom additional changes, trisomy 8 and der(16)t(1;16). Cancer Genet Cytogenet 32 (2): 239-45, 1988.
- Hattinger CM, Rumpler S, Ambros IM, et al.: Demonstration of the translocation der(16)t(1;16)(q12;q11.2) in interphase nuclei of Ewing tumors. Genes Chromosomes Cancer 17 (3): 141-50, 1996.
- Dubois SG, Epling CL, Teague J, et al.: Flow cytometric detection of Ewing sarcoma cells in peripheral blood and bone marrow. Pediatr Blood Cancer 54 (1): 13-8, 2010.
- Schleiermacher G, Peter M, Oberlin O, et al.: Increased risk of systemic relapses associated with bone marrow micrometastasis and circulating tumor cells in localized ewing tumor. J Clin Oncol 21 (1): 85-91, 2003.
- Zoubek A, Ladenstein R, Windhager R, et al.: Predictive potential of testing for bone marrow involvement in Ewing tumor patients by RT-PCR: a preliminary evaluation. Int J Cancer 79 (1): 56-60, 1998.
- Shukla NN, Patel JA, Magnan H, et al.: Plasma DNA-based molecular diagnosis, prognostication, and monitoring of patients with EWSR1 fusion-positive sarcomas. JCO Precis Oncol 2017: , 2017.
- Shulman DS, Klega K, Imamovic-Tuco A, et al.: Detection of circulating tumour DNA is associated with inferior outcomes in Ewing sarcoma and osteosarcoma: a report from the Children's Oncology Group. Br J Cancer 119 (5): 615-621, 2018.
- Krumbholz M, Eiblwieser J, Ranft A, et al.: Quantification of Translocation-Specific ctDNA Provides an Integrating Parameter for Early Assessment of Treatment Response and Risk Stratification in Ewing Sarcoma. Clin Cancer Res 27 (21): 5922-5930, 2021.
- Vo KT, Edwards JV, Epling CL, et al.: Impact of Two Measures of Micrometastatic Disease on Clinical Outcomes in Patients with Newly Diagnosed Ewing Sarcoma: A Report from the Children's Oncology Group. Clin Cancer Res 22 (14): 3643-50, 2016.
- Lerman DM, Monument MJ, McIlvaine E, et al.: Tumoral TP53 and/or CDKN2A alterations are not reliable prognostic biomarkers in patients with localized Ewing sarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (5): 759-65, 2015.
- Shulman DS, Chen S, Hall D, et al.: Adverse prognostic impact of the loss of STAG2 protein expression in patients with newly diagnosed localised Ewing sarcoma: A report from the Children's Oncology Group. Br J Cancer 127 (12): 2220-2226, 2022.
- Parham DM, Hijazi Y, Steinberg SM, et al.: Neuroectodermal differentiation in Ewing's sarcoma family of tumors does not predict tumor behavior. Hum Pathol 30 (8): 911-8, 1999.
- Luksch R, Sampietro G, Collini P, et al.: Prognostic value of clinicopathologic characteristics including neuroectodermal differentiation in osseous Ewing's sarcoma family of tumors in children. Tumori 85 (2): 101-7, 1999 Mar-Apr.
- de Alava E, Kawai A, Healey JH, et al.: EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing's sarcoma. J Clin Oncol 16 (4): 1248-55, 1998.
- van Doorninck JA, Ji L, Schaub B, et al.: Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 28 (12): 1989-94, 2010.
- Le Deley MC, Delattre O, Schaefer KL, et al.: Impact of EWS-ETS fusion type on disease progression in Ewing's sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol 28 (12): 1982-8, 2010.
- Paulussen M, Ahrens S, Dunst J, et al.: Localized Ewing tumor of bone: final results of the cooperative Ewing's Sarcoma Study CESS 86. J Clin Oncol 19 (6): 1818-29, 2001.
- Rosito P, Mancini AF, Rondelli R, et al.: Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: a preliminary report of 6 years of experience. Cancer 86 (3): 421-8, 1999.
- Wunder JS, Paulian G, Huvos AG, et al.: The histological response to chemotherapy as a predictor of the oncological outcome of operative treatment of Ewing sarcoma. J Bone Joint Surg Am 80 (7): 1020-33, 1998.
- Oberlin O, Deley MC, Bui BN, et al.: Prognostic factors in localized Ewing's tumours and peripheral neuroectodermal tumours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer 85 (11): 1646-54, 2001.
- Lozano-Calderón SA, Albergo JI, Groot OQ, et al.: Complete tumor necrosis after neoadjuvant chemotherapy defines good responders in patients with Ewing sarcoma. Cancer 129 (1): 60-70, 2023.
- Ferrari S, Bertoni F, Palmerini E, et al.: Predictive factors of histologic response to primary chemotherapy in patients with Ewing sarcoma. J Pediatr Hematol Oncol 29 (6): 364-8, 2007.
- Hawkins DS, Schuetze SM, Butrynski JE, et al.: [18F]Fluorodeoxyglucose positron emission tomography predicts outcome for Ewing sarcoma family of tumors. J Clin Oncol 23 (34): 8828-34, 2005.
- Denecke T, Hundsdörfer P, Misch D, et al.: Assessment of histological response of paediatric bone sarcomas using FDG PET in comparison to morphological volume measurement and standardized MRI parameters. Eur J Nucl Med Mol Imaging 37 (10): 1842-53, 2010.
- Palmerini E, Colangeli M, Nanni C, et al.: The role of FDG PET/CT in patients treated with neoadjuvant chemotherapy for localized bone sarcomas. Eur J Nucl Med Mol Imaging 44 (2): 215-223, 2017.
- Lin PP, Jaffe N, Herzog CE, et al.: Chemotherapy response is an important predictor of local recurrence in Ewing sarcoma. Cancer 109 (3): 603-11, 2007.
- Bosma SE, Rueten-Budde AJ, Lancia C, et al.: Individual risk evaluation for local recurrence and distant metastasis in Ewing sarcoma: A multistate model: A multistate model for Ewing sarcoma. Pediatr Blood Cancer 66 (11): e27943, 2019.
- Reiter AJ, Huang L, Craig BT, et al.: Survival outcomes in pediatric patients with metastatic Ewing sarcoma who achieve a rapid complete response of pulmonary metastases. Pediatr Blood Cancer 71 (7): e31026, 2024.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
Page Footer
I want to...
Audiences
Secure Member Sites
The Cigna Group Information
Disclaimer
Individual and family medical and dental insurance plans are insured by Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc., and Cigna HealthCare of Texas, Inc. Group health insurance and health benefit plans are insured or administered by CHLIC, Connecticut General Life Insurance Company (CGLIC), or their affiliates (see
All insurance policies and group benefit plans contain exclusions and limitations. For availability, costs and complete details of coverage, contact a licensed agent or Cigna sales representative. This website is not intended for residents of New Mexico.