Genetics of Breast and Gynecologic Cancers (PDQ®): Genetics - Health Professional Information [NCI]
Introduction
General Information
Among women in the United States, breast cancer is the most commonly diagnosed cancer after nonmelanoma skin cancer, and it is the second leading cause of cancer deaths after lung cancer. In 2025, an estimated 319,750 new cases of breast cancer (including 2,800 cases in men) will be diagnosed, and 42,680 deaths (including 510 deaths in men) will occur.[
A possible genetic contribution to both breast and ovarian cancer risk is indicated by the increased incidence of these cancers among women with a family history (refer to the
Risk Factors for Breast Cancer
This section discusses factors that can modify an individual's risk of developing breast cancer. These risk factors can affect women in the general population, women who have a family histories of breast cancer, and women who carry pathogenic variants in breast cancer risk genes. For more information on breast cancer risk factors in the general population, see
The following breast cancer risk factors are discussed in this section:
-
Age . -
Family history of breast cancer . -
Benign breast disease, mammographic density, and background parenchymal enhancement . -
Parity, age at first birth, and breastfeeding . -
Contraceptives . -
Hormone replacement therapy (HRT) . -
Radiation exposure . -
Alcohol and smoking . -
Physical activity .
These factors can increase or decrease breast cancer risk in all women. However, they may affect breast cancer risk differently in women with increased breast cancer susceptibility (i.e., women who have high-risk family histories and/or pathogenic variants in hereditary breast cancer genes). Factors that increase breast cancer risk in the general population may lower breast cancer risk, increase breast cancer risk more than expected, or have no effect on breast cancer risk in women with high breast cancer susceptibility. In some cases, these risk factors may affect high-risk women in the same way that they affect average-risk women. Furthermore, modifying risk factors has a greater effect on the absolute breast cancer risk in women with high breast cancer susceptibility than in women with low breast cancer susceptibility.[
Age
Like other cancer types, breast cancer's cumulative risk increases with age. As individuals age, they encounter more environmental exposures and accumulate genomic changes. Hence, most breast cancers occur after age 50 years.[
Family history of breast cancer
A family history of breast cancer is a well-established, consistent risk factor for breast cancer. Approximately 5% to 10% of women with breast cancer also had a mother or sister with breast cancer in cross-sectional studies. About 10% to 20% of women had a first-degree relative (FDR) or a second-degree relative (SDR) with breast cancer.[
The following factors can increase a woman's breast cancer risk:
- Large number of affected relatives.
- Family members who were diagnosed with breast cancer at young ages.
- Family members with bilateral breast cancers.
- Family members with multiple ipsilateral breast cancers.
- Male relatives with breast cancer.
Furthermore, women with family histories of multiple breast cancers had higher hazard ratios (HRs) (HR, 2.7; 95% CI, 2.6–2.9) than women who had a single breast cancer in their families (HR, 1.8; 95% CI, 1.8–1.9). When women had multiple breast cancers in their families (with one breast cancer occurring before age 40 years), the HR was 3.8 (95% CI, 3.1–4.8). However, breast cancer risk also significantly increased when a relative was diagnosed with breast cancer at 60 years or older, suggesting that having a relative with breast cancer at any age can increase risk.[
Albright et al. addressed how affected third-degree relatives (TDRs) can contribute to an individual's breast cancer risk.[
One of the largest studies of twins ever conducted examined 80,309 monozygotic twins and 123,382 dizygotic twins. This study had a heritability estimate of 31% for breast cancer (95% CI, 11%–51%).[
Benign breast disease, mammographic density, and background parenchymal enhancement
Benign breast disease (BBD)
- BBD is a broad group of conditions characterized by non-cancerous changes in breast tissue. BBD can be divided into three categories: nonproliferative lesions, proliferative lesions without atypia, and atypical hyperplasias. BBD is a consistent risk factor for breast cancer in the general population.[
15 ,16 ] - BBD is also an important risk factor in women who have high breast cancer susceptibility due to family histories of cancer or pathogenic variants in breast cancer risk genes. For example, a study of 17,154 women found that women with a history of BBD have an increased risk of breast cancer that is independent of their underlying familial and genetic risks.[
17 ] However, breast cancer risk associated with personal histories of BBD did not vary between women with BRCA1 pathogenic variants (RR, 1.64; 95% CI, 1.04–2.58), women with BRCA2 pathogenic variants (RR, 1.34; 95% CI, 0.78–2.3), and women who only had family histories of breast cancer (RR, 1.31; 95% CI, 1.13–1.53). In women with high breast cancer susceptibility, BBD can further increase breast cancer risk, because it multiplies their underlying familial and genetic risks.
Mammographic density
- Women with dense breast tissue (assessed by mammogram) also have an increased risk of developing breast cancer.[
15 ,18 ,19 ] Studies have shown that breast density likely has a genetic etiology.[20 ,21 ,22 ] - A systematic review reported that women who had dense breast tissue and an FDR with breast cancer had an increased chance of developing breast cancer.[
23 ] Two retrospective studies also investigated the association between mammographic density and breast cancer risk in BRCA1 and BRCA2 carriers.[24 ,25 ] These retrospective studies had samples of 206 and 691 BRCA pathogenic variant carriers. In these studies, 96 and 248 women developed breast cancer, respectively.[24 ,25 ] The studies found that mammographic density is an independent risk factor for breast cancer in both BRCA1 and BRCA2 pathogenic variant carriers. Associations between breast density and breast cancer risk were similar to those observed in the general population (RR, 2.30 for density ≥50% vs. <50%).
Background parenchymal enhancement (BPE)
- Like breast density (assessed by mammogram), BPE (assessed by breast magnetic resonance imaging [MRI]) may increase breast cancer risk. Data have shown that moderate BPE (odds ratio [OR], 1.6; 95% CI, 1.0–2.6) and mild BPE (OR, 2.1; 95% CI, 1.5–3.0) can increase breast cancer risk in women with high breast cancer susceptibility. However, an association between mild/moderate BPE and breast cancer risk was not found in women with average breast cancer susceptibility.[
26 ]
Parity, age at first birth, and breastfeeding
Parity
- A large prospective study analyzed the relationship between parity and breast cancer risk in female BRCA1 and BRCA2 carriers. Results showed that parity affected breast cancer risk in BRCA1 and BRCA2 carriers differently. Breast cancer risk increased in uniparous BRCA1 carriers and parous BRCA2 carriers.[
27 ] In BRCA1 carriers, there was no overall association between parity and breast cancer risk when compared with nulliparity and breast cancer risk. Uniparous BRCA1 carriers were at an increased risk of breast cancer in the prospective analysis (HRprospective, 1.69; 95% CI, 1.09–2.62) when compared with nulliparous BRCA1 carriers. The results also suggested that uniparous women who breastfed may have decreased breast cancer risk when compared with those who did not breastfeed. In BRCA2 carriers, being parous was associated with a 33% increase in breast cancer risk (HRcombined, 1.33; 95% CI, 1.05–1.69). Multiparity did not decrease breast cancer risk in BRCA2 carriers, unless they had at least four full-term pregnancies (HRcombined, 0.72; 95% CI, 0.54–0.98).
Age at first birth
- In the general population, breast cancer risk increases when women have early menarche and/or late menopause. Breast cancer risk decreases when a woman's first full-term pregnancy occurs at a young age. However, these risk factors can affect women with high breast cancer susceptibility differently than women in the general population. BRCA1 and BRCA2 pathogenic variant carriers who become pregnant prior to age 30 years may have increased breast cancer risk. This effect is even more significant in BRCA1 pathogenic variant carriers.[
28 ,29 ,30 ]BRCA1 and BRCA2 pathogenic variant carriers who developed breast cancer during pregnancy or became pregnant after developing breast cancer did not experience adverse survival outcomes.[31 ]
Breastfeeding
- Breastfeeding can reduce breast cancer risk in BRCA1 (but not BRCA2) pathogenic variant carriers.[
32 ] Breastfeeding for long periods of time was associated with decreased breast cancer risk in BRCA1 carriers (P -trend = .0003).[27 ]
Reproductive history can also affect a woman's risk for ovarian cancer and endometrial cancer. For more information, see the
Contraceptives
Breast cancer risk is one of the factors to consider when prescribing contraceptives, which assist with pregnancy control, abnormal bleeding, and other gynecological symptoms. Oral contraceptives (OCs) may slightly increase breast cancer risk in long-term users, but this appears to be a short-term effect.[
Some studies show that OC use does not further increase breast cancer risk in women with high breast cancer susceptibility. For example, a meta-analysis with data from 54 studies showed that women with family histories of breast cancer did not have increased breast cancer risk from OC use.[
Some studies also suggest that the year an OC was made and a woman's age when beginning OC use may matter. For example, OCs made before 1975 are associated with increased breast cancer risk in BRCA1/2 carriers (summary relative risk [SRR], 1.47; 95% CI, 1.06–2.04).[
Other contraceptive methods have not been studied in women with pathogenic variants in breast cancer risk genes. However, studies have investigated associations between intrauterine devices and breast cancer risk in the general population. A meta-analysis and systematic review of seven studies examined the effect of the levonorgestrel-releasing intrauterine system (LNG-IUS) on breast cancer risk. The meta-analysis included studies that controlled for family history of breast cancer, but associations were not separately evaluated or stratified by family history of breast cancer. In LNG-IUS users, breast cancer risk increased in all women (OR, 1.16; 95% CI, 1.06–1.28), in women younger than 50 years (OR, 1.12; 95% CI, 1.02–1.22), and in women 50 years and older (OR, 1.52; 95% CI, 1.34–1.72).[
Hormone replacement therapy
Both observational studies and randomized clinical trials have examined the association between postmenopausal HRT and breast cancer. Short-term use of HRT for treatment of postmenopausal symptoms appears to confer little or no breast cancer risk.[
Among women with family histories of breast cancer, the associations between HRT and breast cancer risk have not been consistent. Some studies suggested risk was particularly elevated among women with family histories of breast cancer, while others did not report an interaction between these factors.[
The effect of HRT on breast cancer risk among carriers of BRCA1 and BRCA2 pathogenic variants has been studied in the context of bilateral risk-reducing oophorectomy. Short-term HRT use does not seem to alter an oophorectomy's protective effect on breast cancer risk.[
HRT use may also increase a woman's chance of developing endometrial cancer. For more information, see the
Radiation exposure
Radiation exposure can increase an individual's breast cancer risk. This is demonstrated by the survivors of the atomic bombings in Hiroshima and Nagasaki and by women who have received therapeutic radiation treatments to the chest and upper body. However, it is unclear how much radiation exposure affects breast cancer risk in women with high breast cancer susceptibility.
Early data suggested that carriers of BRCA1 and BRCA2 pathogenic variants may have increased sensitivity to radiation, which may contribute to cancer susceptibility.[
It is possible that radiation exposure from diagnostic procedures, including mammography, poses a greater risk to women with high breast cancer susceptibility than to women who are at average risk of developing breast cancer. Therapeutic radiation could also increase cancer risk in women with high breast cancer susceptibility. However, a cohort study of BRCA1 and BRCA2 pathogenic variant carriers treated with breast-conserving therapy did not show evidence of increased radiation sensitivity in participants. Sequelae were not observed in the breasts, lungs, or bone marrow of BRCA carriers.[
Conversely, tumors in women with pathogenic variants in breast cancer risk genes may be more responsive to radiation treatment than tumors in women at average breast cancer risk. Studies examining the impact of radiation exposure in carriers of BRCA1 and BRCA2 pathogenic variants have had conflicting results.[
A retrospective cohort study estimated the effect of adjuvant radiation therapy (for primary breast cancer) on CBC risk in BRCA1 and BRCA2 carriers (N, 691; median follow-up period, 8.6 y).[
Alcohol and smoking
The risk of breast cancer increases by approximately 10% for each 10 g of daily alcohol intake (approximately one drink or less) in the general population.[
Recent studies have evaluated the association between alcohol consumption, tobacco smoking, and breast cancer risk in individuals with BRCA1/2 pathogenic variants or family histories of breast cancer. One study evaluated if tobacco smoking and alcohol consumption are associated with increased breast cancer risk in BRCA1 and BRCA2 carriers using pooled data from an international cohort.[
Physical activity
Increased physical activity has been associated with reduced breast cancer risk in most epidemiological studies. This risk reduction has also been seen in studies of female BRCA1 or BRCA2 pathogenic variant carriers. For example, one study reported a 38% reduction in premenopausal breast cancer risk from moderate physical activity (OR for the top quartile of physical activity compared with the lowest level, 0.62; 95% CI, 0.40–0.96).[
Risk Factors for Ovarian Cancer
Refer to the PDQ summary on
Age
Ovarian cancer incidence rises in a linear fashion from age 30 years to age 50 years and continues to increase, though at a slower rate, thereafter. Before age 30 years, the risk of developing epithelial ovarian cancer is remote, even in hereditary cancer families.[
Family history including inherited cancer genes
Although reproductive, demographic, and lifestyle factors affect risk of ovarian cancer, the single greatest ovarian cancer risk factor is a family history of the disease. A large meta-analysis of 15 published studies estimated an OR of 3.1 for the risk of ovarian cancer associated with at least one FDR with ovarian cancer.[
Reproductive history
Nulliparity is consistently associated with an increased risk of ovarian cancer, including among carriers of BRCA/BRCA2 pathogenic variants, yet a meta-analysis identified a risk reduction only in women with four or more live births.[
Surgical history
Bilateral tubal ligation and hysterectomy are associated with reduced ovarian cancer risk,[
Oral contraceptives (OCs)
Use of OCs for 4 or more years is associated with an approximately 50% reduction in ovarian cancer risk in the general population.[
Risk Factors for Endometrial Cancer
Refer to the PDQ summary on
Age
Age is an important risk factor for endometrial cancer. Most women with endometrial cancer are diagnosed after menopause. Only 15% of women are diagnosed with endometrial cancer before age 50 years, and fewer than 5% are diagnosed before age 40 years.[
Family history including inherited cancer genes
Although the hyperestrogenic state is the most common predisposing factor for endometrial cancer, family history also plays a significant role in a woman's risk for disease. Approximately 3% to 5% of uterine cancer cases are attributable to a hereditary cause,[
Non-Lynch syndrome genes may also contribute to endometrial cancer risk. In an unselected endometrial cancer cohort undergoing multigene panel testing, approximately 3% of patients tested positive for a germline pathogenic variant in non-Lynch syndrome genes, including CHEK2, APC, ATM, BARD1, BRCA1, BRCA2, BRIP1, NBN, PTEN, and RAD51C.[
Reproductive history
Reproductive factors such as multiparity, late menarche, and early menopause decrease the risk of endometrial cancer because of the lower cumulative exposure to estrogen and the higher relative exposure to progesterone.[
Hormones
Hormonal factors that increase the risk of type I endometrial cancer are better understood. All endometrial cancers share a predominance of estrogen relative to progesterone. Prolonged exposure to estrogen or unopposed estrogen increases the risk of endometrial cancer. Endogenous exposure to estrogen can result from obesity, polycystic ovary syndrome, and nulliparity, while exogenous estrogen can result from taking unopposed estrogen or tamoxifen. Unopposed estrogen increases the risk of developing endometrial cancer by twofold to twentyfold, proportional to the duration of use.[
Autosomal Dominant Inheritance of Breast and Gynecologic Cancer Predisposition
Autosomal dominant inheritance of breast and gynecologic cancers is characterized by transmission of cancer predisposition from generation to generation, through either the mother's or the father's side of the family, with the following characteristics:
- Inheritance risk of 50%. When a parent carries an autosomal dominant genetic predisposition, each child has a 50:50 chance of inheriting the predisposition. Although the risk of inheriting the predisposition is 50%, not everyone with the predisposition will develop cancer because of incomplete penetrance and/or gender-restricted or gender-related expression.
- Both males and females can inherit and transmit an autosomal dominant cancer predisposition. A male who inherits a cancer predisposition can still pass the altered gene on to his sons and daughters.
Breast and ovarian cancer are components of several autosomal dominant cancer syndromes. The syndromes most strongly associated with both cancers are the syndromes associated with BRCA1 or BRCA2 pathogenic variants. Breast cancer is also a common feature of
Germline pathogenic variants in the genes responsible for these autosomal dominant cancer syndromes produce different clinical phenotypes of characteristic malignancies and, in some instances, associated nonmalignant abnormalities.
The family characteristics that suggest hereditary cancer predisposition include the following:
- Multiple cancers within a family.
- Cancers typically occur at an earlier age than in sporadic cases (defined as cases not associated with genetic risk).
- Two or more primary cancers in a single individual. These could be multiple primary cancers of the same type (e.g., bilateral breast cancer) or primary cancer of different types (e.g., breast cancer and ovarian cancer in the same individual or endometrial and colon cancer in the same individual).
- Cases of male breast cancer. The inheritance risk for autosomal dominant genetic conditions is 50% for both males and females, but the differing penetrance of the genes may result in some unaffected individuals in the family.
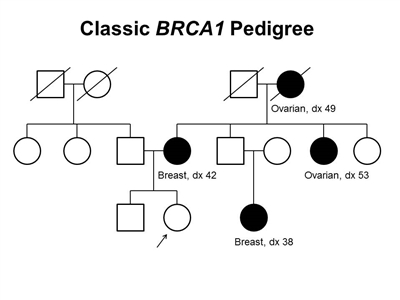
Figure 1. BRCA1 pedigree. This pedigree shows some of the classic features of a family with a BRCA1 pathogenic variant across three generations, including affected family members with breast cancer or ovarian cancer and a young age at onset. BRCA1 families may exhibit some or all of these features. As an autosomal dominant syndrome, a BRCA1 pathogenic variant can be transmitted through maternal or paternal lineages, as depicted in the figure.
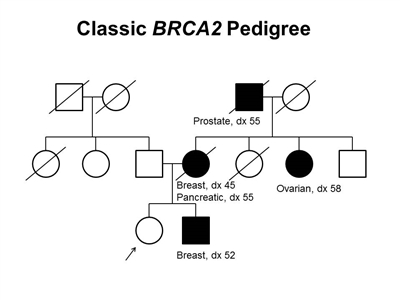
Figure 2. BRCA2 pedigree. This pedigree shows some of the classic features of a family with a BRCA2 pathogenic variant across three generations, including affected family members with breast (including male breast cancer), ovarian, pancreatic, or prostate cancers and a relatively young age at onset. BRCA2 families may exhibit some or all of these features. As an autosomal dominant syndrome, a BRCA2 pathogenic variant can be transmitted through maternal or paternal lineages, as depicted in the figure.
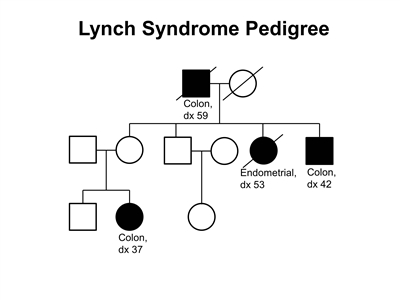
Figure 3. Lynch syndrome pedigree. This pedigree shows some of the classic features of a family with Lynch syndrome, including affected family members with colon cancer or endometrial cancer, a young age at onset in some individuals, and incomplete penetrance. Lynch syndrome families may exhibit some or all of these features. Lynch syndrome families may also include individuals with other gastrointestinal, gynecologic, and genitourinary cancers, or other extracolonic cancers. As an autosomal dominant syndrome, Lynch syndrome can be transmitted through maternal or paternal lineages, as depicted in the figure. Because the cancer risk is not 100%, individuals who have Lynch syndrome may not develop cancer, such as the mother of the female with colon cancer diagnosed at age 37 years in this pedigree (called incomplete penetrance).
There are no pathognomonic features distinguishing breast and ovarian cancers occurring in carriers of BRCA1 or BRCA2 pathogenic variants from those occurring in noncarriers. Breast cancers occurring in carriers of BRCA1 pathogenic variants are more likely to be ER-negative, progesterone receptor (PR)–negative, human epidermal growth factor receptor two (HER2/neu)–negative (i.e., triple-negative breast cancers [TNBC]), and have a basal phenotype. BRCA1-associated ovarian cancers are more likely to be high-grade and of serous histopathology. (Refer to the
Some pathologic features distinguish carriers of Lynch syndrome–associated pathogenic variants from noncarriers. The hallmark feature of endometrial cancers occurring in Lynch syndrome is mismatch repair (MMR) deficiencies, including the presence of microsatellite instability (MSI), and the absence of specific MMR proteins. In addition to these molecular changes, there are also histologic changes including tumor-infiltrating lymphocytes, peritumoral lymphocytes, undifferentiated tumor histology, lower uterine segment origin, and synchronous tumors.
Considerations in Risk Assessment and in Identifying a Family History of Breast and Ovarian Cancer Risk
The accuracy and completeness of family histories must be considered when they are used to assess risk. A reported family history may be erroneous, or a person may be unaware of relatives affected with cancer. In addition, small family sizes and premature deaths may limit the information obtained from a family history. Breast or ovarian cancer on the paternal side of the family usually involves more distant relatives than does breast or ovarian cancer on the maternal side, so information may be more difficult to obtain. When self-reported information is compared with independently verified cases, the sensitivity of a history of breast cancer is relatively high, at 83% to 97%, but lower for ovarian cancer, at 60%.[
Models for Prediction of Breast and Gynecologic Cancer Risk
Models to predict an individual's risk of developing breast and/or gynecologic cancer are available.[
- Calibration: How well the model predicts what will happen. When calibration statistics are close to 1, this means that the predicted value is similar to the actual value.
- Discrimination: How well the model can differentiate between those with and without the outcome. When only case-control data are available, the discrimination of the model (which is often assessed by measuring the area under the receiver operator curve, AUROC or AUC for short) can be assessed but the calibration cannot. An AUC of 1.0 means that the model has perfect discriminatory accuracy. AUCs closer to 0.50 show that the model is poor at discrimination. Generally, an AUC of 0.80 or higher is good to excellent, while AUCs between 0.70 and 0.80 are poor.
There are several items to consider when using models, including (1) time horizon for the prediction, (2) variables included in the model, and (3) whether models can also predict the probability of carrying a pathogenic variant in breast cancer susceptibility genes like BRCA1 and BRCA2.
- Time horizon of models: Most models can predict an individual's lifetime risk of developing a specific cancer over a short time horizon (e.g., 1 year, 5 years, and 10 years). Although some clinical guidelines refer to lifetime risk cutoffs when assessing higher versus lower cancer risks, no model has been validated to predict full lifetime risk, since that would require following cohorts for a lifetime.[
134 ] Using a shorter time horizon improved model performance, particularly for women under age 50 years, since many factors for risk models change over time.[135 ] For example, data from a large family-based cohort (n = 14,657 women; median follow-up of 10 years), showed that the 5-year incidence for breast cancer almost always had a higher specificity (i.e., fewer false positives) than that of lifetime risk from birth. For women aged 20 to 39 years, 5-year risk performed better than lifetime risk from birth. For women aged 40 years or older, receiver-operating characteristic curves were similar or superior for 5-year risk than for lifetime risk in multiple breast cancer models. Classifications based on remaining lifetime risk were inferior to 5-year risk estimates.
- Variables included in models: In addition to a lack of validation for lifetime risk, cancer risk models are limited by the factors added to the models to help predict risk. Unlike risk models for diseases with shorter induction times (e.g., cardiovascular disease), cancer's longer induction times can make updating models (based on known risk factors) lengthy, since prospective validation is needed to calibrate the models. Most breast cancer risk models include established reproductive risk factors for breast cancer (e.g., age at menarche, parity, etc.). Many risk models also include established risk factors like alcohol consumption and body size. Few risk models assess whether cessation or change in risk factors over time lead to a change in cancer risk.
- Prediction of cancer susceptibility genes: In addition, models can predict an individual's likelihood of having a pathogenic variant in BRCA1, BRCA2, or one of the MMR genes associated with Lynch syndrome. Not all models can be applied to all patients. Each model is appropriate only when the patient's characteristics and family history are similar to those from the study population the model was based on. Different models may provide widely varying risk estimates for the same clinical scenario, and validation of these estimates has not been performed for many models.[
131 ,136 ,137 ] For more information, see theModels for prediction of the likelihood of a BRCA1 or BRCA2 pathogenic variant section.
Limitations of risk models: Risk models only use a subset of risk factors for breast, ovarian, and endometrial cancer risk. Additionally, risk models are limited by moderate discrimination for these cancer types. Moderate discrimination means that when clinical cutoffs are used to define high- and low-risk individuals (e.g., individuals with >20% lifetime risk are defined as high-risk), people will be misclassified. This means that there will be both false positives (people at lower risk who follow high-risk protocols) and false negatives (people at higher risk who follow low-risk protocols).
Breast cancer risk assessment models
In general, breast cancer risk assessment models are designed for two types of populations: (1) women without pathogenic variants in breast cancer susceptibility genes or strong family histories of breast/ovarian cancer, and (2) women at higher risk because of personal or family histories of breast/ovarian cancer.[
Models of the first type designed for women (e.g., the Gail model, which is the basis for the Breast Cancer Risk Assessment Tool [BCRAT] [
Models designed for women at higher risk require more detailed information about personal and family cancer histories of breast and ovarian cancers, including ages at onset of cancer and/or carrier status of specific breast cancer-susceptibility alleles. The genetic factors used by the latter models differ, with some assuming one risk locus (e.g., the Claus model [
The models also differ in whether they include information about nongenetic risk factors. Three models (Gail/BCRAT, Pfeiffer,[
Breast cancer risk models have limited the ability to discriminate between individuals who are affected or unaffected with cancer. A model with high discrimination would be close to 1, and a model with little discrimination would be close to 0.5. Model discrimination is rarely above an AUC of 0.70.[
In the United States, the BRCAPRO, Claus,[
In addition to statistical and regression-based models, risk-assessment models are being developed based on artificial intelligence (AI), using imaging (primarily from mammography) and other clinical data from the electronic health record. Risk-assessment based on machine learning and AI algorithms (when applied to mammographic images) have produced AUCs in a similar or even higher range than some of the pedigree and regression-based risk models.[
Additional considerations for clinical use of breast cancer risk assessment models
The Gail model is the basis for the
- Women who do not adhere to mammography screening recommendations.[
155 ,156 ] - Women in the highest-risk strata (e.g., those with breast cancer family histories, particularly if FDRs are older when diagnosed with breast cancer).[
158 ]
The Gail/BCRAT model is valid for women aged 35 years and older. The model was primarily developed for White women.[
Generally, the Gail/BCRAT model should not be the sole model used for families with one or more of the following characteristics:
- Multiple affected individuals with breast cancer or ovarian cancer (especially when one or more breast cancers are diagnosed before age 50 y).
- A woman with both breast and ovarian cancer.
- Ashkenazi Jewish ancestry with at least one case of breast or ovarian cancer (as these families are more likely to have a hereditary cancer susceptibility syndrome).
Commonly used models that incorporate family history include the IBIS, BOADICEA/CanRisk, and BRCAPRO models. The IBIS/Tyrer-Cuzick model incorporates both genetic and nongenetic factors.[
In addition, readily available models that provide information about an individual woman's risk in relation to the population-level risk depending on her risk factors may be useful in a clinical setting (e.g.,
Although most breast cancer risk models have been shown to be well calibrated overall, model performance can be different for subgroups of women. In particular, independent, prospective validation of risk models for women who tested negative for BRCA1 or BRCA2 pathogenic variants supported that the most commonly used clinical risk models underpredicted risk for this group of women.[
Risk models in older individuals: As individuals age, the chance to have competing risks from other outcomes increases (e.g., cardiovascular disease). Some risk models incorporate the concept of competing risk into their calculations (e.g., BCRAT), while others do not (e.g., BOADICEA/CanRisk). Differences that occur due to competing risk are particularly important to consider, especially in older women with other comorbidities.
Ovarian cancer risk assessment models
Model development for prediction of ovarian cancer risk has been similar to that of breast cancer risk models with pedigree-based models and nonpedigree-based models.
Endometrial cancer risk assessment models
Endometrial cancer risk models also can be divided into regression-based models, pedigree-based models, and AI-based models. The Pfeiffer model has been used to predict endometrial cancer risk in the general population.[
Regression-based models differ from pedigree-based models, which require detailed information on the number of relatives with cancer, types of cancer, and ages of cancer diagnoses in family members. MMRpredict, PREMM5 (PREdiction Model for gene Mutations), and MMRpro are three quantitative predictive models used to identify individuals who may potentially have Lynch syndrome.[
AI-based models have been used for risk-stratification and prognosis in endometrial cancer cases, but they have not been used to predict risk of endometrial cancer onset.[
Models for Predicting the Likelihood of aBRCA1/BRCA2Pathogenic Variant
Many models have been developed to predict the probability of identifying germline BRCA1/BRCA2 pathogenic variants in individuals or families. These models include those using logistic regression,[
In addition to BOADICEA, BRCAPRO is commonly used for genetic counseling in the clinical setting. BRCAPRO and BOADICEA predict the probability of being a carrier and produce estimates of breast cancer risk (refer to
BOADICEA is a polygenetic model that uses complex segregation analysis to examine both breast cancer risk and the probability of having a BRCA1 or BRCA2 pathogenic variant.[
The performance of the models can vary in specific ethnic groups. The BRCAPRO model appeared to best fit a series of French Canadian families.[
The power of several of the models has been compared in different studies.[
| | Myriad Prevalence Tables[ |
BRCAPRO[ |
BOADICEA (now CanRisk)[ |
Tyrer-Cuzick[ |
|---|---|---|---|---|
| AJ = Ashkenazi Jewish; BOADICEA = Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm; FDR = first-degree relatives; SDR = second-degree relatives. | ||||
| Method | Empiric data from Myriad Genetics based on personal and family history reported on requisition forms | Statistical model, assumes autosomal dominant inheritance | Statistical model, assumes polygenic risk | Statistical model, assumes autosomal dominant inheritance |
| Features of the model | Probandmay or may not have breast or ovarian cancer | Proband may or may not have breast or ovarian cancer | Proband may or may not have breast or ovarian cancer | Proband must beunaffected |
| Considers age of breast cancer diagnosis as <50 y, >50 y | Considers exact age at breast and ovarian cancer diagnosis | Considers exact age at breast and ovarian cancer diagnosis | Also includes reproductive factors and body mass index to estimate breast cancer risk | |
| Considers breast cancer in ≥1 affected relative only if diagnosed <50 y | Considers prior genetic testing in family (i.e.,BRCA1/BRCA2 pathogenic variant–negative relatives) | Includes allFDRandSDRwith and without cancer | ||
| Considers ovarian cancer in ≥1 relative at any age | Considers oophorectomy status | Includes AJ ancestry | ||
| Includes AJ ancestry | Includes all FDR and SDR with and without cancer | |||
| Very easy to use | Includes AJ ancestry | |||
| Limitations | Simplified/limited consideration of family structure | Requires computer software and time-consuming data entry | Requires computer software and time-consuming data entry | Designed for individuals unaffected with breast cancer |
| Incorporates only FDR and SDR; may need to change proband to best capture risk and to account for disease in the paternal lineage | ||||
| May overestimate risk in bilateral breast cancer[ |
||||
| Early age of breast cancer onset | May perform better in White populations than in racial and ethnic minority populations[ |
Incorporates only FDR and SDR; may need to change proband to best capture risk | ||
| May underestimate risk ofBRCApathogenic variant in high-grade serous ovarian cancers but overestimate the risk for other histologies[ |
||||
Genetic testing for BRCA1 and BRCA2 pathogenic variants has been available to the public since 1996. As more individuals have undergone testing, risk assessment models have improved. This, in turn, gives providers better data to estimate an individual patient's risk of carrying a pathogenic variant, but risk assessment continues to be an art. There are factors that might limit the ability to provide an accurate risk assessment (i.e., small family size, paucity of women, or ethnicity) including the specific circumstances of the individual patient (such as history of disease or risk-reducing surgeries).
Considerations When Conducting Genetic Testing
Indications for hereditary breast and gynecologic cancers genetic testing
Several professional organizations and expert panels—including the American Society of Clinical Oncology,[
In 2019, the American Society of Breast Surgeons published a recommendation to make genetic testing for "BRCA1/BRCA2, and PALB2, with other genes as appropriate for the clinical scenario and family history" available to all breast cancer patients.[
Other studies have also found that the NCCN criteria have good sensitivity when predicting BRCA1/BRCA2 variants; however, less is known about many other genes. For example, one study showed that the NCCN criteria were able to detect 88.9% of the BRCA1/BRCA2 pathogenic variant carriers [
As the cost of genetic testing continues to decrease, there is a need for unbiased evidence to guide indications for testing, including the cost-benefit impact on screening, prevention, and treatment. Efforts to generate less biased evidence include a single institution study of 3,907 unselected women with breast cancer tested for nine breast cancer genes, including BRCA1/BRCA2, ATM, CDH1, CHEK2, NF1, PALB2, PTEN, and TP53.[
Another study to assess frequency of pathogenic or likely pathogenic variants among breast cancer patients included a nested case-control study conducted through the WHI cohort among women with (cases) and without (controls) invasive breast cancer. Participants were tested for pathogenic or likely pathogenic variants in ten breast cancer–associated genes, including BRCA1/BRCA2.[
Benefits of offering genetic testing at the time of cancer diagnosis
At the time of a new cancer diagnosis, genetic testing for inherited cancer predisposition may guide patient care including decisions about surgery, chemotherapy and other biologics, and radiation treatment.[
Breast cancer diagnosis
Benefits of offering genetic testing at the time of breast cancer diagnosis include, but are not limited to, the following:
- Surgery: The identification of inherited susceptibility to breast cancer may influence surgical treatment decisions. As an example, the high risk of a second primary breast cancer among BRCA pathogenic variant carriers, particularly those diagnosed at an early age, may influence their decision to choose a bilateral mastectomy (versus a lumpectomy or unilateral/subtotal mastectomy) for surgical treatment of their breast cancer.[
223 ] Discussion of RRSO is indicated,[224 ] and referral to a gynecologic provider may be considered. - Chemotherapy and other biologics: Medical treatments may be guided by the identification of a pathogenic variant in an inherited cancer predisposing gene. As an example, among BRCA pathogenic variant carriers, breast cancer treatment may include the use of platinum-based agents.[
225 ] Furthermore, novel agents such as PARP inhibitors may be used in the treatment of metastatic breast cancer.[226 ] - Radiation therapy: Decisions about the use of radiation treatment may be guided by the presence of a pathogenic variant in an inherited breast cancer susceptibility gene. In particular, the poorer wound healing in irradiated breasts is an important consideration for those who may consider risk-reducing mastectomy with reconstruction. As an example, individuals with a pathogenic variant in TP53 may experience higher risks from radiation, including increased risks for subsequent new cancers.[
227 ,228 ] Thus, identification of TP53 carriers in the context of an active breast cancer diagnosis may influence radiation treatment decisions and reconstruction options.
Ovarian cancer diagnosis
Benefits of offering genetic testing at the time of ovarian cancer diagnosis include, but are not limited to, the following:
- Surgery: In most cases, the decision for ovarian cancer surgery is made on the basis of an adnexal mass or abdominal symptoms. When possible, considering the likelihood of a heritable genetic variant at the time of diagnosis may add value to surgical decision-making. The identification of inherited susceptibility to ovarian/fallopian tube cancer may influence surgical treatment decisions. For a questionable adnexal mass in a younger woman who is at risk of carrying a pathogenic variant of a highly penetrant ovarian cancer gene, knowledge of this information may help guide a decision for risk-reducing or therapeutic surgery.[
229 ,230 ] For women who may be considering fertility preservation surgery, genetic knowledge may motivate consideration of bilateral salpingo-oophorectomy, and in the case of carriers of BRCA1 pathogenic variants, a more detailed discussion regarding aggressive uterine cancer risk. - Chemotherapy and other biologics: First-line chemotherapy for ovarian cancer still relies on a backbone of platinum and taxane chemotherapy. Current treatment options for optimally resected stage III ovarian carcinoma include intravenous (IV) chemotherapy, dose-dense IV chemotherapy, and a combination of IV paclitaxel plus intraperitoneal (IP) cisplatin, followed by IP paclitaxel 1 week later. Carriers of BRCA1 and BRCA2 pathogenic variants are considered more platinum sensitive, with longer progression-free survival times compared with BRCA1 and BRCA2 wild-type patients,[
231 ,232 ] so it is unclear whether a particular treatment strategy is driven more by antiangiogenesis effects, peritoneal dose intensity, or platinum dose intensity. The advent of PARP as a biologic target (in combination with chemotherapy or as maintenance) may also increase the armory of first-line treatment of ovarian cancer.[233 ] (Refer to theOvarian Cancer Treatment Strategies section in BRCA1 and BRCA2: Cancer Risks and Management for more information about PARP inhibitors in ovarian cancer treatment.)
Endometrial cancer diagnosis
Benefits of offering genetic testing at the time of endometrial cancer diagnosis include, but are not limited to, the following:
- Surgery: The most common treatment for a newly diagnosed endometrial cancer includes hysterectomy with removal of the ovaries and fallopian tubes, as well as assessment of lymph nodes.[
234 ] An exception to this practice might apply to a younger woman who wishes to retain fertility or retain her adnexa. IHC of endometrial sampling may allow for an assessment of the likelihood of a heritable genetic variant at the time of diagnosis, which may add value to the surgical decision-making process. For a young woman who is found to have Lynch syndrome, knowledge of this information may help guide a decision for hormonal management of endometrial cancer to allow future childbearing, or RRSO if her risk of ovarian cancer is deemed high enough on the basis of a specific genetic variant. For a young woman who is found to carry a pathogenic variant in BRCA1/BRCA2, or one of the other homologous recombination deficiencies increasing ovarian cancer risk, she may wish to decide between salpingo-oophorectomy or, at least, salpingectomy. - Chemotherapy and other biologics: Immune checkpoint inhibitors are now approved for use in endometrial cancers that have MSI or MMR deficiency.[
235 ] While MSI and MMR status can be assessed at either the time of diagnosis or recurrent disease, it may be beneficial to perform tumor testing at diagnosis with the primary pathology processing, usually at the time of hysterectomy.
Multigene (panel) testing
Since the availability of next-generation sequencing and the Supreme Court of the United States ruling that human genes cannot be patented, several clinical laboratories now offer genetic testing through multigene panels at a cost comparable to that of single-gene testing. Even testing for BRCA1 and BRCA2 is a limited panel test of two genes. Approximately 25% of all ovarian/fallopian tube/peritoneal cancers are caused by a heritable genetic condition. Of these, about one-quarter (6% of all ovarian/fallopian tube/peritoneal cancers) are caused by genes other than BRCA1 and BRCA2, including many genes associated with the Fanconi anemia pathway or otherwise involved with homologous recombination.[
In general, multigene panel testing increases the yield of non-BRCA pathogenic variants across a variety of populations.[
Multi-gene panel testing was conducted as part of two large efforts led by the worldwide Breast Cancer Association Consortium (BCAC) [
NCCN recommends that women diagnosed with TNBC undergo BRCA1/BRCA2, CDH1, PALB2, PTEN, STK11, and TP53 testing to guide treatment decisions at any age.[
Multi-gene panel testing studies were conducted in women from the United States who had African ancestry, and results showed that certain genes were associated with increased breast cancer risk in this population. These genes were similar to the breast cancer risk genes found in individuals from the United States with European ancestry. A case-control study of 10,047 women with African ancestry found a pathogenic variant frequency of 10.3% in those with ER-negative breast cancer, 5.2% in those with ER-positive breast cancer, and 2.3% in those without breast cancer. BRCA1 (OR, 47), BRCA2 (OR, 7.25) and PALB2 (OR, 8.54) were associated with the highest breast cancer risks.[
There are caveats of multigene testing. Genes identified as part of multigene panel testing can be associated with varied breast cancer risk or confer no known risk.[
(Refer to the
References:
- American Cancer Society: Cancer Facts and Figures 2025. American Cancer Society, 2025.
Available online . Last accessed January 16, 2025. - Ravdin PM, Cronin KA, Howlader N, et al.: The decrease in breast-cancer incidence in 2003 in the United States. N Engl J Med 356 (16): 1670-4, 2007.
- Quante AS, Herz J, Whittemore AS, et al.: Assessing absolute changes in breast cancer risk due to modifiable risk factors. Breast Cancer Res Treat 152 (1): 193-7, 2015.
- Feuer EJ, Wun LM, Boring CC, et al.: The lifetime risk of developing breast cancer. J Natl Cancer Inst 85 (11): 892-7, 1993.
- Yang Q, Khoury MJ, Rodriguez C, et al.: Family history score as a predictor of breast cancer mortality: prospective data from the Cancer Prevention Study II, United States, 1982-1991. Am J Epidemiol 147 (7): 652-9, 1998.
- Colditz GA, Willett WC, Hunter DJ, et al.: Family history, age, and risk of breast cancer. Prospective data from the Nurses' Health Study. JAMA 270 (3): 338-43, 1993.
- Slattery ML, Kerber RA: A comprehensive evaluation of family history and breast cancer risk. The Utah Population Database. JAMA 270 (13): 1563-8, 1993.
- Johnson N, Lancaster T, Fuller A, et al.: The prevalence of a family history of cancer in general practice. Fam Pract 12 (3): 287-9, 1995.
- Pharoah PD, Day NE, Duffy S, et al.: Family history and the risk of breast cancer: a systematic review and meta-analysis. Int J Cancer 71 (5): 800-9, 1997.
- Bevier M, Sundquist K, Hemminki K: Risk of breast cancer in families of multiple affected women and men. Breast Cancer Res Treat 132 (2): 723-8, 2012.
- Kharazmi E, Chen T, Narod S, et al.: Effect of multiplicity, laterality, and age at onset of breast cancer on familial risk of breast cancer: a nationwide prospective cohort study. Breast Cancer Res Treat 144 (1): 185-92, 2014.
- Reiner AS, Sisti J, John EM, et al.: Breast Cancer Family History and Contralateral Breast Cancer Risk in Young Women: An Update From the Women's Environmental Cancer and Radiation Epidemiology Study. J Clin Oncol 36 (15): 1513-1520, 2018.
- Albright FS, Kohlmann W, Neumayer L, et al.: Population-based relative risks for specific family history constellations of breast cancer. Cancer Causes Control 30 (6): 581-590, 2019.
- Mucci LA, Hjelmborg JB, Harris JR, et al.: Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315 (1): 68-76, 2016.
- Chen J, Pee D, Ayyagari R, et al.: Projecting absolute invasive breast cancer risk in white women with a model that includes mammographic density. J Natl Cancer Inst 98 (17): 1215-26, 2006.
- Dupont WD, Page DL, Parl FF, et al.: Long-term risk of breast cancer in women with fibroadenoma. N Engl J Med 331 (1): 10-5, 1994.
- Zeinomar N, Phillips KA, Daly MB, et al.: Benign breast disease increases breast cancer risk independent of underlying familial risk profile: Findings from a Prospective Family Study Cohort. Int J Cancer 145 (2): 370-379, 2019.
- Boyd NF, Byng JW, Jong RA, et al.: Quantitative classification of mammographic densities and breast cancer risk: results from the Canadian National Breast Screening Study. J Natl Cancer Inst 87 (9): 670-5, 1995.
- Byrne C, Schairer C, Wolfe J, et al.: Mammographic features and breast cancer risk: effects with time, age, and menopause status. J Natl Cancer Inst 87 (21): 1622-9, 1995.
- Pankow JS, Vachon CM, Kuni CC, et al.: Genetic analysis of mammographic breast density in adult women: evidence of a gene effect. J Natl Cancer Inst 89 (8): 549-56, 1997.
- Boyd NF, Lockwood GA, Martin LJ, et al.: Mammographic densities and risk of breast cancer among subjects with a family history of this disease. J Natl Cancer Inst 91 (16): 1404-8, 1999.
- Vachon CM, King RA, Atwood LD, et al.: Preliminary sibpair linkage analysis of percent mammographic density. J Natl Cancer Inst 91 (20): 1778-9, 1999.
- Vachon CM, van Gils CH, Sellers TA, et al.: Mammographic density, breast cancer risk and risk prediction. Breast Cancer Res 9 (6): 217, 2007.
- Mitchell G, Antoniou AC, Warren R, et al.: Mammographic density and breast cancer risk in BRCA1 and BRCA2 mutation carriers. Cancer Res 66 (3): 1866-72, 2006.
- Ramón Y Cajal T, Chirivella I, Miranda J, et al.: Mammographic density and breast cancer in women from high risk families. Breast Cancer Res 17: 93, 2015.
- Thompson CM, Mallawaarachchi I, Dwivedi DK, et al.: The Association of Background Parenchymal Enhancement at Breast MRI with Breast Cancer: A Systematic Review and Meta-Analysis. Radiology 292 (3): 552-561, 2019.
- Terry MB, Liao Y, Kast K, et al.: The Influence of Number and Timing of Pregnancies on Breast Cancer Risk for Women With BRCA1 or BRCA2 Mutations. JNCI Cancer Spectr 2 (4): pky078, 2018.
- Johannsson O, Loman N, Borg A, et al.: Pregnancy-associated breast cancer in BRCA1 and BRCA2 germline mutation carriers. Lancet 352 (9137): 1359-60, 1998.
- Jernström H, Lerman C, Ghadirian P, et al.: Pregnancy and risk of early breast cancer in carriers of BRCA1 and BRCA2. Lancet 354 (9193): 1846-50, 1999.
- Friebel TM, Domchek SM, Rebbeck TR: Modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: systematic review and meta-analysis. J Natl Cancer Inst 106 (6): dju091, 2014.
- Valentini A, Lubinski J, Byrski T, et al.: The impact of pregnancy on breast cancer survival in women who carry a BRCA1 or BRCA2 mutation. Breast Cancer Res Treat 142 (1): 177-85, 2013.
- Jernström H, Lubinski J, Lynch HT, et al.: Breast-feeding and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst 96 (14): 1094-8, 2004.
- Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Collaborative Group on Hormonal Factors in Breast Cancer. Lancet 347 (9017): 1713-27, 1996.
- Iodice S, Barile M, Rotmensz N, et al.: Oral contraceptive use and breast or ovarian cancer risk in BRCA1/2 carriers: a meta-analysis. Eur J Cancer 46 (12): 2275-84, 2010.
- Schrijver LH, Olsson H, Phillips KA, et al.: Oral Contraceptive Use and Breast Cancer Risk: Retrospective and Prospective Analyses From a BRCA1 and BRCA2 Mutation Carrier Cohort Study. JNCI Cancer Spectr 2 (2): pky023, 2018.
- Huber D, Seitz S, Kast K, et al.: Use of oral contraceptives in BRCA mutation carriers and risk for ovarian and breast cancer: a systematic review. Arch Gynecol Obstet 301 (4): 875-884, 2020.
- Kotsopoulos J, Lubinski J, Moller P, et al.: Timing of oral contraceptive use and the risk of breast cancer in BRCA1 mutation carriers. Breast Cancer Res Treat 143 (3): 579-86, 2014.
- Conz L, Mota BS, Bahamondes L, et al.: Levonorgestrel-releasing intrauterine system and breast cancer risk: A systematic review and meta-analysis. Acta Obstet Gynecol Scand 99 (8): 970-982, 2020.
- Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Collaborative Group on Hormonal Factors in Breast Cancer. Lancet 350 (9084): 1047-59, 1997.
- Gorsky RD, Koplan JP, Peterson HB, et al.: Relative risks and benefits of long-term estrogen replacement therapy: a decision analysis. Obstet Gynecol 83 (2): 161-6, 1994.
- Writing Group for the Women's Health Initiative Investigators: Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. JAMA 288 (3): 321-33, 2002.
- Chlebowski RT, Hendrix SL, Langer RD, et al.: Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women's Health Initiative Randomized Trial. JAMA 289 (24): 3243-53, 2003.
- Beral V; Million Women Study Collaborators: Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 362 (9382): 419-27, 2003.
- Anderson GL, Limacher M, Assaf AR, et al.: Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA 291 (14): 1701-12, 2004.
- Schuurman AG, van den Brandt PA, Goldbohm RA: Exogenous hormone use and the risk of postmenopausal breast cancer: results from The Netherlands Cohort Study. Cancer Causes Control 6 (5): 416-24, 1995.
- Steinberg KK, Thacker SB, Smith SJ, et al.: A meta-analysis of the effect of estrogen replacement therapy on the risk of breast cancer. JAMA 265 (15): 1985-90, 1991.
- Sellers TA, Mink PJ, Cerhan JR, et al.: The role of hormone replacement therapy in the risk for breast cancer and total mortality in women with a family history of breast cancer. Ann Intern Med 127 (11): 973-80, 1997.
- Stanford JL, Weiss NS, Voigt LF, et al.: Combined estrogen and progestin hormone replacement therapy in relation to risk of breast cancer in middle-aged women. JAMA 274 (2): 137-42, 1995.
- Colditz GA, Egan KM, Stampfer MJ: Hormone replacement therapy and risk of breast cancer: results from epidemiologic studies. Am J Obstet Gynecol 168 (5): 1473-80, 1993.
- Rebbeck TR, Friebel T, Wagner T, et al.: Effect of short-term hormone replacement therapy on breast cancer risk reduction after bilateral prophylactic oophorectomy in BRCA1 and BRCA2 mutation carriers: the PROSE Study Group. J Clin Oncol 23 (31): 7804-10, 2005.
- Kotsopoulos J, Gronwald J, Karlan BY, et al.: Hormone Replacement Therapy After Oophorectomy and Breast Cancer Risk Among BRCA1 Mutation Carriers. JAMA Oncol 4 (8): 1059-1065, 2018.
- Gordhandas S, Norquist BM, Pennington KP, et al.: Hormone replacement therapy after risk reducing salpingo-oophorectomy in patients with BRCA1 or BRCA2 mutations; a systematic review of risks and benefits. Gynecol Oncol 153 (1): 192-200, 2019.
- Helzlsouer KJ, Harris EL, Parshad R, et al.: Familial clustering of breast cancer: possible interaction between DNA repair proficiency and radiation exposure in the development of breast cancer. Int J Cancer 64 (1): 14-7, 1995.
- Helzlsouer KJ, Harris EL, Parshad R, et al.: DNA repair proficiency: potential susceptibility factor for breast cancer. J Natl Cancer Inst 88 (11): 754-5, 1996.
- Abbott DW, Thompson ME, Robinson-Benion C, et al.: BRCA1 expression restores radiation resistance in BRCA1-defective cancer cells through enhancement of transcription-coupled DNA repair. J Biol Chem 274 (26): 18808-12, 1999.
- Abbott DW, Freeman ML, Holt JT: Double-strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. J Natl Cancer Inst 90 (13): 978-85, 1998.
- Easton DF: Cancer risks in A-T heterozygotes. Int J Radiat Biol 66 (6 Suppl): S177-82, 1994.
- Kleihues P, Schäuble B, zur Hausen A, et al.: Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol 150 (1): 1-13, 1997.
- Pierce LJ, Strawderman M, Narod SA, et al.: Effect of radiotherapy after breast-conserving treatment in women with breast cancer and germline BRCA1/2 mutations. J Clin Oncol 18 (19): 3360-9, 2000.
- Narod SA, Lubinski J, Ghadirian P, et al.: Screening mammography and risk of breast cancer in BRCA1 and BRCA2 mutation carriers: a case-control study. Lancet Oncol 7 (5): 402-6, 2006.
- Andrieu N, Easton DF, Chang-Claude J, et al.: Effect of chest X-rays on the risk of breast cancer among BRCA1/2 mutation carriers in the international BRCA1/2 carrier cohort study: a report from the EMBRACE, GENEPSO, GEO-HEBON, and IBCCS Collaborators' Group. J Clin Oncol 24 (21): 3361-6, 2006.
- Goldfrank D, Chuai S, Bernstein JL, et al.: Effect of mammography on breast cancer risk in women with mutations in BRCA1 or BRCA2. Cancer Epidemiol Biomarkers Prev 15 (11): 2311-3, 2006.
- Gronwald J, Pijpe A, Byrski T, et al.: Early radiation exposures and BRCA1-associated breast cancer in young women from Poland. Breast Cancer Res Treat 112 (3): 581-4, 2008.
- Pijpe A, Andrieu N, Easton DF, et al.: Exposure to diagnostic radiation and risk of breast cancer among carriers of BRCA1/2 mutations: retrospective cohort study (GENE-RAD-RISK). BMJ 345: e5660, 2012.
- Giannakeas V, Lubinski J, Gronwald J, et al.: Mammography screening and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers: a prospective study. Breast Cancer Res Treat 147 (1): 113-8, 2014.
- Drooger J, Akdeniz D, Pignol JP, et al.: Adjuvant radiotherapy for primary breast cancer in BRCA1 and BRCA2 mutation carriers and risk of contralateral breast cancer with special attention to patients irradiated at younger age. Breast Cancer Res Treat 154 (1): 171-80, 2015.
- Reiner AS, Robson ME, Mellemkjær L, et al.: Radiation Treatment, ATM, BRCA1/2, and CHEK2*1100delC Pathogenic Variants and Risk of Contralateral Breast Cancer. J Natl Cancer Inst 112 (12): 1275-1279, 2020.
- Smith-Warner SA, Spiegelman D, Yaun SS, et al.: Alcohol and breast cancer in women: a pooled analysis of cohort studies. JAMA 279 (7): 535-40, 1998.
- Hamajima N, Hirose K, Tajima K, et al.: Alcohol, tobacco and breast cancer--collaborative reanalysis of individual data from 53 epidemiological studies, including 58,515 women with breast cancer and 95,067 women without the disease. Br J Cancer 87 (11): 1234-45, 2002.
- McGuire V, John EM, Felberg A, et al.: No increased risk of breast cancer associated with alcohol consumption among carriers of BRCA1 and BRCA2 mutations ages <50 years. Cancer Epidemiol Biomarkers Prev 15 (8): 1565-7, 2006.
- Dennis J, Ghadirian P, Little J, et al.: Alcohol consumption and the risk of breast cancer among BRCA1 and BRCA2 mutation carriers. Breast 19 (6): 479-83, 2010.
- Cybulski C, Lubinski J, Huzarski T, et al.: Prospective evaluation of alcohol consumption and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res Treat 151 (2): 435-41, 2015.
- Brunet JS, Ghadirian P, Rebbeck TR, et al.: Effect of smoking on breast cancer in carriers of mutant BRCA1 or BRCA2 genes. J Natl Cancer Inst 90 (10): 761-6, 1998.
- Ghadirian P, Lubinski J, Lynch H, et al.: Smoking and the risk of breast cancer among carriers of BRCA mutations. Int J Cancer 110 (3): 413-6, 2004.
- Li H, Terry MB, Antoniou AC, et al.: Alcohol Consumption, Cigarette Smoking, and Risk of Breast Cancer for BRCA1 and BRCA2 Mutation Carriers: Results from The BRCA1 and BRCA2 Cohort Consortium. Cancer Epidemiol Biomarkers Prev 29 (2): 368-378, 2020.
- Zeinomar N, Knight JA, Genkinger JM, et al.: Alcohol consumption, cigarette smoking, and familial breast cancer risk: findings from the Prospective Family Study Cohort (ProF-SC). Breast Cancer Res 21 (1): 128, 2019.
- Lammert J, Lubinski J, Gronwald J, et al.: Physical activity during adolescence and young adulthood and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res Treat 169 (3): 561-571, 2018.
- Kehm RD, Genkinger JM, MacInnis RJ, et al.: Recreational Physical Activity Is Associated with Reduced Breast Cancer Risk in Adult Women at High Risk for Breast Cancer: A Cohort Study of Women Selected for Familial and Genetic Risk. Cancer Res 80 (1): 116-125, 2020.
- Amos CI, Struewing JP: Genetic epidemiology of epithelial ovarian cancer. Cancer 71 (2 Suppl): 566-72, 1993.
- Stratton JF, Pharoah P, Smith SK, et al.: A systematic review and meta-analysis of family history and risk of ovarian cancer. Br J Obstet Gynaecol 105 (5): 493-9, 1998.
- Whittemore AS, Harris R, Itnyre J: Characteristics relating to ovarian cancer risk: collaborative analysis of 12 US case-control studies. II. Invasive epithelial ovarian cancers in white women. Collaborative Ovarian Cancer Group. Am J Epidemiol 136 (10): 1184-203, 1992.
- Brinton LA, Lamb EJ, Moghissi KS, et al.: Ovarian cancer risk after the use of ovulation-stimulating drugs. Obstet Gynecol 103 (6): 1194-203, 2004.
- Gronwald J, Byrski T, Huzarski T, et al.: Influence of selected lifestyle factors on breast and ovarian cancer risk in BRCA1 mutation carriers from Poland. Breast Cancer Res Treat 95 (2): 105-9, 2006.
- McLaughlin JR, Risch HA, Lubinski J, et al.: Reproductive risk factors for ovarian cancer in carriers of BRCA1 or BRCA2 mutations: a case-control study. Lancet Oncol 8 (1): 26-34, 2007.
- Antoniou AC, Rookus M, Andrieu N, et al.: Reproductive and hormonal factors, and ovarian cancer risk for BRCA1 and BRCA2 mutation carriers: results from the International BRCA1/2 Carrier Cohort Study. Cancer Epidemiol Biomarkers Prev 18 (2): 601-10, 2009.
- Narod SA, Sun P, Ghadirian P, et al.: Tubal ligation and risk of ovarian cancer in carriers of BRCA1 or BRCA2 mutations: a case-control study. Lancet 357 (9267): 1467-70, 2001.
- Kotsopoulos J, Lubinski J, Gronwald J, et al.: Factors influencing ovulation and the risk of ovarian cancer in BRCA1 and BRCA2 mutation carriers. Int J Cancer 137 (5): 1136-46, 2015.
- Rodriguez C, Patel AV, Calle EE, et al.: Estrogen replacement therapy and ovarian cancer mortality in a large prospective study of US women. JAMA 285 (11): 1460-5, 2001.
- Riman T, Dickman PW, Nilsson S, et al.: Hormone replacement therapy and the risk of invasive epithelial ovarian cancer in Swedish women. J Natl Cancer Inst 94 (7): 497-504, 2002.
- Lacey JV, Mink PJ, Lubin JH, et al.: Menopausal hormone replacement therapy and risk of ovarian cancer. JAMA 288 (3): 334-41, 2002.
- Anderson GL, Judd HL, Kaunitz AM, et al.: Effects of estrogen plus progestin on gynecologic cancers and associated diagnostic procedures: the Women's Health Initiative randomized trial. JAMA 290 (13): 1739-48, 2003.
- Tortolero-Luna G, Mitchell MF: The epidemiology of ovarian cancer. J Cell Biochem Suppl 23: 200-7, 1995.
- Hankinson SE, Hunter DJ, Colditz GA, et al.: Tubal ligation, hysterectomy, and risk of ovarian cancer. A prospective study. JAMA 270 (23): 2813-8, 1993.
- Rutter JL, Wacholder S, Chetrit A, et al.: Gynecologic surgeries and risk of ovarian cancer in women with BRCA1 and BRCA2 Ashkenazi founder mutations: an Israeli population-based case-control study. J Natl Cancer Inst 95 (14): 1072-8, 2003.
- Kauff ND, Satagopan JM, Robson ME, et al.: Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med 346 (21): 1609-15, 2002.
- Rebbeck TR, Lynch HT, Neuhausen SL, et al.: Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med 346 (21): 1616-22, 2002.
- Kotsopoulos J, Huzarski T, Gronwald J, et al.: Bilateral Oophorectomy and Breast Cancer Risk in BRCA1 and BRCA2 Mutation Carriers. J Natl Cancer Inst 109 (1): , 2017.
- John EM, Whittemore AS, Harris R, et al.: Characteristics relating to ovarian cancer risk: collaborative analysis of seven U.S. case-control studies. Epithelial ovarian cancer in black women. Collaborative Ovarian Cancer Group. J Natl Cancer Inst 85 (2): 142-7, 1993.
- Narod SA, Risch H, Moslehi R, et al.: Oral contraceptives and the risk of hereditary ovarian cancer. Hereditary Ovarian Cancer Clinical Study Group. N Engl J Med 339 (7): 424-8, 1998.
- Whittemore AS, Balise RR, Pharoah PD, et al.: Oral contraceptive use and ovarian cancer risk among carriers of BRCA1 or BRCA2 mutations. Br J Cancer 91 (11): 1911-5, 2004.
- McGuire V, Felberg A, Mills M, et al.: Relation of contraceptive and reproductive history to ovarian cancer risk in carriers and noncarriers of BRCA1 gene mutations. Am J Epidemiol 160 (7): 613-8, 2004.
- Modan B, Hartge P, Hirsh-Yechezkel G, et al.: Parity, oral contraceptives, and the risk of ovarian cancer among carriers and noncarriers of a BRCA1 or BRCA2 mutation. N Engl J Med 345 (4): 235-40, 2001.
- Soliman PT, Oh JC, Schmeler KM, et al.: Risk factors for young premenopausal women with endometrial cancer. Obstet Gynecol 105 (3): 575-80, 2005.
- Vasen HF, Stormorken A, Menko FH, et al.: MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol 19 (20): 4074-80, 2001.
- Daniels MS: Genetic testing by cancer site: uterus. Cancer J 18 (4): 338-42, 2012 Jul-Aug.
- Dunlop MG, Farrington SM, Nicholl I, et al.: Population carrier frequency of hMSH2 and hMLH1 mutations. Br J Cancer 83 (12): 1643-5, 2000.
- de la Chapelle A: The incidence of Lynch syndrome. Fam Cancer 4 (3): 233-7, 2005.
- Ring KL, Bruegl AS, Allen BA, et al.: Germline multi-gene hereditary cancer panel testing in an unselected endometrial cancer cohort. Mod Pathol 29 (11): 1381-1389, 2016.
- Shu CA, Pike MC, Jotwani AR, et al.: Uterine Cancer After Risk-Reducing Salpingo-oophorectomy Without Hysterectomy in Women With BRCA Mutations. JAMA Oncol 2 (11): 1434-1440, 2016.
- de Jonge MM, de Kroon CD, Jenner DJ, et al.: Endometrial Cancer Risk in Women With Germline BRCA1 or BRCA2 Mutations: Multicenter Cohort Study. J Natl Cancer Inst 113 (9): 1203-1211, 2021.
- McPherson CP, Sellers TA, Potter JD, et al.: Reproductive factors and risk of endometrial cancer. The Iowa Women's Health Study. Am J Epidemiol 143 (12): 1195-202, 1996.
- Dossus L, Allen N, Kaaks R, et al.: Reproductive risk factors and endometrial cancer: the European Prospective Investigation into Cancer and Nutrition. Int J Cancer 127 (2): 442-51, 2010.
- Shapiro S, Kelly JP, Rosenberg L, et al.: Risk of localized and widespread endometrial cancer in relation to recent and discontinued use of conjugated estrogens. N Engl J Med 313 (16): 969-72, 1985.
- Ziel HK, Finkle WD: Increased risk of endometrial carcinoma among users of conjugated estrogens. N Engl J Med 293 (23): 1167-70, 1975.
- Fisher B, Costantino JP, Redmond CK, et al.: Endometrial cancer in tamoxifen-treated breast cancer patients: findings from the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14. J Natl Cancer Inst 86 (7): 527-37, 1994.
- Pike MC, Peters RK, Cozen W, et al.: Estrogen-progestin replacement therapy and endometrial cancer. J Natl Cancer Inst 89 (15): 1110-6, 1997.
- Fournier A, Dossus L, Mesrine S, et al.: Risks of endometrial cancer associated with different hormone replacement therapies in the E3N cohort, 1992-2008. Am J Epidemiol 180 (5): 508-17, 2014.
- Weiss NS, Sayvetz TA: Incidence of endometrial cancer in relation to the use of oral contraceptives. N Engl J Med 302 (10): 551-4, 1980.
- Soini T, Hurskainen R, Grénman S, et al.: Cancer risk in women using the levonorgestrel-releasing intrauterine system in Finland. Obstet Gynecol 124 (2 Pt 1): 292-9, 2014.
- Lindor NM, McMaster ML, Lindor CJ, et al.: Concise handbook of familial cancer susceptibility syndromes - second edition. J Natl Cancer Inst Monogr (38): 1-93, 2008.
- Vasen HF, Offerhaus GJ, den Hartog Jager FC, et al.: The tumour spectrum in hereditary non-polyposis colorectal cancer: a study of 24 kindreds in the Netherlands. Int J Cancer 46 (1): 31-4, 1990.
- Watson P, Lynch HT: Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer 71 (3): 677-85, 1993.
- Watson P, Vasen HF, Mecklin JP, et al.: The risk of endometrial cancer in hereditary nonpolyposis colorectal cancer. Am J Med 96 (6): 516-20, 1994.
- Aarnio M, Mecklin JP, Aaltonen LA, et al.: Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer 64 (6): 430-3, 1995.
- Raymond VM, Everett JN, Furtado LV, et al.: Adrenocortical carcinoma is a lynch syndrome-associated cancer. J Clin Oncol 31 (24): 3012-8, 2013.
- Raymond VM, Mukherjee B, Wang F, et al.: Elevated risk of prostate cancer among men with Lynch syndrome. J Clin Oncol 31 (14): 1713-8, 2013.
- Suspiro A, Fidalgo P, Cravo M, et al.: The Muir-Torre syndrome: a rare variant of hereditary nonpolyposis colorectal cancer associated with hMSH2 mutation. Am J Gastroenterol 93 (9): 1572-4, 1998.
- Kerber RA, Slattery ML: Comparison of self-reported and database-linked family history of cancer data in a case-control study. Am J Epidemiol 146 (3): 244-8, 1997.
- Parent ME, Ghadirian P, Lacroix A, et al.: The reliability of recollections of family history: implications for the medical provider. J Cancer Educ 12 (2): 114-20, 1997 Summer.
- Ready K, Litton JK, Arun BK: Clinical application of breast cancer risk assessment models. Future Oncol 6 (3): 355-65, 2010.
- Amir E, Freedman OC, Seruga B, et al.: Assessing women at high risk of breast cancer: a review of risk assessment models. J Natl Cancer Inst 102 (10): 680-91, 2010.
- Rosner BA, Colditz GA, Webb PM, et al.: Mathematical models of ovarian cancer incidence. Epidemiology 16 (4): 508-15, 2005.
- Pfeiffer RM, Park Y, Kreimer AR, et al.: Risk prediction for breast, endometrial, and ovarian cancer in white women aged 50 y or older: derivation and validation from population-based cohort studies. PLoS Med 10 (7): e1001492, 2013.
- Quante AS, Whittemore AS, Shriver T, et al.: Practical problems with clinical guidelines for breast cancer prevention based on remaining lifetime risk. J Natl Cancer Inst 107 (7): , 2015.
- MacInnis RJ, Knight JA, Chung WK, et al.: Comparing 5-Year and Lifetime Risks of Breast Cancer using the Prospective Family Study Cohort. J Natl Cancer Inst 113 (6): 785-791, 2021.
- Gail MH, Mai PL: Comparing breast cancer risk assessment models. J Natl Cancer Inst 102 (10): 665-8, 2010.
- Quante AS, Whittemore AS, Shriver T, et al.: Breast cancer risk assessment across the risk continuum: genetic and nongenetic risk factors contributing to differential model performance. Breast Cancer Res 14 (6): R144, 2012.
- Gail MH, Brinton LA, Byar DP, et al.: Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst 81 (24): 1879-86, 1989.
- Colditz GA, Rosner B: Cumulative risk of breast cancer to age 70 years according to risk factor status: data from the Nurses' Health Study. Am J Epidemiol 152 (10): 950-64, 2000.
- Claus EB, Risch N, Thompson WD: Autosomal dominant inheritance of early-onset breast cancer. Implications for risk prediction. Cancer 73 (3): 643-51, 1994.
- Tyrer J, Duffy SW, Cuzick J: A breast cancer prediction model incorporating familial and personal risk factors. Stat Med 23 (7): 1111-30, 2004.
- Parmigiani G, Berry D, Aguilar O: Determining carrier probabilities for breast cancer-susceptibility genes BRCA1 and BRCA2. Am J Hum Genet 62 (1): 145-58, 1998.
- Antoniou AC, Pharoah PD, McMullan G, et al.: A comprehensive model for familial breast cancer incorporating BRCA1, BRCA2 and other genes. Br J Cancer 86 (1): 76-83, 2002.
- Antoniou AC, Pharoah PP, Smith P, et al.: The BOADICEA model of genetic susceptibility to breast and ovarian cancer. Br J Cancer 91 (8): 1580-90, 2004.
- Antoniou AC, Cunningham AP, Peto J, et al.: The BOADICEA model of genetic susceptibility to breast and ovarian cancers: updates and extensions. Br J Cancer 98 (8): 1457-66, 2008.
- Mavaddat N, Ficorella L, Carver T, et al.: Incorporating Alternative Polygenic Risk Scores into the BOADICEA Breast Cancer Risk Prediction Model. Cancer Epidemiol Biomarkers Prev 32 (3): 422-427, 2023.
- Carver T, Hartley S, Lee A, et al.: CanRisk Tool-A Web Interface for the Prediction of Breast and Ovarian Cancer Risk and the Likelihood of Carrying Genetic Pathogenic Variants. Cancer Epidemiol Biomarkers Prev 30 (3): 469-473, 2021.
- Anothaisintawee T, Teerawattananon Y, Wiratkapun C, et al.: Risk prediction models of breast cancer: a systematic review of model performances. Breast Cancer Res Treat 133 (1): 1-10, 2012.
- Amir E, Evans DG, Shenton A, et al.: Evaluation of breast cancer risk assessment packages in the family history evaluation and screening programme. J Med Genet 40 (11): 807-14, 2003.
- Laitman Y, Simeonov M, Keinan-Boker L, et al.: Breast cancer risk prediction accuracy in Jewish Israeli high-risk women using the BOADICEA and IBIS risk models. Genet Res (Camb) 95 (6): 174-7, 2013.
- MacInnis RJ, Bickerstaffe A, Apicella C, et al.: Prospective validation of the breast cancer risk prediction model BOADICEA and a batch-mode version BOADICEACentre. Br J Cancer 109 (5): 1296-301, 2013.
- Vilmun BM, Vejborg I, Lynge E, et al.: Impact of adding breast density to breast cancer risk models: A systematic review. Eur J Radiol 127: 109019, 2020.
- Terry MB, Liao Y, Whittemore AS, et al.: 10-year performance of four models of breast cancer risk: a validation study. Lancet Oncol 20 (4): 504-517, 2019.
- Claus EB, Risch N, Thompson WD: The calculation of breast cancer risk for women with a first degree family history of ovarian cancer. Breast Cancer Res Treat 28 (2): 115-20, 1993.
- Bondy ML, Lustbader ED, Halabi S, et al.: Validation of a breast cancer risk assessment model in women with a positive family history. J Natl Cancer Inst 86 (8): 620-5, 1994.
- Spiegelman D, Colditz GA, Hunter D, et al.: Validation of the Gail et al. model for predicting individual breast cancer risk. J Natl Cancer Inst 86 (8): 600-7, 1994.
- Rockhill B, Spiegelman D, Byrne C, et al.: Validation of the Gail et al. model of breast cancer risk prediction and implications for chemoprevention. J Natl Cancer Inst 93 (5): 358-66, 2001.
- Costantino JP, Gail MH, Pee D, et al.: Validation studies for models projecting the risk of invasive and total breast cancer incidence. J Natl Cancer Inst 91 (18): 1541-8, 1999.
- Bondy ML, Newman LA: Breast cancer risk assessment models: applicability to African-American women. Cancer 97 (1 Suppl): 230-5, 2003.
- Schonfeld SJ, Pee D, Greenlee RT, et al.: Effect of changing breast cancer incidence rates on the calibration of the Gail model. J Clin Oncol 28 (14): 2411-7, 2010.
- Gail MH, Costantino JP, Pee D, et al.: Projecting individualized absolute invasive breast cancer risk in African American women. J Natl Cancer Inst 99 (23): 1782-92, 2007.
- Gail MH: Discriminatory accuracy from single-nucleotide polymorphisms in models to predict breast cancer risk. J Natl Cancer Inst 100 (14): 1037-41, 2008.
- Gail MH: Value of adding single-nucleotide polymorphism genotypes to a breast cancer risk model. J Natl Cancer Inst 101 (13): 959-63, 2009.
- Barlow WE, White E, Ballard-Barbash R, et al.: Prospective breast cancer risk prediction model for women undergoing screening mammography. J Natl Cancer Inst 98 (17): 1204-14, 2006.
- Tice JA, Cummings SR, Ziv E, et al.: Mammographic breast density and the Gail model for breast cancer risk prediction in a screening population. Breast Cancer Res Treat 94 (2): 115-22, 2005.
- Lee AJ, Cunningham AP, Tischkowitz M, et al.: Incorporating truncating variants in PALB2, CHEK2, and ATM into the BOADICEA breast cancer risk model. Genet Med 18 (12): 1190-1198, 2016.
- MacInnis RJ, Liao Y, Knight JA, et al.: Considerations When Using Breast Cancer Risk Models for Women with Negative BRCA1/BRCA2 Mutation Results. J Natl Cancer Inst 112 (4): 418-422, 2020.
- Liang X, Harris HR, Hendryx M, et al.: Sleep Characteristics and Risk of Ovarian Cancer Among Postmenopausal Women. Cancer Prev Res (Phila) 14 (1): 55-64, 2021.
- Jiang Y, Wang C, Zhou S: Artificial intelligence-based risk stratification, accurate diagnosis and treatment prediction in gynecologic oncology. Semin Cancer Biol 96: 82-99, 2023.
- Shi J, Kraft P, Rosner BA, et al.: Risk prediction models for endometrial cancer: development and validation in an international consortium. J Natl Cancer Inst 115 (5): 552-559, 2023.
- Barnetson RA, Tenesa A, Farrington SM, et al.: Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med 354 (26): 2751-63, 2006.
- Kastrinos F, Uno H, Ukaegbu C, et al.: Development and Validation of the PREMM5 Model for Comprehensive Risk Assessment of Lynch Syndrome. J Clin Oncol 35 (19): 2165-2172, 2017.
- Chen S, Wang W, Lee S, et al.: Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA 296 (12): 1479-87, 2006.
- Khan O, Blanco A, Conrad P, et al.: Performance of Lynch syndrome predictive models in a multi-center US referral population. Am J Gastroenterol 106 (10): 1822-7; quiz 1828, 2011.
- Mercado RC, Hampel H, Kastrinos F, et al.: Performance of PREMM(1,2,6), MMRpredict, and MMRpro in detecting Lynch syndrome among endometrial cancer cases. Genet Med 14 (7): 670-80, 2012.
- Shazly SA, Coronado PJ, Yılmaz E, et al.: Endometrial Cancer Individualized Scoring System (ECISS): A machine learning-based prediction model of endometrial cancer prognosis. Int J Gynaecol Obstet 161 (3): 760-768, 2023.
- Couch FJ, DeShano ML, Blackwood MA, et al.: BRCA1 mutations in women attending clinics that evaluate the risk of breast cancer. N Engl J Med 336 (20): 1409-15, 1997.
- Shattuck-Eidens D, Oliphant A, McClure M, et al.: BRCA1 sequence analysis in women at high risk for susceptibility mutations. Risk factor analysis and implications for genetic testing. JAMA 278 (15): 1242-50, 1997.
- Frank TS, Manley SA, Olopade OI, et al.: Sequence analysis of BRCA1 and BRCA2: correlation of mutations with family history and ovarian cancer risk. J Clin Oncol 16 (7): 2417-25, 1998.
- Frank TS, Deffenbaugh AM, Reid JE, et al.: Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol 20 (6): 1480-90, 2002.
- Evans DG, Eccles DM, Rahman N, et al.: A new scoring system for the chances of identifying a BRCA1/2 mutation outperforms existing models including BRCAPRO. J Med Genet 41 (6): 474-80, 2004.
- Apicella C, Dowty JG, Dite GS, et al.: Validation study of the LAMBDA model for predicting the BRCA1 or BRCA2 mutation carrier status of North American Ashkenazi Jewish women. Clin Genet 72 (2): 87-97, 2007.
- Risch HA, McLaughlin JR, Cole DE, et al.: Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet 68 (3): 700-10, 2001.
- Malone KE, Daling JR, Doody DR, et al.: Prevalence and predictors of BRCA1 and BRCA2 mutations in a population-based study of breast cancer in white and black American women ages 35 to 64 years. Cancer Res 66 (16): 8297-308, 2006.
- Struewing JP, Abeliovich D, Peretz T, et al.: The carrier frequency of the BRCA1 185delAG mutation is approximately 1 percent in Ashkenazi Jewish individuals. Nat Genet 11 (2): 198-200, 1995.
- Oddoux C, Struewing JP, Clayton CM, et al.: The carrier frequency of the BRCA2 6174delT mutation among Ashkenazi Jewish individuals is approximately 1%. Nat Genet 14 (2): 188-90, 1996.
- Warner E, Foulkes W, Goodwin P, et al.: Prevalence and penetrance of BRCA1 and BRCA2 gene mutations in unselected Ashkenazi Jewish women with breast cancer. J Natl Cancer Inst 91 (14): 1241-7, 1999.
- Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Anglian Breast Cancer Study Group. Br J Cancer 83 (10): 1301-8, 2000.
- Euhus DM, Smith KC, Robinson L, et al.: Pretest prediction of BRCA1 or BRCA2 mutation by risk counselors and the computer model BRCAPRO. J Natl Cancer Inst 94 (11): 844-51, 2002.
- de la Hoya M, Díez O, Pérez-Segura P, et al.: Pre-test prediction models of BRCA1 or BRCA2 mutation in breast/ovarian families attending familial cancer clinics. J Med Genet 40 (7): 503-10, 2003.
- Katki HA: Incorporating medical interventions into carrier probability estimation for genetic counseling. BMC Med Genet 8: 13, 2007.
- Weitzel JN, Lagos VI, Cullinane CA, et al.: Limited family structure and BRCA gene mutation status in single cases of breast cancer. JAMA 297 (23): 2587-95, 2007.
- Jervis S, Song H, Lee A, et al.: A risk prediction algorithm for ovarian cancer incorporating BRCA1, BRCA2, common alleles and other familial effects. J Med Genet 52 (7): 465-75, 2015.
- Oros KK, Ghadirian P, Maugard CM, et al.: Application of BRCA1 and BRCA2 mutation carrier prediction models in breast and/or ovarian cancer families of French Canadian descent. Clin Genet 70 (4): 320-9, 2006.
- Vogel KJ, Atchley DP, Erlichman J, et al.: BRCA1 and BRCA2 genetic testing in Hispanic patients: mutation prevalence and evaluation of the BRCAPRO risk assessment model. J Clin Oncol 25 (29): 4635-41, 2007.
- Kurian AW, Gong GD, John EM, et al.: Performance of prediction models for BRCA mutation carriage in three racial/ethnic groups: findings from the Northern California Breast Cancer Family Registry. Cancer Epidemiol Biomarkers Prev 18 (4): 1084-91, 2009.
- Kurian AW, Gong GD, Chun NM, et al.: Performance of BRCA1/2 mutation prediction models in Asian Americans. J Clin Oncol 26 (29): 4752-8, 2008.
- Berry DA, Parmigiani G, Sanchez J, et al.: Probability of carrying a mutation of breast-ovarian cancer gene BRCA1 based on family history. J Natl Cancer Inst 89 (3): 227-38, 1997.
- Barcenas CH, Hosain GM, Arun B, et al.: Assessing BRCA carrier probabilities in extended families. J Clin Oncol 24 (3): 354-60, 2006.
- Kang HH, Williams R, Leary J, et al.: Evaluation of models to predict BRCA germline mutations. Br J Cancer 95 (7): 914-20, 2006.
- Antoniou AC, Hardy R, Walker L, et al.: Predicting the likelihood of carrying a BRCA1 or BRCA2 mutation: validation of BOADICEA, BRCAPRO, IBIS, Myriad and the Manchester scoring system using data from UK genetics clinics. J Med Genet 45 (7): 425-31, 2008.
- Fischer C, Kuchenbäcker K, Engel C, et al.: Evaluating the performance of the breast cancer genetic risk models BOADICEA, IBIS, BRCAPRO and Claus for predicting BRCA1/2 mutation carrier probabilities: a study based on 7352 families from the German Hereditary Breast and Ovarian Cancer Consortium. J Med Genet 50 (6): 360-7, 2013.
- Biswas S, Tankhiwale N, Blackford A, et al.: Assessing the added value of breast tumor markers in genetic risk prediction model BRCAPRO. Breast Cancer Res Treat 133 (1): 347-55, 2012.
- Tai YC, Chen S, Parmigiani G, et al.: Incorporating tumor immunohistochemical markers in BRCA1 and BRCA2 carrier prediction. Breast Cancer Res 10 (2): 401, 2008.
- Ready KJ, Vogel KJ, Atchley DP, et al.: Accuracy of the BRCAPRO model among women with bilateral breast cancer. Cancer 115 (4): 725-30, 2009.
- Huo D, Senie RT, Daly M, et al.: Prediction of BRCA Mutations Using the BRCAPRO Model in Clinic-Based African American, Hispanic, and Other Minority Families in the United States. J Clin Oncol 27 (8): 1184-90, 2009.
- Daniels MS, Babb SA, King RH, et al.: Underestimation of risk of a BRCA1 or BRCA2 mutation in women with high-grade serous ovarian cancer by BRCAPRO: a multi-institution study. J Clin Oncol 32 (12): 1249-55, 2014.
- Robson ME, Storm CD, Weitzel J, et al.: American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol 28 (5): 893-901, 2010.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2.2024. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023.
Available online with free registration. Last accessed September 18, 2024. - Statement of the American Society of Human Genetics on genetic testing for breast and ovarian cancer predisposition. Am J Hum Genet 55 (5): i-iv, 1994.
- Hampel H, Bennett RL, Buchanan A, et al.: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 17 (1): 70-87, 2015.
- Owens DK, Davidson KW, Krist AH, et al.: Risk Assessment, Genetic Counseling, and Genetic Testing for BRCA-Related Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 322 (7): 652-665, 2019.
- Lancaster JM, Powell CB, Kauff ND, et al.: Society of Gynecologic Oncologists Education Committee statement on risk assessment for inherited gynecologic cancer predispositions. Gynecol Oncol 107 (2): 159-62, 2007.
- Manahan ER, Kuerer HM, Sebastian M, et al.: Consensus Guidelines on Genetic` Testing for Hereditary Breast Cancer from the American Society of Breast Surgeons. Ann Surg Oncol 26 (10): 3025-3031, 2019.
- Beitsch PD, Whitworth PW, Hughes K, et al.: Underdiagnosis of Hereditary Breast Cancer: Are Genetic Testing Guidelines a Tool or an Obstacle? J Clin Oncol 37 (6): 453-460, 2019.
- Cropper C, Woodson A, Arun B, et al.: Evaluating the NCCN Clinical Criteria for Recommending BRCA1 and BRCA2 Genetic Testing in Patients With Breast Cancer. J Natl Compr Canc Netw 15 (6): 797-803, 2017.
- Grindedal EM, Heramb C, Karsrud I, et al.: Current guidelines for BRCA testing of breast cancer patients are insufficient to detect all mutation carriers. BMC Cancer 17 (1): 438, 2017.
- Yadav S, Hu C, Hart SN, et al.: Evaluation of Germline Genetic Testing Criteria in a Hospital-Based Series of Women With Breast Cancer. J Clin Oncol 38 (13): 1409-1418, 2020.
- Kurian AW, Bernhisel R, Larson K, et al.: Prevalence of Pathogenic Variants in Cancer Susceptibility Genes Among Women With Postmenopausal Breast Cancer. JAMA 323 (10): 995-997, 2020.
- Couch FJ, Nathanson KL, Offit K: Two decades after BRCA: setting paradigms in personalized cancer care and prevention. Science 343 (6178): 1466-70, 2014.
- Theobald KA, Susswein LR, Marshall ML, et al.: Utility of Expedited Hereditary Cancer Testing in the Surgical Management of Patients with a New Breast Cancer Diagnosis. Ann Surg Oncol 25 (12): 3556-3562, 2018.
- Gornick MC, Kurian AW, An LC, et al.: Knowledge regarding and patterns of genetic testing in patients newly diagnosed with breast cancer participating in the iCanDecide trial. Cancer 124 (20): 4000-4009, 2018.
- Chiba A, Hoskin TL, Hallberg EJ, et al.: Impact that Timing of Genetic Mutation Diagnosis has on Surgical Decision Making and Outcome for BRCA1/BRCA2 Mutation Carriers with Breast Cancer. Ann Surg Oncol 23 (10): 3232-8, 2016.
- Obermair A, Youlden DR, Baade PD, et al.: The impact of risk-reducing hysterectomy and bilateral salpingo-oophorectomy on survival in patients with a history of breast cancer--a population-based data linkage study. Int J Cancer 134 (9): 2211-22, 2014.
- Byrski T, Dent R, Blecharz P, et al.: Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast Cancer Res 14 (4): R110, 2012.
- Robson M, Im SA, Senkus E, et al.: Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med 377 (6): 523-533, 2017.
- Heymann S, Delaloge S, Rahal A, et al.: Radio-induced malignancies after breast cancer postoperative radiotherapy in patients with Li-Fraumeni syndrome. Radiat Oncol 5: 104, 2010.
- Bougeard G, Renaux-Petel M, Flaman JM, et al.: Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol 33 (21): 2345-52, 2015.
- Rebbeck TR, Kauff ND, Domchek SM: Meta-analysis of risk reduction estimates associated with risk-reducing salpingo-oophorectomy in BRCA1 or BRCA2 mutation carriers. J Natl Cancer Inst 101 (2): 80-7, 2009.
- Kotsopoulos J, Lubinski J, Lynch HT, et al.: Oophorectomy after menopause and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Cancer Epidemiol Biomarkers Prev 21 (7): 1089-96, 2012.
- Cass I, Baldwin RL, Varkey T, et al.: Improved survival in women with BRCA-associated ovarian carcinoma. Cancer 97 (9): 2187-95, 2003.
- Tan DS, Rothermundt C, Thomas K, et al.: "BRCAness" syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol 26 (34): 5530-6, 2008.
- Moore K, Colombo N, Scambia G, et al.: Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med 379 (26): 2495-2505, 2018.
- Practice Bulletin No. 149: Endometrial cancer. Obstet Gynecol 125 (4): 1006-26, 2015.
- Ott PA, Bang YJ, Berton-Rigaud D, et al.: Safety and Antitumor Activity of Pembrolizumab in Advanced Programmed Death Ligand 1-Positive Endometrial Cancer: Results From the KEYNOTE-028 Study. J Clin Oncol 35 (22): 2535-2541, 2017.
- Walsh T, Casadei S, Lee MK, et al.: Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A 108 (44): 18032-7, 2011.
- Frey MK, Kim SH, Bassett RY, et al.: Rescreening for genetic mutations using multi-gene panel testing in patients who previously underwent non-informative genetic screening. Gynecol Oncol 139 (2): 211-5, 2015.
- Desmond A, Kurian AW, Gabree M, et al.: Clinical Actionability of Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Risk Assessment. JAMA Oncol 1 (7): 943-51, 2015.
- Couch FJ, Shimelis H, Hu C, et al.: Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol 3 (9): 1190-1196, 2017.
- Weitzel JN, Neuhausen SL, Adamson A, et al.: Pathogenic and likely pathogenic variants in PALB2, CHEK2, and other known breast cancer susceptibility genes among 1054 BRCA-negative Hispanics with breast cancer. Cancer 125 (16): 2829-2836, 2019.
- Frey MK, Kopparam RV, Ni Zhou Z, et al.: Prevalence of nonfounder BRCA1/2 mutations in Ashkenazi Jewish patients presenting for genetic testing at a hereditary breast and ovarian cancer center. Cancer 125 (5): 690-697, 2019.
- Eoh KJ, Kim JE, Park HS, et al.: Detection of Germline Mutations in Patients with Epithelial Ovarian Cancer Using Multi-gene Panels: Beyond BRCA1/2. Cancer Res Treat 50 (3): 917-925, 2018.
- Tung N, Lin NU, Kidd J, et al.: Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J Clin Oncol 34 (13): 1460-8, 2016.
- Buys SS, Sandbach JF, Gammon A, et al.: A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 123 (10): 1721-1730, 2017.
- Pritzlaff M, Summerour P, McFarland R, et al.: Male breast cancer in a multi-gene panel testing cohort: insights and unexpected results. Breast Cancer Res Treat 161 (3): 575-586, 2017.
- Yadav S, Reeves A, Campian S, et al.: Outcomes of retesting BRCA negative patients using multigene panels. Fam Cancer 16 (3): 319-328, 2017.
- Crawford B, Adams SB, Sittler T, et al.: Multi-gene panel testing for hereditary cancer predisposition in unsolved high-risk breast and ovarian cancer patients. Breast Cancer Res Treat 163 (2): 383-390, 2017.
- Kurian AW, Ward KC, Howlader N, et al.: Genetic Testing and Results in a Population-Based Cohort of Breast Cancer Patients and Ovarian Cancer Patients. J Clin Oncol 37 (15): 1305-1315, 2019.
- Carter NJ, Marshall ML, Susswein LR, et al.: Germline pathogenic variants identified in women with ovarian tumors. Gynecol Oncol 151 (3): 481-488, 2018.
- Dorling L, Carvalho S, Allen J, et al.: Breast Cancer Risk Genes - Association Analysis in More than 113,000 Women. N Engl J Med 384 (5): 428-439, 2021.
- Hu C, Hart SN, Gnanaolivu R, et al.: A Population-Based Study of Genes Previously Implicated in Breast Cancer. N Engl J Med 384 (5): 440-451, 2021.
- Shimelis H, LaDuca H, Hu C, et al.: Triple-Negative Breast Cancer Risk Genes Identified by Multigene Hereditary Cancer Panel Testing. J Natl Cancer Inst 110 (8): 855-862, 2018.
- Palmer JR, Polley EC, Hu C, et al.: Contribution of Germline Predisposition Gene Mutations to Breast Cancer Risk in African American Women. J Natl Cancer Inst 112 (12): 1213-1221, 2020.
- Díaz-Zabala H, Guo X, Ping J, et al.: Evaluating breast cancer predisposition genes in women of African ancestry. Genet Med 24 (7): 1468-1475, 2022.
- Southey MC, Goldgar DE, Winqvist R, et al.: PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J Med Genet 53 (12): 800-811, 2016.
- Kurian AW, Ward KC, Hamilton AS, et al.: Uptake, Results, and Outcomes of Germline Multiple-Gene Sequencing After Diagnosis of Breast Cancer. JAMA Oncol 4 (8): 1066-1072, 2018.
Penetrance of Inherited Susceptibility to Hereditary Breast and / or Gynecologic Cancers
The proportion of individuals carrying a pathogenic variant who will manifest a certain disease is referred to as penetrance. In general, common genetic variants that are associated with cancer susceptibility have a lower penetrance than rare genetic variants. This is depicted in
Figure 4. Genetic architecture of cancer risk. This graph depicts the general finding of a low relative risk associated with common, low-penetrance genetic variants, such as single-nucleotide polymorphisms identified in genome-wide association studies, and a higher relative risk associated with rare, high-penetrance genetic variants, such as pathogenic variants in the BRCA1/BRCA2 genes associated with hereditary breast and ovarian cancer and the mismatch repair genes associated with Lynch syndrome.
Throughout this summary, we discuss studies that report on relative and absolute risks (ARs). These are two important but different concepts. Relative risk (RR) refers to an estimate of risk relative to another group (e.g., risk of an outcome like breast cancer for women who are exposed to a risk factor relative to the risk of breast cancer for women who are unexposed to the same risk factor). RR measures that are greater than 1 mean that the risk for those captured in the numerator (i.e., the exposed) is higher than the risk for those captured in the denominator (i.e., the unexposed). RR measures that are less than 1 mean that the risk for those captured in the numerator (i.e., the exposed) is lower than the risk for those captured in the denominator (i.e., the unexposed). Measures with similar relative interpretations include the odds ratio (OR), hazard ratio, and risk ratio.
AR measures consider the number of people who have a particular outcome, the number of people in a population who could have the outcome, and person-time (the period of time during which an individual was at risk of having the outcome). AR measures also reflect the absolute burden of an outcome in a population. Absolute measures include risks and rates and can be expressed over a specific time frame (e.g., 1 year, 5 years) or overall lifetime. Cumulative risk is a measure of risk that occurs over a defined time period. For example, overall lifetime risk is a type of cumulative risk that is usually calculated on the basis of a given life expectancy (e.g., 80 or 90 years). Cumulative risk can also be presented over other time frames (e.g., up to age 50 years).
Large RR measures do not mean that there will be large effects in the actual number of individuals at a population level because the disease outcome may be quite rare. For example, the RR for smoking is much higher for lung cancer than for heart disease, but the absolute difference between smokers and nonsmokers is greater for heart disease, the more-common outcome, than for lung cancer, the more-rare outcome.
Therefore, in evaluating the effect of exposures and biological markers on disease prevention across the continuum, it is important to recognize the differences between relative and absolute effects in weighing the overall impact of a given risk factor. For example, the magnitude is in the range of 30% (e.g., ORs or RRs of 1.3) for many breast cancer risk factors, which means that women with a risk factor (e.g., alcohol consumption, late age at first birth, oral contraceptive use, postmenopausal body size) have a 30% relative increase in breast cancer in comparison with what they would have if they did not have that risk factor. But the absolute increase in risk is based on the underlying AR of disease.
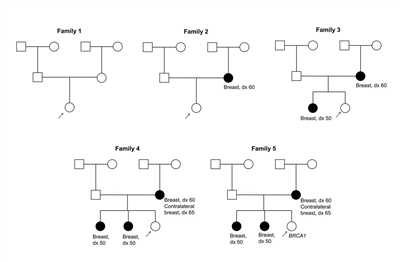
Figure 5. These five pedigrees depict probands with varying degrees of family history. Table 2 accompanies this figure.
| Family History | Lifetime Risk (%) | Lifetime Risk After Risk Factor Modification (%) | Absolute Risk Difference (%) | Relative Risk |
|---|---|---|---|---|
| a Refer to |
||||
| Low (Family 1) | 10.9 | 8.4 | 2.50 | 1.29 (29% increased risk) |
| Moderate (Family 2) | 21.6 | 16.8 | 4.80 | 1.28 (28% increased risk) |
| Moderate/high (Family 3) | 27.1 | 21.3 | 5.80 | 1.27 (27% increased risk) |
| High (Family 4) | 32.0 | 25.3 | 6.70 | 1.26 (26% increased risk) |
| BRCA1pathogenic variant (Family 5) | 53.7 | 44.2 | 9.50 | 1.21 (21% increased risk) |
With the increasing use of multigene panel tests, a framework for cancer risk management among individuals with pathogenic variants detected in novel genes has been described [
References:
- Quante AS, Herz J, Whittemore AS, et al.: Assessing absolute changes in breast cancer risk due to modifiable risk factors. Breast Cancer Res Treat 152 (1): 193-7, 2015.
- Tung N, Domchek SM, Stadler Z, et al.: Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol 13 (9): 581-8, 2016.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
Page Footer
I want to...
Audiences
Secure Member Sites
The Cigna Group Information
Disclaimer
Individual and family medical and dental insurance plans are insured by Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc., and Cigna HealthCare of Texas, Inc. Group health insurance and health benefit plans are insured or administered by CHLIC, Connecticut General Life Insurance Company (CGLIC), or their affiliates (see
All insurance policies and group benefit plans contain exclusions and limitations. For availability, costs and complete details of coverage, contact a licensed agent or Cigna sales representative. This website is not intended for residents of New Mexico.