Genetics of Colorectal Cancer (PDQ®): Genetics - Health Professional Information [NCI]
Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer in both men and women.
Estimated new cases and deaths from CRC in 2024 in the United States:[
- New cases: 152,810.
- Deaths: 53,010.
About 75% of patients with CRC have sporadic disease with no apparent evidence of having inherited the disorder. The remaining 10% to 30% of patients have a family history of CRC that suggests a hereditary contribution, common exposures or shared risk factors among family members, or a combination of both.[
In addition, pathogenic variants in lower-penetrance genes may contribute to familial colon cancer risk. In such cases, gene-gene and gene-environment interactions may contribute to the development of CRC.
(Refer to the PDQ summaries on
Colorectal Polyps as Precursors to Colorectal Cancer (CRC)
Colorectal tumors present with a broad spectrum of neoplasms, ranging from benign growths to invasive cancer, and are predominantly epithelial-derived tumors (i.e., adenomas or adenocarcinomas).
Transformation of any polyp into cancer goes through the adenoma-carcinoma sequence. Polyps that have traditionally been considered nonneoplastic include those of the hyperplastic, juvenile, hamartomatous, inflammatory, and lymphoid types. However, in certain circumstances, hamartomatous and juvenile polyps can progress into cancer.
Research, however, does suggest a substantial risk of colon cancer in individuals with juvenile polyposis syndrome and Peutz-Jeghers syndrome, although the nonadenomatous polyps associated with these syndromes have historically been viewed as nonneoplastic.[
Epidemiological studies have shown that a personal history of colon adenomas places one at an increased risk of developing colon cancer.[
Two complementary interpretations of this observation are as follows:
- The adenoma may reflect an innate or acquired tendency of the colon to form tumors.
- Adenomas are the primary precursor lesion of colon cancer.
More than 95% of CRCs are carcinomas, and about 95% of these are adenocarcinomas. It is well recognized that adenomatous polyps are benign tumors that may undergo malignant transformation. They have been classified into three histological types, with increasing malignant potential: tubular, tubulovillous, and villous. Adenocarcinomas are generally considered to arise from adenomas,[
- Benign and malignant tissue occur within colorectal tumors.[
14 ] - When patients with adenomas were followed for 20 years, the risk of cancer at the site of the adenoma was 25%, a rate much higher than that expected in the normal population.[
15 ]
The following three characteristics of adenomas are highly correlated with the potential to transform into cancer:[
- Larger size.
- Villous pathology.
- The degree of dysplasia within the adenoma.
In addition, removal of adenomatous polyps is associated with reduced CRC incidence.[
Family History as a Risk Factor for CRC
Some of the earliest studies of family history of CRC were those of Utah families that reported a higher percentage of deaths from CRC (3.9%) among the first-degree relatives (FDRs) of patients who had died from CRC than among sex-matched and age-matched controls (1.2%).[
A systematic review and meta-analysis of familial CRC risk has been reported.[
The number of affected family members and age at cancer diagnosis correlated with the CRC risk. In studies reporting more than one FDR with CRC, the RR was 3.76 (95% CI, 2.56–5.51). The highest RR was observed when the index case was diagnosed in individuals younger than 45 years (RR, 3.87; 95% CI, 2.40–6.22) compared with family members of index cases diagnosed at ages 45 to 59 years (RR, 2.25; 95% CI, 1.85–2.72), and to family members of index cases diagnosed at age 60 years or older (RR, 1.82; 95% CI, 1.47–2.25). In this meta-analysis, the familial risk of CRC associated with adenoma in an FDR was analyzed. The pooled analysis demonstrated an RR for CRC of 1.99 (95% CI, 1.55–2.55) in individuals who had an FDR with an adenoma.[
| Family History | Relative Risk of CRC[ |
Absolute Risk (%) of CRC by Age 79 ya |
|---|---|---|
| CI = confidence interval; FDR = first-degree relative. | ||
| a Data from the Surveillance, Epidemiology, and End Results Program database. | ||
| b The absolute risks of CRC for individuals with affected relatives was calculated using the relative risks for CRC[ |
||
| No family history | 1 | 4a |
| One FDR with CRC | 2.3 (95% CI, 2.0–2.5) | 9b |
| More than one FDR with CRC | 4.3 (95% CI, 3.0–6.1) | 16b |
| One affected FDR diagnosed with CRC before age 45 y | 3.9 (95% CI, 2.4–6.2) | 15b |
| One FDR with colorectal adenoma | 2.0 (95% CI, 1.6–2.6) | 8b |
When the family history includes two or more relatives with CRC, the possibility of a genetic syndrome is increased substantially. The first step in this evaluation is a detailed review of the family history to determine the number of relatives affected, their relationship to each other, the age at which the CRC was diagnosed, the presence of multiple primary CRCs, and the presence of any other cancers (e.g., endometrial) consistent with an inherited CRC syndrome. (Refer to the
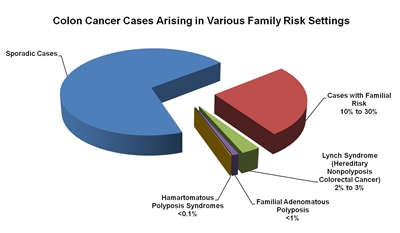
Figure 1. The fractions of colon cancer cases that arise in various family risk settings. Reprinted from Gastroenterology, Vol. 119, No. 3, Randall W. Burt, Colon Cancer Screening, Pages 837-853, Copyright (2000), with permission from Elsevier.
Inheritance of CRC Predisposition
Several genes associated with CRC risk have been identified; these are described in detail in the
- Vertical transmission of cancer predisposition in autosomal dominant conditions. (Vertical transmission refers to the presence of a genetic predisposition in sequential generations.)
- Inheritance risk of 50% for both male and female children. When a parent carries an autosomal dominant genetic predisposition, each child has a 50% chance of inheriting the predisposition. The risk is the same for both male and female children.
- Other clinical characteristics also suggest the presence of a hereditary CRC syndrome:
- Cancers in people with a hereditary predisposition typically occur at an earlier age than in sporadic cases.[
38 ] - A predisposition to CRC may include a predisposition to other cancers, such as endometrial cancer, as detailed in the
Major Genetic Syndromes section of this summary. - In addition, two or more primary cancers may occur in a single individual. These could be multiple primary cancers of the same type (e.g., two separate primary CRCs) or primary cancer of different types (e.g., colorectal and endometrial cancer in the same individual).
- The presence of nonneoplastic extracolonic features may suggest a hereditary colon cancer predisposition syndrome (e.g., congenital hypertrophy of the retinal pigment epithelium and desmoid tumors in familial adenomatous polyposis [FAP]).
- An uncommon tumor (e.g., adrenocortical carcinoma, sebaceous adenoma or carcinoma, and trichilemmoma) may serve as a clue to the presence of a hereditary cancer syndrome.
- The presence of multiple polyps may suggest a hereditary colon cancer predisposition syndrome. As susceptibility to oligopolyposis (as few as 10–15 polyps) has become apparent, clinicians, and gastrointestinal endoscopists in particular, may consider multigene (panel) testing of an ever-expanding list of genes associated with CRC. (Refer to
Table 2 , Genes Associated with a High Susceptibility of Colorectal Cancer, for more information.) Because oligopolyposis also involves diverse pathology (including hamartomas, sessile serrated polyps, and sessile serrated adenomas), careful attention to polyp count and polyp histologies helps to determine whether genetic testing and/or further clinical evaluation is appropriate.
- Cancers in people with a hereditary predisposition typically occur at an earlier age than in sporadic cases.[
The two most common causes of hereditary CRC are FAP (including AFAP), due to germline pathogenic variants in the APC gene,[
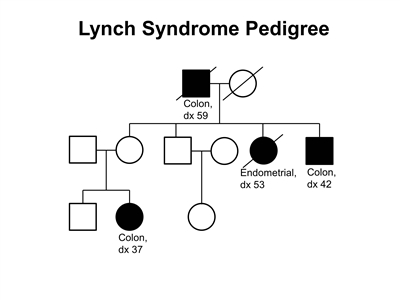
Figure 2. Lynch syndrome pedigree. This pedigree shows some of the classic features of a family with Lynch syndrome, including affected family members with colon cancer or endometrial cancer, a young age at onset in some individuals, and incomplete penetrance. Lynch syndrome families may exhibit some or all of these features. Lynch syndrome families may also include individuals with other gastrointestinal, gynecologic, and genitourinary cancers, or other extracolonic cancers. As an autosomal dominant syndrome, Lynch syndrome can be transmitted through maternal or paternal lineages, as depicted in the figure. Because the cancer risk is not 100%, individuals who have Lynch syndrome may not develop cancer, such as the mother of the female with colon cancer diagnosed at age 37 years in this pedigree (called incomplete penetrance).
Identification of Individuals at High Genetic Risk of CRC
National Comprehensive Cancer Network (NCCN) guidelines are updated annually to help identify patients who are appropriate for referral to cancer genetic counseling services. Furthermore, NCCN also provides cancer surveillance recommendations for hereditary cancer syndromes. The American College of Medical Genetics and Genomics and the National Society of Genetic Counselors have published a comprehensive set of personal/family history criteria to help identify at-risk individuals for referral to cancer genetics risk consultations.[
When such persons are identified, options tailored to the patient situation are considered. (Refer to the
At this time, the use of pathogenic variant testing to identify genetic susceptibility to CRC is not recommended as a screening measure in the general population. The rarity of pathogenic variants in CRC-associated genes and the limited sensitivity of current testing strategies render general population testing potentially misleading and not cost-effective.
Rather detailed recommendations for surveillance in FAP and Lynch syndrome have been provided by several organizations representing various medical specialties and societies. These organizations include the following:
- American Cancer Society.[
53 ] - United States Multisociety (American Gastroenterological Association and American Society for Gastrointestinal Endoscopy) Task Force on Colorectal Cancer.[
54 ] - American Society of Colon and Rectal Surgeons.[
55 ] - NCCN.[
56 ] -
Gene Reviews . - American College of Gastroenterology.[
57 ] -
Society of Gynecologic Oncology and American College of Obstetrics & Gynecology .
The evidence bases for recommendations are generally included within the statements or guidelines. In many instances, these guidelines reflect expert opinion resting on studies that are rarely randomized prospective trials.
Early-onset CRC
The epidemiology of CRC with regard to age at diagnosis is shifting, with individuals increasingly being diagnosed before age 55 years,[
In the absence of an additional family or personal history suggestive of Lynch syndrome, isolated cases of CRC diagnosed before age 36 years are uncommonly associated with MMR gene pathogenic variants. One study found MMR pathogenic variants in only 6.5% of such individuals,[
The use of polygenic risk scores (PRS) is being studied in the context of early-onset CRC in individuals who have tested negative for common CRC susceptibility variants (NCT02863107), with data from one large analysis [
Difficulties in Identifying a Family History of CRC Risk
The accuracy and completeness of family history data must be considered when using family history to assess individual risk in clinical practice and when identifying families appropriate for cancer research. A reported family history may be erroneous, or a person may be unaware of relatives with cancer.[
Accuracy of patient-reported family history of colon cancer has been shown to be good, but it is not optimal. Patient report should be verified by obtaining medical records whenever possible, especially for reproductive tract cancers that may be relevant in identifying risk of Lynch syndrome and less reliably reported by some patients. (Refer to the
Several approaches are available to evaluate a patient with newly diagnosed CRC who may or may not be suspected of having a cancer genetics syndrome. The clinician may suspect a potential inherited disposition based on the family history and physical exam, and genetic tests are available to confirm these suspicions. The American College of Medical Genetics and Genomics has published guidelines for evaluating patients with suspected colon cancer susceptibility syndromes.[
A priori risk-assessment testing (which models risk based on a variety of factors, such as age at cancer onset and the spectrum of tumors in the family) may be an appropriate alternative in many cases. Application of such risk models does anticipate the use of multigene (panel) testing; however, their exact role remains to be established.
Molecular Events Associated With Colon Carcinogenesis
Much of our initial understanding of the molecular pathogenesis of CRC derived from rare hereditary CRC syndromes and revealed heterogeneity of CRC both molecularly and clinically. It is well accepted that most CRCs develop from adenomas. The transition from normal epithelium to adenoma to carcinoma is associated with acquired molecular events.[
Chromosomal instability (CIN) pathway
Most CRCs develop through the CIN pathway. Key changes in CIN cancers include widespread alterations in chromosome number (aneuploidy) and frequent detectable losses at the molecular level of portions of chromosomes (loss of heterozygosity), such as 5q, 18q, and 17p; and pathogenic variants of the KRAS oncogene. The important genes involved in these chromosome losses are APC (5q), DCC/MADH2/MADH4 (18q), and TP53 (17p).[
Microsatellite instability (MSI) pathway
Soon thereafter, a subset (10%–15%) of CRCs was identified that lacked evidence of chromosomal instability but exhibited aberrations in microsatellite repeat sequences,[
The key characteristics of MSI cancers are that they have a largely intact chromosome complement and, as a result of defects in the DNA MMR system, more readily acquire pathogenic variants in important and often unique cancer-associated genes. These types of cancers are detectable at the molecular level by alterations in repeating units of DNA that occur normally throughout the genome, known as DNA microsatellites.
The rate of adenoma-to-carcinoma progression appears to be faster in microsatellite-unstable tumors than in microsatellite-stable tumors.[
The knowledge derived from the study of inherited CRC syndromes has provided important clues regarding the molecular events that mediate tumor initiation and tumor progression in people without germline abnormalities. Among the earliest events in the colorectal tumor progression pathway (both MSI and CIN) is loss of function of the APC gene product.
CpG island methylator phenotype (CIMP) and the serrated polyposis pathway
Beginning in the 1980s, studies began reporting an increased risk of CRC in patients with hyperplastic polyposis syndrome (HPS), now referred to as serrated polyposis syndrome (SPS).[
Further histological characterization of serrated polyps led to subtypes: traditional serrated adenomas (TSA), mixed serrated polyps (MP), and more recently, sessile serrated adenoma/sessile serrated polyp (SSA/SSP).[
In colonoscopy screening studies, large serrated polyps were strongly and independently associated with the development of advanced colorectal neoplasms, while left-sided HPs were not. The term SSA has been a concern to clinicians as these characteristically lack nuclear atypia, the traditional hallmark of adenomas, but rather are termed adenomas due to other architectural features. The classification of SSA is supported by the knowledge that the molecular characteristics denote an increased cancer risk.[
While APs in Lynch syndrome patients can exhibit MSI, sporadic adenomas rarely do. However, serrated polyps with dysplasia can exhibit MSI with hypermethylation of the MLH1 promoter. Large (>1 cm) serrated polyps carry greater cancer risk than do conventional hyperplastic polyps and, when developing into cancers, characteristically exhibit MSI.[
The MSI seen in sporadic CRCs is due to hypermethylation of the promoter of MLH1, which abrogates its expression. As promoter regions of other tumor suppressor genes were "silenced" through hypermethylation, cancer genome studies of CRC ensued. These showed a consistent pattern of hypermethylation in the evaluated genes in approximately 50% of CRCs.[
CIMP-high CRCs were much more likely (82.1%; P < .0001) to express MSI than were microsatellite-stable CRCs (24.4%; P < .0001).[
Studies of polyps revealed CIMP-positive polyps in SPS patients and most frequently in right-sided SSAs.[
Conclusion
The characterization of CIMP CRCs and evidence that MSI occurs later in the adenoma-carcinoma sequence leads to modification of the previous colorectal tumorigenesis model, which was comprised of two pathways: MSI (MIN) and CIN. There is much overlap between the MSI and CIMP pathways. At the heart of the CIMP pathway are serrated polyps harboring BRAF pathogenic variants. The CIN pathway is characterized by AP precursors of which the vast majority harbor APC pathogenic variants that occur early in the pathway.
References:
- American Cancer Society: Cancer Facts and Figures 2024. American Cancer Society, 2024.
Available online . Last accessed December 30, 2024. - Kanth P, Grimmett J, Champine M, et al.: Hereditary Colorectal Polyposis and Cancer Syndromes: A Primer on Diagnosis and Management. Am J Gastroenterol 112 (10): 1509-1525, 2017.
- Lynch HT, Smyrk TC, Watson P, et al.: Genetics, natural history, tumor spectrum, and pathology of hereditary nonpolyposis colorectal cancer: an updated review. Gastroenterology 104 (5): 1535-49, 1993.
- Rustgi AK: The genetics of hereditary colon cancer. Genes Dev 21 (20): 2525-38, 2007.
- Howe JR, Mitros FA, Summers RW: The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol 5 (8): 751-6, 1998.
- Jeevaratnam P, Cottier DS, Browett PJ, et al.: Familial giant hyperplastic polyposis predisposing to colorectal cancer: a new hereditary bowel cancer syndrome. J Pathol 179 (1): 20-5, 1996.
- Rashid A, Houlihan PS, Booker S, et al.: Phenotypic and molecular characteristics of hyperplastic polyposis. Gastroenterology 119 (2): 323-32, 2000.
- Neugut AI, Jacobson JS, DeVivo I: Epidemiology of colorectal adenomatous polyps. Cancer Epidemiol Biomarkers Prev 2 (2): 159-76, 1993 Mar-Apr.
- Shinya H, Wolff WI: Morphology, anatomic distribution and cancer potential of colonic polyps. Ann Surg 190 (6): 679-83, 1979.
- Fenoglio CM, Lane N: The anatomical precursor of colorectal carcinoma. Cancer 34 (3): suppl:819-23, 1974.
- Morson B: President's address. The polyp-cancer sequence in the large bowel. Proc R Soc Med 67 (6): 451-7, 1974.
- Muto T, Bussey HJ, Morson BC: The evolution of cancer of the colon and rectum. Cancer 36 (6): 2251-70, 1975.
- Stryker SJ, Wolff BG, Culp CE, et al.: Natural history of untreated colonic polyps. Gastroenterology 93 (5): 1009-13, 1987.
- O'Brien MJ, Winawer SJ, Zauber AG, et al.: The National Polyp Study. Patient and polyp characteristics associated with high-grade dysplasia in colorectal adenomas. Gastroenterology 98 (2): 371-9, 1990.
- Winawer SJ, Stewart ET, Zauber AG, et al.: A comparison of colonoscopy and double-contrast barium enema for surveillance after polypectomy. National Polyp Study Work Group. N Engl J Med 342 (24): 1766-72, 2000.
- Winawer SJ, Zauber AG, Ho MN, et al.: Prevention of colorectal cancer by colonoscopic polypectomy. The National Polyp Study Workgroup. N Engl J Med 329 (27): 1977-81, 1993.
- Müller AD, Sonnenberg A: Prevention of colorectal cancer by flexible endoscopy and polypectomy. A case-control study of 32,702 veterans. Ann Intern Med 123 (12): 904-10, 1995.
- O'brien MJ, Winawer SJ, Zauber AG, et al.: Flat adenomas in the National Polyp Study: is there increased risk for high-grade dysplasia initially or during surveillance? Clin Gastroenterol Hepatol 2 (10): 905-11, 2004.
- Zauber AG, O'Brien MJ, Winawer SJ: On finding flat adenomas: is the search worth the gain? Gastroenterology 122 (3): 839-40, 2002.
- Rembacken BJ, Fujii T, Cairns A, et al.: Flat and depressed colonic neoplasms: a prospective study of 1000 colonoscopies in the UK. Lancet 355 (9211): 1211-4, 2000.
- Woolf CM: A genetic study of carcinoma of the large intestine. Am J Hum Genet 10 (1): 42-7, 1958.
- Fuchs CS, Giovannucci EL, Colditz GA, et al.: A prospective study of family history and the risk of colorectal cancer. N Engl J Med 331 (25): 1669-74, 1994.
- Slattery ML, Kerber RA: Family history of cancer and colon cancer risk: the Utah Population Database. J Natl Cancer Inst 86 (21): 1618-26, 1994.
- Negri E, Braga C, La Vecchia C, et al.: Family history of cancer and risk of colorectal cancer in Italy. Br J Cancer 77 (1): 174-9, 1998.
- St John DJ, McDermott FT, Hopper JL, et al.: Cancer risk in relatives of patients with common colorectal cancer. Ann Intern Med 118 (10): 785-90, 1993.
- Duncan JL, Kyle J: Family incidence of carcinoma of the colon and rectum in north-east Scotland. Gut 23 (2): 169-71, 1982.
- Rozen P, Fireman Z, Figer A, et al.: Family history of colorectal cancer as a marker of potential malignancy within a screening program. Cancer 60 (2): 248-54, 1987.
- Johns LE, Houlston RS: A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol 96 (10): 2992-3003, 2001.
- Cottet V, Pariente A, Nalet B, et al.: Colonoscopic screening of first-degree relatives of patients with large adenomas: increased risk of colorectal tumors. Gastroenterology 133 (4): 1086-92, 2007.
- Winawer SJ, Zauber AG, Gerdes H, et al.: Risk of colorectal cancer in the families of patients with adenomatous polyps. National Polyp Study Workgroup. N Engl J Med 334 (2): 82-7, 1996.
- Ahsan H, Neugut AI, Garbowski GC, et al.: Family history of colorectal adenomatous polyps and increased risk for colorectal cancer. Ann Intern Med 128 (11): 900-5, 1998.
- Win AK, Macinnis RJ, Hopper JL, et al.: Risk prediction models for colorectal cancer: a review. Cancer Epidemiol Biomarkers Prev 21 (3): 398-410, 2012.
- Chen S, Wang W, Lee S, et al.: Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA 296 (12): 1479-87, 2006.
- Balmaña J, Stockwell DH, Steyerberg EW, et al.: Prediction of MLH1 and MSH2 mutations in Lynch syndrome. JAMA 296 (12): 1469-78, 2006.
- Barnetson RA, Tenesa A, Farrington SM, et al.: Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med 354 (26): 2751-63, 2006.
- Burt RW: Colon cancer screening. Gastroenterology 119 (3): 837-53, 2000.
- Burt RW, Petersen GM: Familial colorectal cancer: diagnosis and management. In: Young GP, Rozen P, Levin B, eds.: Prevention and Early Detection of Colorectal Cancer. WB Saunders, 1996, pp 171-194.
- Mork ME, You YN, Ying J, et al.: High Prevalence of Hereditary Cancer Syndromes in Adolescents and Young Adults With Colorectal Cancer. J Clin Oncol 33 (31): 3544-9, 2015.
- Kinzler KW, Nilbert MC, Su LK, et al.: Identification of FAP locus genes from chromosome 5q21. Science 253 (5020): 661-5, 1991.
- Groden J, Thliveris A, Samowitz W, et al.: Identification and characterization of the familial adenomatous polyposis coli gene. Cell 66 (3): 589-600, 1991.
- Leppert M, Burt R, Hughes JP, et al.: Genetic analysis of an inherited predisposition to colon cancer in a family with a variable number of adenomatous polyps. N Engl J Med 322 (13): 904-8, 1990.
- Spirio L, Olschwang S, Groden J, et al.: Alleles of the APC gene: an attenuated form of familial polyposis. Cell 75 (5): 951-7, 1993.
- Brensinger JD, Laken SJ, Luce MC, et al.: Variable phenotype of familial adenomatous polyposis in pedigrees with 3' mutation in the APC gene. Gut 43 (4): 548-52, 1998.
- Soravia C, Berk T, Madlensky L, et al.: Genotype-phenotype correlations in attenuated adenomatous polyposis coli. Am J Hum Genet 62 (6): 1290-301, 1998.
- Pedemonte S, Sciallero S, Gismondi V, et al.: Novel germline APC variants in patients with multiple adenomas. Genes Chromosomes Cancer 22 (4): 257-67, 1998.
- Sieber OM, Lamlum H, Crabtree MD, et al.: Whole-gene APC deletions cause classical familial adenomatous polyposis, but not attenuated polyposis or "multiple" colorectal adenomas. Proc Natl Acad Sci U S A 99 (5): 2954-8, 2002.
- Leach FS, Nicolaides NC, Papadopoulos N, et al.: Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 75 (6): 1215-25, 1993.
- Papadopoulos N, Nicolaides NC, Wei YF, et al.: Mutation of a mutL homolog in hereditary colon cancer. Science 263 (5153): 1625-9, 1994.
- Nicolaides NC, Papadopoulos N, Liu B, et al.: Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 371 (6492): 75-80, 1994.
- Miyaki M, Konishi M, Tanaka K, et al.: Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet 17 (3): 271-2, 1997.
- Hampel H, Bennett RL, Buchanan A, et al.: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 17 (1): 70-87, 2015.
- Bashford MT, Kohlman W, Everett J, et al.: Addendum: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 21 (12): 2844, 2019.
- Smith RA, Cokkinides V, Eyre HJ: American Cancer Society guidelines for the early detection of cancer, 2006. CA Cancer J Clin 56 (1): 11-25; quiz 49-50, 2006 Jan-Feb.
- Winawer S, Fletcher R, Rex D, et al.: Colorectal cancer screening and surveillance: clinical guidelines and rationale-Update based on new evidence. Gastroenterology 124 (2): 544-60, 2003.
- Church J, Simmang C; Standards Task Force, et al.: Practice parameters for the treatment of patients with dominantly inherited colorectal cancer (familial adenomatous polyposis and hereditary nonpolyposis colorectal cancer). Dis Colon Rectum 46 (8): 1001-12, 2003.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, and Gastric. Version 3.2024. Plymouth Meeting, PA: National Comprehensive Cancer Network, 2024.
Available with free registration. Last accessed December 13, 2024. - Syngal S, Brand RE, Church JM, et al.: ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 110 (2): 223-62; quiz 263, 2015.
- Pearlman R, Frankel WL, Swanson B, et al.: Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol 3 (4): 464-471, 2017.
- Jasperson KW, Vu TM, Schwab AL, et al.: Evaluating Lynch syndrome in very early onset colorectal cancer probands without apparent polyposis. Fam Cancer 9 (2): 99-107, 2010.
- Goel A, Nagasaka T, Spiegel J, et al.: Low frequency of Lynch syndrome among young patients with non-familial colorectal cancer. Clin Gastroenterol Hepatol 8 (11): 966-71, 2010.
- Archambault AN, Su YR, Jeon J, et al.: Cumulative Burden of Colorectal Cancer-Associated Genetic Variants Is More Strongly Associated With Early-Onset vs Late-Onset Cancer. Gastroenterology 158 (5): 1274-1286.e12, 2020.
- Glanz K, Grove J, Le Marchand L, et al.: Underreporting of family history of colon cancer: correlates and implications. Cancer Epidemiol Biomarkers Prev 8 (7): 635-9, 1999.
- Fearon ER, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell 61 (5): 759-67, 1990.
- Vogelstein B, Kinzler KW: The multistep nature of cancer. Trends Genet 9 (4): 138-41, 1993.
- Lengauer C, Kinzler KW, Vogelstein B: Genetic instabilities in human cancers. Nature 396 (6712): 643-9, 1998.
- Kinzler KW, Vogelstein B: Colorectal tumors. In: Vogelstein B, Kinzler KW, eds.: The Genetic Basis of Human Cancer. 2nd ed. McGraw-Hill, 2002, pp 583-612.
- Thibodeau SN, Bren G, Schaid D: Microsatellite instability in cancer of the proximal colon. Science 260 (5109): 816-9, 1993.
- Ionov Y, Peinado MA, Malkhosyan S, et al.: Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 363 (6429): 558-61, 1993.
- Peltomäki P, Lothe RA, Aaltonen LA, et al.: Microsatellite instability is associated with tumors that characterize the hereditary non-polyposis colorectal carcinoma syndrome. Cancer Res 53 (24): 5853-5, 1993.
- Jass JR, Cottier DS, Pokos V, et al.: Mixed epithelial polyps in association with hereditary non-polyposis colorectal cancer providing an alternative pathway of cancer histogenesis. Pathology 29 (1): 28-33, 1997.
- Jass JR: Serrated route to colorectal cancer: back street or super highway? J Pathol 193 (3): 283-5, 2001.
- Wynter CV, Walsh MD, Higuchi T, et al.: Methylation patterns define two types of hyperplastic polyp associated with colorectal cancer. Gut 53 (4): 573-80, 2004.
- Bengoechea O, Martínez-Peñuela JM, Larrínaga B, et al.: Hyperplastic polyposis of the colorectum and adenocarcinoma in a 24-year-old man. Am J Surg Pathol 11 (4): 323-7, 1987.
- Hyman NH, Anderson P, Blasyk H: Hyperplastic polyposis and the risk of colorectal cancer. Dis Colon Rectum 47 (12): 2101-4, 2004.
- Leggett BA, Devereaux B, Biden K, et al.: Hyperplastic polyposis: association with colorectal cancer. Am J Surg Pathol 25 (2): 177-84, 2001.
- McCann BG: A case of metaplastic polyposis of the colon associated with focal adenomatous change and metachronous adenocarcinomas. Histopathology 13 (6): 700-2, 1988.
- Place RJ, Simmang CL: Hyperplastic-adenomatous polyposis syndrome. J Am Coll Surg 188 (5): 503-7, 1999.
- Koide N, Saito Y, Fujii T, et al.: A case of hyperplastic polyposis of the colon with adenocarcinomas in hyperplastic polyps after long-term follow-up. Endoscopy 34 (6): 499-502, 2002.
- Torlakovic E, Snover DC: Serrated adenomatous polyposis in humans. Gastroenterology 110 (3): 748-55, 1996.
- Torlakovic EE, Gomez JD, Driman DK, et al.: Sessile serrated adenoma (SSA) vs. traditional serrated adenoma (TSA). Am J Surg Pathol 32 (1): 21-9, 2008.
- Snover DC, Jass JR, Fenoglio-Preiser C, et al.: Serrated polyps of the large intestine: a morphologic and molecular review of an evolving concept. Am J Clin Pathol 124 (3): 380-91, 2005.
- Lash RH, Genta RM, Schuler CM: Sessile serrated adenomas: prevalence of dysplasia and carcinoma in 2139 patients. J Clin Pathol 63 (8): 681-6, 2010.
- Torlakovic E, Skovlund E, Snover DC, et al.: Morphologic reappraisal of serrated colorectal polyps. Am J Surg Pathol 27 (1): 65-81, 2003.
- Jass JR, Baker K, Zlobec I, et al.: Advanced colorectal polyps with the molecular and morphological features of serrated polyps and adenomas: concept of a 'fusion' pathway to colorectal cancer. Histopathology 49 (2): 121-31, 2006.
- Goldstein NS: Small colonic microsatellite unstable adenocarcinomas and high-grade epithelial dysplasias in sessile serrated adenoma polypectomy specimens: a study of eight cases. Am J Clin Pathol 125 (1): 132-45, 2006.
- Lu FI, van Niekerk de W, Owen D, et al.: Longitudinal outcome study of sessile serrated adenomas of the colorectum: an increased risk for subsequent right-sided colorectal carcinoma. Am J Surg Pathol 34 (7): 927-34, 2010.
- Schreiner MA, Weiss DG, Lieberman DA: Proximal and large hyperplastic and nondysplastic serrated polyps detected by colonoscopy are associated with neoplasia. Gastroenterology 139 (5): 1497-502, 2010.
- Toyota M, Ahuja N, Ohe-Toyota M, et al.: CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 96 (15): 8681-6, 1999.
- Ahuja N, Mohan AL, Li Q, et al.: Association between CpG island methylation and microsatellite instability in colorectal cancer. Cancer Res 57 (16): 3370-4, 1997.
- Samowitz WS, Albertsen H, Herrick J, et al.: Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology 129 (3): 837-45, 2005.
- Yamashita K, Dai T, Dai Y, et al.: Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell 4 (2): 121-31, 2003.
- Weisenberger DJ, Siegmund KD, Campan M, et al.: CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 38 (7): 787-93, 2006.
- Chan AO, Issa JP, Morris JS, et al.: Concordant CpG island methylation in hyperplastic polyposis. Am J Pathol 160 (2): 529-36, 2002.
- Kambara T, Simms LA, Whitehall VL, et al.: BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut 53 (8): 1137-44, 2004.
- O'Brien MJ, Yang S, Clebanoff JL, et al.: Hyperplastic (serrated) polyps of the colorectum: relationship of CpG island methylator phenotype and K-ras mutation to location and histologic subtype. Am J Surg Pathol 28 (4): 423-34, 2004.
- Yang S, Farraye FA, Mack C, et al.: BRAF and KRAS Mutations in hyperplastic polyps and serrated adenomas of the colorectum: relationship to histology and CpG island methylation status. Am J Surg Pathol 28 (11): 1452-9, 2004.
- Chan TL, Zhao W, Leung SY, et al.: BRAF and KRAS mutations in colorectal hyperplastic polyps and serrated adenomas. Cancer Res 63 (16): 4878-81, 2003.
- Rajagopalan H, Bardelli A, Lengauer C, et al.: Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 418 (6901): 934, 2002.
- Yuen ST, Davies H, Chan TL, et al.: Similarity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res 62 (22): 6451-5, 2002.
- Deng G, Bell I, Crawley S, et al.: BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res 10 (1 Pt 1): 191-5, 2004.
- McGivern A, Wynter CV, Whitehall VL, et al.: Promoter hypermethylation frequency and BRAF mutations distinguish hereditary non-polyposis colon cancer from sporadic MSI-H colon cancer. Fam Cancer 3 (2): 101-7, 2004.
- Wang L, Cunningham JM, Winters JL, et al.: BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res 63 (17): 5209-12, 2003.
Major Genetic Syndromes
Introduction
Originally described in the 1800s and 1900s by their clinical findings, the colon cancer susceptibility syndrome names often reflected the physician or patient and family associated with the syndrome (e.g., Gardner syndrome, Turcot syndrome, Muir-Torre syndrome,
With the development of the Human Genome Project and the identification in 1990 of the adenomatous polyposis coli (APC) gene on chromosome 5q, overlap and differences between these familial syndromes became apparent. Gardner syndrome and familial adenomatous polyposis (FAP) were shown to be synonymous, both caused by pathogenic variants in the APC gene. Attenuated FAP (AFAP) was recognized as a syndrome with less adenomas and extraintestinal manifestations due to an APC pathogenic variant at the 3' or 5' ends of the gene. MUTYH-associated polyposis (MAP) was recognized as a separate adenomatous polyp syndrome with autosomal recessive inheritance. Once the pathogenic variants were identified, the absolute risk of colorectal cancer (CRC) could be better assessed for carriers of pathogenic variants (refer to
| Syndrome | Absolute Risk of CRC in Carriers of a Pathogenic Variant |
|---|---|
| FAP = familial adenomatous polyposis; JPS = juvenile polyposis syndrome; PJS = Peutz-Jeghers syndrome. | |
| a Cancer risk estimates quoted here predate the widespread use ofsurveillanceand prophylactic surgery. | |
| |
90% by age 45 y[ |
| |
69% by age 80 y[ |
| |
10% to 56% by age 75 y, depending on the gene involved[ |
| |
35% to 53%[ |
| |
39% by age 70 y[ |
| |
17% to 68% by age 60 y[ |
With these discoveries genetic testing and risk management became possible. Genetic testing refers to searching for variants in known cancer susceptibility genes using a variety of techniques. Comprehensive genetic testing includes sequencing the entire coding region of a gene, the intron -exon boundaries (splice sites), and assessment of rearrangements, deletions, or other changes in copy number (with techniques such as multiplex ligation-dependent probe amplification [MLPA] or Southern blot). Despite extensive accumulated experience that helps distinguish pathogenic variants from benign variants and polymorphisms, genetic testing sometimes identifies variants of uncertain significance (VUS) that cannot be used for predictive purposes.
Familial Adenomatous Polyposis (FAP)
Introduction
By 1900, several reports had demonstrated that patients with a large number of polyps (later subclassified as adenomas) were at very high risk of CRC and that the pattern of transmission in families was autosomal dominant. In the 20th century, the adenoma-to-carcinoma progression was confirmed, and FAP was recognized as the prototypical model for this progression.[
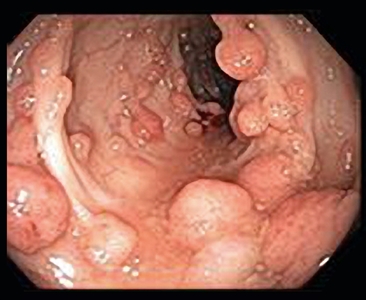
Figure 3. Familial adenomatous polyposis is characterized by multiple (>100) adenomatous polyps in the colon and rectum developing after the first decade of life.
There is also a subset of classic FAP that has an attenuated phenotype. AFAP is a heterogeneous clinical entity characterized by fewer adenomatous polyps in the colon and rectum than in classic FAP. (Refer to the
FAP is one of the most clearly defined and well understood of the inherited colon cancer syndromes.[
In addition to a high risk of colon adenomas in FAP patients, various extracolonic manifestations have also been described, including upper gastrointestinal (GI) tract adenomas and adenocarcinomas; fundic gland stomach polyps; nonepithelial benign tumors (osteomas, epidermal cysts, dental abnormalities); desmoid tumors; congenital hypertrophy of retinal pigment epithelium (CHRPE); and malignant tumors (thyroid and brain tumors, hepatoblastoma). Refer to
| Malignancy | Relative Risk | Absolute Lifetime Risk (%) |
|---|---|---|
| Adapted from Giardiello et al.,[ |
||
| a The Leeds Castle Polyposis Group. | ||
| Desmoid tumor | 852.0 | 15.0 |
| Duodenal tumors and cancer | 330.8 | 5.0–12.0 |
| Thyroid cancer | 7.6 | 2.0 |
| Brain cancer | 7.0 | 2.0 |
| Ampullary cancer | 123.7 | 1.7 |
| Pancreatic cancer | 4.5 | 1.7 |
| Hepatoblastoma | 847.0 | 1.6 |
| Gastric cancer | Not defined | 0.6a |
FAP has also been known as familial polyposis coli or adenomatous polyposis coli (APC). Gardner syndrome was previously the diagnosis for FAP patients who manifested with colorectal polyposis, osteomas, and soft tissue tumors. However, Gardner syndrome has been shown genetically to be a variant of FAP, and thus the term Gardner syndrome is essentially obsolete in clinical practice.[
Clinical phenotype
Colon adenomas and CRC
Individuals who inherit a pathogenic variant in the APC gene have a very high likelihood of developing colonic adenomas; the risk has been estimated to be more than 90%.[
Extracolonic manifestations
Congenital hypertrophy of the retinal pigment epithelium (CHRPE)
CHRPE are flat, darkly pigmented lesions in the retina that are present in approximately 75% of patients with FAP [
Desmoid tumors
Desmoid tumors are proliferative, locally invasive, nonmetastasizing, fibromatous tumors in a collagen matrix. Although they do not metastasize, they can grow very aggressively and be life threatening.[
Most studies have found that 10% of FAP patients develop desmoids, with reported ranges of 8% to 38%. The incidence varies with the means of ascertainment and the location of the pathogenic variant in the APC gene.[
A desmoid risk factor scale has been described in an attempt to identify patients who are likely to develop desmoid tumors.[
The natural history of desmoids is variable. Some authors have proposed a model for desmoid tumor formation whereby abnormal fibroblast function leads to mesenteric, plaque-like desmoid precursor lesions, which in some cases occur before surgery and progress to mesenteric fibromatosis after surgical trauma, ultimately giving rise to desmoid tumors.[
The desmoids in FAP are often intra-abdominal, may present early, and can lead to intestinal obstruction or infarction and/or obstruction of the ureters.[
These data suggest that genetic testing could be of value in the medical management of patients with FAP and/or multiple desmoid tumors. Those with APC genotypes predisposing to desmoid formation (e.g., at the 3' end or codon 1445 of the APC gene) appear to be at high risk of developing desmoids after any surgery, including risk-reducing colectomy and surgical surveillance procedures such as laparoscopy.[
Stomach tumors
The most common FAP-related gastric polyps are fundic gland polyps (FGPs). FGPs are often diffuse and not amenable to endoscopic removal. The incidence of FGPs has been estimated to be as high as 60% in patients with FAP, compared with 0.8% to 1.9% in the general population.[
The hyperplastic surface epithelium is, by definition, nonneoplastic. Accordingly, FGPs have not been considered precancerous. However, case reports of stomach cancer appearing to arise from FGPs have led to a reexamination of this issue.[
Complicating the issue of differential diagnosis, FGPs have been increasingly recognized in non-FAP patients consuming proton pump inhibitors (PPIs).[
Gastric adenomas also occur in patients with FAP. The incidence of gastric adenomas in Western patients is reported to be between 2% and 12%, whereas in Japan, incidence is reported to be between 39% and 50%.[
More recently, a rise in incidence of gastric adenocarcinoma was observed in a Western FAP database.[
Duodenum/small bowel tumors
Whereas the incidence of duodenal adenomas is only 0.4% in unselected patients undergoing upper GI endoscopy,[
Approximately 1.7% of all duodenal cancer cases are attributed to FAP.[
FAP patients with particularly severe duodenal polyposis, sometimes called dense polyposis, or with histologically advanced duodenal adenomas appear to be at the highest risk of developing duodenal adenocarcinoma.[
The predictive utility of the Spigelman classification has been called into question. The point system for dysplasia classifies dysplasia as mild, moderate, or severe, yet pathologists do not customarily attempt to distinguish moderate dysplasia from low-grade. There are no studies validating interobserver concordance in classifying a villous component or interpretation of the degree of dysplasia. A study from the Cleveland Clinic comparing Spigelman classification and its components in patients with FAP with and without cancer found neither adenoma count nor villous component to be predictive of cancer risk.[
| Points | Polyp Number | Polyp Size (mm) | Histology | Dysplasia |
|---|---|---|---|---|
| Stage I, 1–4 points; Stage II, 5–6 points; Stage III, 7–8 points; Stage IV, 9–12 points.[ |
||||
| 1 | 1–4 | 1–4 | Tubular | Mild |
| 2 | 5–20 | 5–10 | Tubulovillous | Moderate |
| 3 | >20 | >10 | Villous | Severe |
Other tumors
Other extracolonic tumors arising in FAP patients include papillary thyroid cancer, adrenal tumors, hepatoblastoma, and brain tumors.
Papillary thyroid cancer (cribriform morular type) has been reported to affect 1% to 2% of patients with FAP.[
Adrenal tumors have been reported in FAP patients, and one study demonstrated LOH at the APClocus in an adrenocortical carcinoma (ACC) in an FAP patient.[
Hepatoblastoma is a rare, rapidly progressive, and usually fatal childhood malignancy that, if confined to the liver, can be cured by radical surgical resection. Multiple cases of hepatoblastoma have been described in children with an APC pathogenic variant.[
The constellation of CRC and brain tumors has been referred to as Turcot syndrome; however, Turcot syndrome is molecularly heterogeneous. Molecular studies have demonstrated that colon polyposis and medulloblastoma are associated with pathogenic variants in APC (thus representing FAP), while colon cancer and glioblastoma are associated with pathogenic variants in mismatch repair (MMR) genes (thus representing Lynch syndrome).[
Medulloblastoma, a highly malignant embryonal central nervous system tumor, accounts for approximately 80% of the brain tumors found in FAP and primarily occurs in children with 70% diagnosed before age 16 years. High-grade astrocytomas and ependymomas have also been described in FAP patients. Although the relative lifetime risk of any brain tumor among members of an FAP family is increased 7-fold and that of medulloblastoma 90-fold, the absolute lifetime risk of any brain tumor is approximately 1% to 2%.[
Genetics of FAP
APCgene
The APC gene on chromosome 5q21 encodes a 2,843-amino acid protein that is important in cell adhesion and signal transduction; the main function of the APC protein is to regulate intracellular concentrations of beta-catenin, a major mediator of the Wnt signal transduction pathway. APC is a tumor suppressor gene, and the loss of APC is among the earliest events in the chromosomal instability colorectal tumor pathway. FAP and AFAP can be diagnosed genetically by testing for germline pathogenic variants in the APC gene in DNA from peripheral blood leukocytes. More than 300 different disease-associated pathogenic variants of the APC gene have been reported.[
Genotype-phenotype correlations
Most APC pathogenic variants that occur between codon 169 and codon 1249 result in the classic FAP phenotype.[
- Researchers have found that dense carpeting of colonic polyps, a feature of classic FAP, is seen in most patients with APC pathogenic variants, particularly those variants that occur between codons 1250 and 1464. AFAP is associated with pathogenic variants that occur in or upstream of exon 4 and in the latter two-thirds of exon 15.[
106 ,107 ,108 ,109 ] (Refer to theAttenuated Familial Adenomatous Polyposis [AFAP] section of this summary for more information.) - CHRPE are rarely associated with pathogenic variants that occur before exon 9.[
39 ,108 ] Individuals with exon 9 variants tend not to have duodenal adenomas.[70 ,110 ] - Families with GAPPS, who express numerous, predominantly fundic gland gastric polyps restricted to the body and fundus with regions of dysplasia or gastric adenocarcinoma, and no evidence of colorectal or duodenal polyposis, were found to possess variants in the promoter (1B) of APC.[
70 ] - APC pathogenic variants occurring between codons 1445 and 1578 have been associated with an increased incidence of desmoid tumors in FAP patients.[
34 ,37 ,38 ,39 ]
A low-penetrance APC variant, I1307K, has been studied for its association with CRC. (Refer to the
Genetic testing for FAP
Probands
Individuals who present with a classic FAP phenotype are candidates for APC testing. However, in many probands with a personal or family history of polyposis, multigene panel testing is an appropriate option to consider given the genetic heterogeneity of polyposis conditions and the phenotypic overlap among associated syndromes.
In particular, patients who develop fewer than 100 colorectal adenomatous polyps may pose a diagnostic challenge. The differential diagnosis includes AFAP, MAP, polymerase proofreading–associated polyposis (PPAP), and constitutional mismatch repair deficiency (CMMRD).[
For example, in a large cross-sectional study, pathogenic variants in APC were found in 80% (95% confidence interval [CI], 71%–87%) of individuals with more than 1,000 adenomas, 56% (95% CI, 54%–59%) in those with 100 to 999 adenomas, 10% (95% CI, 9%–11%) in those with 20 to 99 adenomas, and 5% (95% CI, 4%–7%) in those with 10 to 19 adenomas.[
Most commercial laboratories perform not only full gene sequencing but also deletion/duplication analysis of the APC and other genes. However, it is important to verify the testing methodology with each laboratory. Deletion analysis is especially important for individuals with FAP because 8% to 12% of affected individuals have a whole exon deletion or promoter 1B deletion in the APC gene, which would not be detected with sequencing.[
Cascade testing
In families in which a pathogenic variant in the APC gene is identified, predictive testing for at-risk relatives can definitively identify or rule out the variant. Such testing is important to determine whether at-risk relatives need to undergo aggressive screening or whether such procedures are not necessary or can be discontinued (i.e., in relatives who test negative for the familial pathogenic variant).
Most patients with FAP have an affected parent, and a pattern of autosomal dominant inheritance may be observed in the family. Accordingly, cascade genetic counseling and testing may then be extended to at-risk family members. However, it is estimated that 25% of patients with FAP have a de novo pathogenic variant in APC, meaning that the variant does not appear to be inherited from either parent.[
The early appearance of FAP clinical features and the subsequent recommendations for surveillance beginning at puberty raise special considerations relating to the genetic testing of minors.[
Interventions for FAP
Colon surveillance
Individuals at risk of FAP, because of a known APC pathogenic variant in either the family or themselves, are evaluated for onset of polyposis by flexible sigmoidoscopy or colonoscopy. Once an FAP family member is found to manifest polyps, the only effective management to prevent CRC is colectomy. Prophylactic surgery has been shown to improve survival in patients with FAP.[
A Finnish nationwide, population-based, retrospective study evaluating whether surveillance of family members with FAP reduced overall mortality and improved survival demonstrated that family members of probands who were recruited to the screening program had equivalent survival to the general population up to 20 years after diagnosis of FAP.[
Colonoscopic surveillance usually begins at an early age (10–15 y) in individuals with FAP.[
Colorectal surgery
Colon adenomas will develop in nearly 100% of individuals who are APC pathogenic variant positive; risk-reducing surgery comprises the standard of care to prevent CRC after polyps have appeared and are too numerous or histologically advanced to monitor safely using endoscopic resection.
FAP patients and their doctors should have an individualized discussion to decide when surgery will be performed. It is useful to incorporate into the discussion the risk of developing desmoid tumors after surgery, as well as fecundity for women. Timing of risk-reducing surgery usually depends on the number of polyps, their size, histology, and symptomatology.[
Surgical options include restorative proctocolectomy with ileal pouch–anal anastomosis (IPAA), total colectomy with ileorectal anastomosis (IRA), or total proctocolectomy with ileostomy (TPC). TPC is reserved for patients with low rectal cancer in which the sphincter cannot be spared or for patients on whom an IPAA cannot be performed because of technical problems. There is no risk of developing rectal cancer after TPC because the whole mucosa at risk is removed. These procedures can be performed using minimally invasive techniques.
Irrespective of whether a colectomy and an IRA or a restorative proctocolectomy is performed, most experts suggest that periodic and lifelong surveillance of the rectum or the ileal pouch be performed to remove or ablate any polyps. In earlier unselected studies, the risk of rectal cancer after total colectomy 20 years after IRA was reported to be as high as 25%.[
In most cases, the clinical polyp burden in the rectum at the time of surgery dictates the type of surgical intervention, namely, restorative proctocolectomy with IPAA versus IRA. Patients with a mild phenotype (<1,000 colonic adenomas) and fewer than 20 rectal polyps may be candidates for IRA at the time of prophylactic surgery.[
It is important to continue annual surveillance of the ileal pouch in patients who have undergone IPAA because they are at risk of developing neoplasia in the anal transitional zone/residual rectal mucosa and in the ileal pouch. The cumulative risk of developing adenomas in the ileal pouch can be up to 75% for 15 years after surgery has been completed.[
Chemoprevention
Celecoxib, a specific cyclooxygenase 2 (COX-2) inhibitor, and nonspecific COX-2 inhibitors, such as sulindac (a nonsteroidal anti-inflammatory drug [NSAID]), have been associated with a decrease in polyp size and number in FAP patients, suggesting a role for chemopreventive agents in the treatment of this disorder.[
A small, randomized, placebo-controlled, dose-escalation trial of celecoxib in a pediatric population (aged 10–14 y) demonstrated the safety of celecoxib at all dosing levels when administered over a 3-month period.[
Omega-3-polyunsaturated fatty acid eicosapentaenoic acid in the free fatty acid form has been shown to reduce rectal polyp number and size in a small study of patients with FAP after subtotal colectomy.[
It is unclear at present how to incorporate COX-2 inhibitors into the management of FAP patients who have not yet undergone risk-reducing surgery. A double-blind placebo-controlled trial of 41 child and young adult carriers of APC pathogenic variants who had not yet manifested polyposis demonstrated that sulindac may not be effective as a primary treatment in FAP. There were no statistically significant differences between the sulindac and placebo groups over 4 years of treatment in incidence, number, or size of polyps.[
Consistent with the effects of COX-2 inhibitors on colonic polyps, in a randomized, prospective, double-blind, placebo-controlled trial, celecoxib reduced, but did not eliminate, the number of duodenal polyps in 32 patients with FAP after a 6-month course of treatment. Of importance, a statistically significant effect was seen only in individuals who had more than 5% of the duodenum involved with polyps at baseline and with an oral dose of 400 mg, given twice daily.[
Because of the common clustering of adenomatous polyps around the duodenal papilla (where bile enters the intestine) and preclinical data suggesting that ursodeoxycholate inhibits intestinal adenomas in mice that harbor an Apc germline variant,[
Because of reports demonstrating an increase in cardiac-related events in patients taking rofecoxib and celecoxib,[
Level of evidence (celecoxib): 1b
One cohort study has demonstrated regression of colonic and rectal adenomas with sulindac treatment in FAP. The reported outcome of this trial was the number and size of polyps, a surrogate for the clinical outcome of main interest, CRC incidence.[
Level of evidence (sulindac): 1b
Preclinical studies of a small-molecule epidermal growth factor receptor (EGFR) inhibitor and low-dose sulindac in the Apcmin/+ mouse diminished intestinal adenoma development by 87% [
On the basis of the previously modest effects of sulindac and celecoxib on duodenal polyps in patients with FAP [
Level of evidence (sulindac + erlotinib): 1b
Management of extracolonic tumors
Patients who carry APC germline pathogenic variants are at increased risk of other types of malignancies, including desmoid tumors, gastric tumors, duodenal cancer, small bowel cancer, hepatoblastoma, thyroid cancer, and brain tumors. The management of these extracolonic tumors is described below.
Desmoid tumors
The management of desmoids in FAP can be challenging and can complicate prevention efforts. There is no accepted standard treatment for desmoid tumors. Multiple medical treatments have generally been unsuccessful in the management of desmoids. Treatments have included antiestrogens, NSAIDs, chemotherapy, and radiation therapy, among others. Studies have evaluated the use of raloxifene alone, tamoxifen or raloxifene combined with sulindac, and pirfenidone alone.[
Thirteen patients with intra-abdominal desmoids and/or unfavorable response to other medical treatments who had expression of estrogen-alpha receptors in their desmoid tissues were included in a prospective study of raloxifene, given in doses of 120 mg daily.[
A second study of 13 patients with FAP-associated desmoid tumors, who were treated with tamoxifen 120 mg/day or raloxifene 120 mg/day in combination with sulindac 300 mg/day, reported that ten patients had either stable disease (n = 6) or a partial or complete response (n = 4) for more than 6 months and that three patients had stable disease for more than 30 months.[
A third study reported mixed results in 14 patients with FAP-associated desmoid tumors treated with pirfenidone for 2 years.[
There are reports of using imatinib mesylate to treat desmoid tumors in FAP patients with some success.[
Level of evidence: 4
The benefit of the tyrosine kinase inhibitor sorafenib in the treatment of desmoid tumors was demonstrated in a phase III randomized trial comparing sorafenib (400 mg daily) with placebo in 87 patients with unresectable progressive or symptomatic desmoid tumors.[
Level of evidence: 1
Because of the high rates of morbidity and recurrence, in general, surgical resection is not recommended in the treatment of intra-abdominal desmoid tumors. A review of experiences at one hospital suggested that surgical outcomes with intra-abdominal desmoid tumors may be better than previously believed.[
Stomach tumors
It is not clear how to manage gastric adenomas. Only retrospective case series are available and point to a relatively low prevalence of gastric adenocarcinoma development in FAP patients.[
Level of evidence: 5
Duodenum/small bowel tumors
Endoscopic surveillance usually begins between ages 20 to 25 years in patients with FAP. Baseline upper endoscopy may be performed at an earlier age if the patient has a family history of large duodenal adenoma burden or duodenal/ampullary cancer.[
The main advantages of the Spigelman classification are its long-standing familiarity to and usage by those in the field, which allows reasonable standardization of outcome comparisons across studies.[
- Most pathologists do not employ the term moderate dysplasia, preferring a simpler low- versus high-grade dysplasia system.
- Because of the villous nature of normal duodenal epithelium, pathologists commonly disagree over the classification of tubular, tubulovillous, and villous.
- Spigelman staging requires biopsy, which is not always essential when only a few small plaques are present; conversely, for larger adenomas, sampling variation leads to understaging.[
180 ,181 ]
| Spigelman Stage | NCCN (2024)[ |
ESMO (2013)[ |
|---|---|---|
| ESMO = European Society of Medical Oncology; NCCN = National Comprehensive Cancer Network. | ||
| See |
||
| 0 (no polyps) | Endoscopy every 3–5 y | Not specified |
| I | Endoscopy every 2–3 y | Endoscopy every 5 y |
| II | Endoscopy every 1–2 y | Endoscopy every 3 y |
| III | Endoscopy every 6–12 mo | Endoscopy every 1–2 y |
| IV | Expert endoscopic surveillance every 3–6 mo | Endoscopy every 6-12 mo |
| Excision/ablation of resectable large or villous adenomatous polyps and endoscopic ampullectomy are options that may help individuals avoid surgery | ||
| Surgical evaluation and counseling for individuals with high-grade dysplasia, invasive carcinoma, or dense polyposis that cannot be removed endoscopically | Surgical options include duodenotomy with polypectomy, pancreas-sparing duodenectomy and pancreaticoduodenectomy (Whipple procedure) | |
The results of long-term duodenal adenoma surveillance of FAP patients in Nordic countries and the Netherlands revealed significant duodenal cancer risk in FAP patients.[
Level of evidence (screening for duodenum/small bowel tumors): 3
Many factors, including severity of polyposis, comorbidities, patient preferences, and availability of adequately trained physicians, determine whether surgical or endoscopic therapy is selected for polyp management. Endoscopic resection or ablation of large or histologically advanced adenomas appears to be safe and effective in reducing the short-term risk of developing duodenal adenocarcinoma;[
The endoscopic approach to larger and/or flatter adenomas of the duodenum depends on whether the ampulla is involved. Endoscopic mucosal resection (EMR) after submucosal injection of saline, with or without epinephrine and/or dye, such as indigo carmine, can be employed for nonampullary lesions. Ampullary lesions require even greater care including endoscopic ultrasound evaluation for evidence of bile or pancreatic duct involvement. Stenting of the pancreatic duct is commonly performed to prevent stricturing and pancreatitis. The stents require endoscopic removal at an interval of 1 to 4 weeks. Because the ampulla is tethered at the ductal orifices, it typically does not uniformly lift with injection, so injection is commonly not used. Any consideration of EMR or ampullectomy requires great experience and judgment, with careful consideration of the natural history of untreated lesions and an appreciation of the high rate of adenoma recurrence despite aggressive endoscopic intervention.[
Reluctance to consider surgical resection is related to the short-term morbidity and mortality and the long-term complications related to surgery. Although these concerns are likely overstated,[
Level of evidence (treatment of duodenum/small bowel tumors): 4
Other tumors
Although level 1 evidence is lacking for the following surveillance methods, they are based on expert opinion. NCCN recommends baseline thyroid ultrasound beginning in the late teenage years to screen for papillary thyroid cancer in patients with FAP, with a repeat ultrasound every 2 to 5 years if results are normal. When individuals have a family history of thyroid cancer, shorter screening intervals can be used.[
Level of evidence (thyroid cancer ultrasound screening): 4
Although level 1 evidence is lacking for the following surveillance methods, they are based on expert opinion. NCCN has suggested that the following be considered for children with a predisposition to FAP: liver palpation, abdominal ultrasound, and measurement of serum alpha-fetoprotein every 3 to 6 months for the first 5 years of life.[
Level of evidence (hepatoblastoma or adrenal cancer screening): 5
Although level 1 evidence is lacking for the following surveillance methods, they are based on expert opinion. Medulloblastoma is a highly malignant tumor that is usually only symptomatic 6 months or less before diagnosis; annual surveillance of asymptomatic patients may be insufficient. Thus, surveillance by means of regular CT or magnetic resonance imaging (MRI) cannot be advocated. FAP family members who do not yet have polyposis but have signs or symptoms suggestive of a brain tumor should be evaluated with neuroimaging because brain tumors present before polyposis in more than half of FAP patients. Careful evaluation is also important among FAP families in which one member already has a brain tumor because familial clustering occurs. Of such families with FAP-associated brain tumors, 40% had two affected members.[
Attenuated Familial Adenomatous Polyposis (AFAP)
Clinical phenotype
AFAP was first described clinically in 1990 in a large kindred with a variable number of adenomas. The average number of adenomas in this kindred was 30, although they ranged in number from a few to hundreds.[
Genetics of AFAP
AFAP is associated with particular subsets of APC pathogenic variants. Three groups of site-specific APC pathogenic variants causing AFAP have been characterized:[
- Pathogenic variants associated with the 5' end of APC and exon 4 in which patients can manifest 2 to more than 500 adenomas, including the classic FAP phenotype and upper GI polyps. Any pathogenic variant in the first four exons,[
106 ] as there is an internal ribosomal entry site in exon 4 that permits the ribosome to skip premature truncation pathogenic variants.[210 ] - Exon 9–associated phenotypes in which patients may have 1 to 150 adenomas but no upper GI manifestations.
- 3' region pathogenic variants in which patients have very few adenomas (<50).
In the absence of family history of similarly affected relatives, the differential diagnosis may include AFAP (including MAP), Lynch syndrome, CMMRD, germline variants in the DNA polymerase proofreading subunits (POLD1 or POLE), or an otherwise unclassified sporadic or genetic problem. A careful family history may implicate AFAP or Lynch syndrome.
APC testing is an important component of the evaluation of patients suspected of having AFAP.[
Clinical management
Patients found to have an unusually or unacceptably high adenoma count at an age-appropriate colonoscopy pose a differential diagnostic challenge.[
| Organization | Condition | Screening Method | Screening Frequency | Age Screening Initiated | Comment |
|---|---|---|---|---|---|
| FDA = U.S. Food and Drug Administration; IPAA = ileal pouch–anal anastomosis; IRA = ileorectal anastomosis; NCCN = National Comprehensive Cancer Network. | |||||
| a Colonoscopy with polypectomy can adequately remove polyps when individuals have a small adenoma burden, which is defined as fewer than 20 adenomas that do not have advanced histology and are each <1 cm in diameter. | |||||
| Europe Mallorca Group (2008)[ |
AFAP | Colonoscopy | Every 2 y; every 1 y if adenomas are detected | 18–20 y | |
| NCCN (2024)[ |
Personal history of AFAP with small adenoma burdena | High-quality colonoscopy and polypectomy | Every 1–2 y | If patient had colectomy with IRA, endoscopic evaluation every 6–12 mo, depending on the patient's polyp burden | |
| Chemoprevention may be considered in patients with progressive polyp burdens to manage the remaining rectum or pouch postcolectomy; at this time, the FDA has not approved medications for this specific indication; NCCN recommends that patients seek the advice of providers with expertise in FAP/AFAP | |||||
| Personal history of AFAP with adenoma burden that cannot be handled endoscopically | Not applicable | Not applicable | Not applicable | Colectomy with IRA preferred. Consider proctocolectomy with IPAA if patient has dense rectal polyposis | |
| Asymptomatic at-risk family member; familial pathogenic variant known;APCpathogenic variant status positive | High-quality colonoscopy | Every 1–2 y ifAPCpositive | Late teens | If adenomas are found, follow AFAP screening guidelines | |
| Asymptomatic at-risk family member; familial pathogenic variant known;APCpathogenic variant status unknown | High-quality colonoscopy | If genetic testing is not performed, colonoscopy can be done every 2 y; if adenomas are found, follow AFAP screening guidelines; if adenomas are not found on multiple subsequent exams, a prolonged screening interval (>2 y) may be considered | Late teens | Discuss benefits of genetic testing | |
MUTYH-Associated Polyposis (MAP)
MAP is an autosomal recessively inherited polyposis syndrome caused by pathogenic variants in the Mut Y homolog gene. The Mut Y homolog gene, which is known as MUTYH, was initially called MYH, but was subsequently corrected because the myosin heavy chain gene already had that designation. MUTYH is located on chromosome 1p34.3-32.1.[
The MUTYH gene was first linked to polyposis in 2002 in three siblings with multiple colonic adenomas and CRC but no APC pathogenic variant.[
Adenomas, serrated adenomas, and hyperplastic polyps can be seen in MAP patients.[
Although MAP is the only known biallelic (recessive) adenoma cancer predisposition syndrome described to date, there are examples of biallelic cases presenting with childhood tumors in which MMR genes are involved. For more information, see the
| Organization | Condition | Screening Method | Screening Frequency | Age Screening Initiated | Comment |
|---|---|---|---|---|---|
| CRC = colorectal cancer; FDR = first-degree relative; IPAA = ileal pouch–anal anastomosis; IRA = ileorectal anastomosis; NCCN = National Comprehensive Cancer Network. | |||||
| a Colonoscopy with polypectomy can adequately remove polyps when individuals have a small adenoma burden, which is defined as fewer than 20 adenomas that do not have advanced histology and are each <1 cm in diameter. | |||||
| Nieuwenhuis et al. (2012)[ |
OneMUTYHpathogenic variant (monoallelic/MUTYHheterozygote) | Colonoscopy | Every 1–2 y | ||
| NCCN (2024)[ |
BiallelicMUTYHpathogenic variants; personal history of MAP, small adenoma burdena | High-quality colonoscopy and polypectomy | Every 1–2 y | No later than age 25 to 30 y | If patient had colectomy with IRA, endoscopic evaluation every 6–12 mo, depending on the patient's polyp burden |
| Chemoprevention may be considered in certain individuals (especially those with a high polyp burden postcolectomy), but data are limited in patients with MAP; consider referring patients to a center that has experience with MAP to discuss chemoprevention and surgery options | |||||
| BiallelicMUTYHpathogenic variants; personal history of MAP with adenoma burden that cannot be managed endoscopically | Not applicable | Not applicable | Not applicable | Colectomy with IRA. Consider proctocolectomy with IPAA if patient has dense rectal polyposis. If patient had colectomy with IRA, endoscopic evaluation of the rectum may be done every 6–12 mo based on polyp burden | |
| Asymptomatic, at-risk family member; familial pathogenic variant known;MUTYHpathogenic variant status unknown | High-quality colonoscopy | Every 1–2 y | No later than age 25–30 y | Repeat screening every 1–2 years if polyps are not found; the screening interval can be lengthened if an individual does not have polyps on multiple subsequent colonoscopies, based on a provider's judgment; if polyps are found, use MAP screening guidelines. Discuss benefits of genetic testing if the patient's pathogenic variant status is unknown | |
| Asymptomatic, at-risk family member; familial pathogenic variant known; noMUTYHpathogenic variant found | General population screening | ||||
| OneMUTYHpathogenic variant (monoallelic/MUTYH heterozygote); proband has a personal history or anFDRwith CRC/colon polyps | Increased frequency of screening based on the patient's personal or family history of CRC/colon polyps (refer to NCCN guidelines for CRC screening)[ |
||||
| OneMUTYHpathogenic variant (monoallelic/MUTYH heterozygote); proband does not have a personal history or an FDR with CRC/colon polyps | General population screening | ||||
Many extracolonic cancers have been reported in patients with MAP including gastric, small intestinal, endometrial, liver, ovarian, bladder, thyroid, and skin cancers (melanoma, squamous epithelial, and basal cell carcinomas).[
Duodenal polyps in MAP
Similar to FAP, individuals with MAP often develop duodenal adenomas, and are at risk of developing duodenal cancer. Given the relatively recent identification of MAP compared with FAP, the incidence of duodenal polyps and risk of duodenal cancer in MAP is less well defined. Small case series have suggested the incidence of duodenal polyps in MAP to be approximately 30%, considerably lower than that of FAP. In a registry-based study the prevalence of duodenal polyps was 17%; however, only 50% of individuals in this study had undergone an upper GI endoscopy, suggesting the incidence of duodenal polyps was likely underestimated. The lifetime risk of duodenal cancer was estimated to be 4%.[
A registry study from the United Kingdom and the Netherlands explored incidence of duodenal polyps and duodenal cancer in a group of patients with MAP who were undergoing regular duodenal surveillance.[
Because MAP has an autosomal recessive inheritance pattern, siblings of an affected patient have a 25% chance of also carrying biallelic MUTYH pathogenic variants and should be offered genetic testing. Similarly, testing can be offered to the partner of an affected patient so that the risk in their children can be assessed.
The clinical phenotype of monoallelic MUTYH pathogenic variants is less well characterized with respect to incidence and associated clinical phenotypes, and its role in susceptibility to polyposis and colorectal carcinoma remains unclear. Approximately 1% to 2% of the general population carry a pathogenic variant in MUTYH.[
MMR genes may interact with MUTYH and increase the risk of CRC. An association between MUTYH and MSH6 has been reported. Both proteins interact together in base excision repair processes. A study reported a significant increase of MSH6 pathogenic variants in carriers of monoallelic MUTYH pathogenic variants with CRC compared with noncarriers with CRC (11.5% vs. 0%; P = .037).[
Lynch Syndrome
Introduction
Lynch syndrome is the most common inherited CRC syndrome and accounts for approximately 3% of all newly diagnosed cases of CRC. It is an autosomal dominant condition caused by pathogenic variants in the MMR genes MLH1 (mutL homolog 1), MSH2 (mutS homolog 2), MSH6 (mutS homolog 6), and PMS2 (postmeiotic segregation 2), as well as the gene EPCAM (epithelial cellular adhesion molecule, formerly known as TACSTD1), in which deletions in EPCAM cause epigenetic silencing of MSH2. Lynch syndrome is also associated with a predisposition for developing several extracolonic manifestations, including sebaceous adenomas and cancers of the endometrium and ovaries, stomach, small intestine, transitional cell carcinoma of the ureters and renal pelvis, hepatobiliary system, pancreas, and brain. Lynch syndrome–associated cancers exhibit MSI; therefore, tumor testing is a key component in the diagnosis of Lynch syndrome, in addition to family history. Universal tumor testing of all CRCs is now recommended as a strategy to screen for Lynch syndrome and identify those individuals who may subsequently benefit from germline genetic testing. Intensive cancer screening and surveillance strategies, including frequent colonoscopy, along with risk-reducing surgeries, are mainstays in patients with Lynch syndrome.
History of Lynch syndrome
Between 1913 and 1993, numerous case reports of families with apparent increases in CRC were reported. As series of such reports accumulated, certain characteristic clinical features emerged: early age at onset of CRC; high risk of synchronous (and metachronous) colorectal tumors; preferential involvement of the right colon; improved clinical outcome; and a range of associated extracolonic sites including the endometrium, ovaries, other sites in the GI tract, uroepithelium, brain, and skin (sebaceous tumors). Terms such as cancer family syndrome and hereditary nonpolyposis colorectal cancer (HNPCC) were used to describe this entity.[
The term Lynch syndrome replaced HNPCC and is applied to cases in which the genetic basis can be confidently linked to a germline pathogenic variant in a DNA MMR gene. Moreover, HNPCC is misleading as many patients have polyps and many have tumors other than CRC.
With the increased recognition of families that were considered to have a genetic predisposition to the development of CRC, research for a causative etiology led to the development of the
In 1987, a chromosomal deletion of a small segment of 5q led to the detection of a genetic linkage between FAP and this genomic region,[
In 2009, a germline deletion in the EPCAM gene was identified as another cause of MSH2 inactivation in the absence of a germline pathogenic variant in MSH2. The variant in EPCAM led to hypermethylation of the MSH2 promoter. Thus, EPCAM, which is not a DNA MMR gene, is also implicated in Lynch syndrome and is now routinely tested in at-risk patients along with the DNA MMR genes listed above.
Defining Lynch syndrome families
Families with a preponderance of CRC and a possible genetic predisposition were initially categorized as having Lynch syndrome based on family history criteria, as well as personal history of young-onset CRC. With the advent of molecular tumor diagnostic testing and the discovery of the germline alterations associated with Lynch syndrome, the clinical criteria have currently fallen out of favor due to their underperformance. However, their use, or the risk estimates provided by the Lynch syndrome prediction models, may be applicable among individuals without personal history of cancer but with a family history suggestive of Lynch syndrome, or for those individuals with CRC but without available tumor for molecular diagnostic testing. (Refer to the
The first criteria for defining Lynch syndrome families were established by the International Collaborative Group meeting in Amsterdam in 1990 and are known as the Amsterdam criteria.[
Amsterdam criteria I (1990):
- One family member diagnosed with CRC before age 50 years.
- Two affected generations.
- Three affected relatives, one of them an FDR of the other two.
- FAP should be excluded.
- Tumors should be verified by pathological examination.
Amsterdam criteria II (1999):
- Same as Amsterdam criteria I, but tumors of the endometrium, small bowel, ureter, or renal pelvis can be used to substitute an otherwise qualifying CRC.
These criteria were subsequently used beyond research purposes to identify potential candidates for microsatellite and germline testing. However, the Amsterdam criteria failed to identify a substantial proportion of Lynch syndrome kindreds; families that fulfilled Amsterdam criteria I but did not have evidence of MSI and were without a pathogenic germline variant in a DNA MMR gene, were referred to as familial colorectal cancer type X (FCCX).
With the hallmark feature of MSI associated with Lynch syndrome tumors, and the limitations of the Amsterdam criteria related to low sensitivity, the Bethesda guidelines were introduced in 1997. The Bethesda guidelines are a combination of clinical, histopathologic, and family cancer history features that identify cases of CRC that warrant MSI tumor screening. The Bethesda guidelines (with a subsequent revision in 2004) were formulated to target patients in whom evaluation of CRC tumors for MMR deficiency should be considered, and to improve the sensitivity of clinical criteria used to identify individuals who are candidates for mutational DNA analysis.[
Bethesda guidelines (1997):
- Cancer in families that meet the Amsterdam criteria.
- The presence of two Lynch syndrome–related cancers, including synchronous and metachronous CRCs or associated extracolonic cancers. Endometrial, ovarian, gastric, hepatobiliary, or small-bowel cancer or transitional cell carcinoma of the renal pelvis or ureter.
- The presence of CRC and an FDR with CRC and/or Lynch syndrome–related extracolonic cancer and/or a colorectal adenoma; one of the cancers diagnosed before age 45 years, and the adenoma diagnosed before age 40 years.
- CRC or endometrial cancer diagnosed before age 45 years.
- Right-sided CRC with an undifferentiated pattern (solid/cribriform) on histopathology diagnosed before age 45 years. Solid/cribriform defined as poorly differentiated or undifferentiated carcinoma composed of irregular, solid sheets of large eosinophilic cells and containing small gland-like spaces.
- Signet-ring–cell CRC diagnosed before age 45 years. Composed of more than 50% signet ring cells.
- Adenomas diagnosed before age 40 years.
Revised Bethesda guidelines (2004)*:
- CRC diagnosed in an individual younger than 50 years.
- Presence of synchronous, metachronous colorectal, or other Lynch syndrome–associated tumors.**
- CRC with MSI-high (MSI-H) pathological associated features diagnosed in an individual younger than 60 years. Presence of tumor-infiltrating lymphocytes, Crohn-like lymphocytic reaction, mucinous/signet-ring differentiation, or medullary growth pattern.
- CRC or Lynch syndrome–associated tumor** diagnosed in at least one FDR younger than 50 years.
- CRC or Lynch syndrome–associated tumor** diagnosed at any age in two FDRs or second-degree relatives (SDRs).
*One criterion must be met for the tumor to be considered for MSI testing.
**Lynch syndrome–associated tumors include colorectal, endometrial, stomach, ovarian, pancreatic, ureter and renal pelvis, biliary tract, and brain tumors; sebaceous gland adenomas and keratoacanthomas in Muir-Torre syndrome; and carcinoma of the small bowel.[
Although the Bethesda guidelines were able to identify a higher proportion of Lynch syndrome carriers than the Amsterdam criteria, they still missed approximately 30% of Lynch syndrome families.[
With the advent of alternative approaches, including universal testing of all newly diagnosed cases of CRC for MSI (regardless of age at diagnosis or family history of cancer), clinical criteria for Lynch syndrome have been rendered obsolete. While the Bethesda guidelines were intended for individuals with cancer, their performance in individuals unaffected by cancer may still be of use. Given the limited modalities available to assess unaffected individuals for Lynch syndrome, family history and the use of clinical criteria may be appropriate in identifying those who warrant further genetic evaluation and testing.
Clinical risk assessment models that predict the likelihood of an MMR gene pathogenic variant
Because health care providers ineffectively use clinical criteria to select individuals with CRC for genetic referral and evaluation for Lynch syndrome, computer-based clinical prediction models were developed and introduced in 2006 as alternative modalities to provide systematic genetic risk assessment for Lynch syndrome. The risk models include the
Four models (PREMM[1,2,6], PREMMplus, MMRpredict, and MMRpro) quantify an individual's probability of carrying a pathogenic variant in one of the following MMR genes: MLH1, MSH2, and MSH6. The PREMM(1,2,6) model was subsequently extended to include prediction of pathogenic PMS2 and EPCAM variants (PREMM5).[
While the models were all created for the same purpose, they differ in the way they were developed and the variables used to predict risk. In addition, the populations in which they were validated reveal each model's specific characteristics that may impact accuracy.[
Overall, there is ample evidence that each of the models has superior performance characteristics of sensitivity, specificity, and positive and negative predictive values that support their use when compared with the existing clinical guidelines for diagnosis and evaluation of Lynch syndrome. Because of the diverse clinical settings in which a health care provider could assess an individual for Lynch syndrome, prediction models offer a potentially feasible and useful strategy to systematically identify at-risk individuals, whether or not they are affected with CRC.
Summary
In conclusion, the presence of tumor MSI in CRCs, along with a compelling personal and family history of cancer, warrants germline genetic testing for Lynch syndrome, and most clinical practice guidelines provide for such an approach. These guidelines combine genetic counseling and testing strategies with clinical screening and treatment measures. Providers and patients alike can use these guidelines to better understand available options and key decisions. (Refer to
Genetics of Lynch syndrome
The genetics of both the tumor and the germline have an important role in the development and diagnosis of Lynch syndrome. Tumor DNA in Lynch syndrome–associated tumors exhibits characteristic MSI, and in these cases, there is typically loss of IHC expression for one or more of the proteins associated with the MMR genes. Molecular testing with MSI and/or IHC has been adopted as a universal screen for diagnosis of Lynch syndrome in newly diagnosed patients with CRC and endometrial cancer. IHC testing results can potentially direct gene-specific germline testing. Many genetic testing laboratories offer multigene (panel) tests that simultaneously test for pathogenic variants in all of the Lynch syndrome–associated genes (and often additional genes associated with inherited cancer susceptibility).
Genetic and molecular testing for Lynch syndrome
MSI
The presence of MSI in colorectal tumor specimens is a hallmark feature of Lynch syndrome and can be cause for suspicion of a germline pathogenic MMR gene variant. Microsatellites are short, repetitive sequences of DNA (mononucleotides, dinucleotides, trinucleotides, or tetranucleotides) located throughout the genome, primarily in intronic or intergenic sequences.[
Certain histopathologic features are strongly suggestive of MSI phenotype, including the presence of tumor-infiltrating lymphocytes (refer to
Figure 4. Tumor-infiltrating lymphocytes are a histopathologic feature suggestive of microsatellite instability.
Initial designation of a colorectal adenocarcinoma as microsatellite unstable was based on the detection of a specified percentage of unstable loci from a panel of three dinucleotide and two mononucleotide repeats that were selected at a National Institutes of Health (NIH) Consensus Conference and referred to as the Bethesda panel. If more than 30% of a tumor's markers were unstable, it was scored as MSI-H; if at least one, but fewer than 30% of markers were unstable, the tumor was designated MSI-low (MSI-L). If no loci were unstable, the tumor was designated microsatellite stable (MSS). Most tumors arising in the setting of Lynch syndrome will be MSI-H.[
The original Bethesda panel has been replaced by a pentaplex panel of five mononucleotide repeats,[
(Refer to the
(Refer to the
IHC
IHC methods are cheaper, easier to understand, and more widely available as a surrogate for MSI and, for these reasons, have replaced polymerase chain reaction (PCR)–based MSI testing in most institutions. IHC is performed in the colorectal or endometrial tumor (or metastatic sites) [
MSI can lead to nucleotide-pairing slippage (looping) in which single nucleotide mispairs are introduced. Heterodimers of MMR proteins are formed to identify the errors and bind the DNA at these sites.[
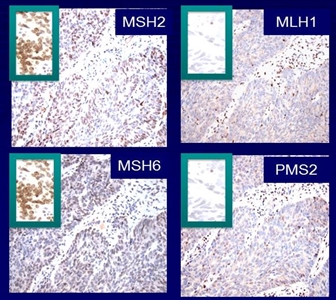
Figure 5. Immunohistochemical tumor testing for protein expression of the mismatch repair genes associated with Lynch syndrome, depicted for a single patient with colorectal cancer. Protein expression is preserved for MSH2 and MSH6 (inset) and absent for MLH1 and PMS2 (inset). Absence of MMR protein expression is suggestive of Lynch syndrome and warrants additional evaluation.
As a result, when the germline pathogenic variant is in the MSH2 gene, the tumor IHC may not express both MSH2 and MSH6, as the latter protein requires binding to MSH2 for stability. In this case, if no pathogenic variant is found in either gene, germline pathogenic variant testing for EPCAM should be considered if it was not already included. Approximately 20% of patients with absence of MSH2 and MSH6 protein expression by IHC and no MSH2 or MSH6 pathogenic variant identified will have germline deletions in EPCAM.[
In patients with no variants in any of these genes, tumor sequencing may reveal biallelic, somatic MSH2 variants. For more information, see the
Similarly, the loss of MLH1 (either by germline pathogenic variant or hypermethylation of the MLH1 promoter) results in the absence of expression of both MLH1 and PMS2 proteins in the tumor. The most common abnormal IHC pattern for DNA MMR proteins in colorectal adenocarcinomas is loss of expression of MLH1 and PMS2. PMS2 and MLH1 function as a stable heterodimer known as MutLα. MutLα binds to MutSβ and guides excision repair of the newly synthesized DNA strand.[
Unlike MLH1 and MSH2 (which both dimerize with other proteins or have other binding partners), germline pathogenic variants in MSH6 and PMS2 result in the isolated loss of those specific proteins by IHC. However, tumors from MSH6 pathogenic variant carriers may not display the MSI phenotype at a frequency as high as MLH1 and MSH2 carriers (despite an inactive DNA MMR system), as there are pathogenic missense variants that do not completely abrogate protein expression yielding false negative results by IHC testing.[
In some cases, tumors manifest MSI and/or IHC shows loss of DNA MMR protein expression, but no germline pathogenic variant is identified. This tumor phenotype is predominantly due to biallelic somatic inactivation of the DNA MMR genes and is not a pathogenic germline alteration. This phenomenon has been labeled Lynch-like syndrome (sometimes called LLS in the literature), although this terminology may cause confusion since this term represents a mechanism of sporadic carcinogenesis, rather than a stand-alone genetic syndrome. For more information, see the
| Loss of Protein Expression | Germline MMR Defect Predicted by IHC Protein Expression Loss | ||||
|---|---|---|---|---|---|
| | MLH1 | MSH2 | MSH6 | PMS2 | EPCAM |
| IHC = immunohistochemistry; MMR = mismatch repair. | |||||
| MLH1/PMS2 | X | ||||
| MSH2/MSH6 | X | X | |||
| MSH6 | X | ||||
| PMS2 | X | X | |||
| MLH1 | X | X | |||
| MSH2 | X | X | |||
SomaticMLH1hypermethylation
It is important to recognize that hypermethylation of the MLH1 promoter, a somatic event confined to the tumor, can lead to abnormal protein expression of MLH1 on IHC. Approximately 10% to 15% of sporadic CRC cases have a microsatellite unstable tumor phenotype due to MLH1 hypermethylation and are not heritable. These sporadic MSI colon cancers [
BRAF pathogenic variants have been detected in 68% of CRC tumors with MLH1 promoter hypermethylation and very rarely, if ever, in CRC from patients with Lynch syndrome.[
Somatic biallelic mismatch repair deficiency (sometimes called Lynch-like syndrome)
Prior to the widespread use of somatic tumor sequencing, somatic MLH1 hypermethylation was thought to be the only major sporadic pathway for a tumor to have MSI-H/MMR-deficient (MMR-D) biology. It was presumed that any MSI-H/MMR-D CRC or endometrial cancer with patterns of MMR deficiency (other than MLH1/PMS2 loss) was due to Lynch syndrome, even if a germline pathogenic variant in an MMR gene could not be identified. However, as IHC is more routinely performed for all CRCs and endometrial cancers, it has become clear that other patterns of MMR deficiency can arise via sporadic mechanisms, typically by somatic biallelic MMR gene inactivation.[
Work from the Ohio Colorectal Cancer Prevention Initiative found that 88.4% of CRCs and 100% of endometrial cancers with unexplained MMR-D statuses (i.e., cancers did not have a germline pathogenic variant in an MMR gene or MLH1 promoter hypermethylation) had somatic biallelic MMR deficiency found on somatic tumor sequencing.[
When CRCs, endometrial cancers, or other tumors have unexplained MMR-deficiency, in which somatic sequencing fails to identify somatic biallelic MMR deficiency, it is possible that a small fraction of these cases could represent occult Lynch syndrome (or other forms of unidentifiable inherited cancer risk). In these cases, it is recommended that patients' clinical and family histories be carefully examined for possible inherited cancer risk. This will help determine if patients and their families will be clinically managed as if they have suspected Lynch syndrome. In these cases, careful scrutiny of any germline MMR gene VUS is also warranted to assess whether these VUSs could harbor pathogenicity. For more information about VUS genetic test results, see the
Some studies have attempted to define the risk of CRC and other Lynch syndrome–associated malignancies in cohorts of individuals with Lynch-like syndrome. However, the ability to clinically interpret these data may be limited since these populations presumably include a mix of individuals with sporadic MMR-D cancers and others with inherited cancer risk.[
IHC in constitutional mismatch repair deficiency (CMMRD) syndrome
In contrast with Lynch syndrome (which is defined by the presence of a single, germline, heterozygous, deleterious variant in an MMR gene), rarely, individuals can have deleterious germline variants in both alleles of the same MMR gene. These cases can present with homozygous or compound heterozygous genotypes. This is termed biallelic mismatch repair deficiency (BMMRD) or constitutional mismatch repair deficiency (CMMRD). The likelihood of CMMRD involving homozygous MMR gene pathogenic variants will inevitably be higher among consanguineous unions. Rates of consanguinity may be higher in rural and geographically and/or culturally isolated populations.[
Tumor studies yield characteristic abnormalities. In a series of 28 patients with CMMRD,[
The PMS2 gene is markedly overrepresented in cases of CMMRD. It has been suggested that the presence of homozygosity in other MMR gene variants is a prenatally lethal state, while milder expression of PMS2 variants is consistent with survival when present in both parental alleles.
For more information about CMMRD's clinical phenotype, see the
| Clinical Phenotype | Pathogenic Germline Variant in DNA MMR | Somatic Inactivation of DNA MMR | Tumor Phenotype |
|---|---|---|---|
| CMMRD = constitutional mismatch repair deficiency; FCCX = familial colorectal cancer type X; MMR = mismatch repair; MSI = microsatellite instability; MSS = microsatellite stable. | |||
| a Adapted from Carethers et al.[ |
|||
| Lynch syndrome | Present in one allele | Present in one allele | MSI |
| Sporadic CRC with hypermethylation ofMLH1promoter | Absent | +BRAF | MSI |
| CMMRD | Present in two alleles | Absent | MSI (tumor and normal tissue) |
| Somatic biallelic MMR deficiency (sometimes called Lynch-like syndrome) | Absent | Present in two alleles | MSI |
| FCCX | Absent | Absent | MSS |
Constitutional epimutation
While somatic hypermethylation of the MLH1 promoter is acquired and not uncommon, examples of MLH1 promoter hypermethylation have been described in the germline and are generally not associated with a stable Mendelian inheritance. This constitutional methylation of MMR genes occurs most often in MLH1 and, to a lesser extent, MSH2 and is termed constitutional epimutation.[
Interpreting molecular alterations in tumors and distinguishing the likely primary epimutation cases from those of sporadic MSI poses significant challenges. Most instances of absence of MLH1 expression are caused by the sporadic hypermethylation of the MLH1 promoter. Rare instances of a de novo constitutional epimutation in MLH1[
Such MLH1-predominant primary epimutations are to be distinguished from secondary epimutations such as those occurring when MSH2 is methylated as a consequence of inherited variants in the upstream EPCAM gene. (Refer to the
Molecular diagnostic tumor testing to screen for Lynch syndrome in clinical practice
While many molecular pathology laboratories can assess both MSI and IHC, an approach that uses IHC testing as the initial screen for defective MMR activity has been favored because it is less labor intensive and more cost-effective.[
Universal tumor testing to screen for Lynch syndrome
Use of MSI and/or IHC testing in all newly diagnosed cases of CRC, regardless of the age at diagnosis or family history of cancer, increases the sensitivity of the initial screen for Lynch syndrome. This approach is more sensitive than existing clinical criteria, as many individuals with Lynch syndrome are diagnosed at older ages (>50 y) and have less striking family histories of CRC than previously appreciated. This universal testing of colorectal (and
Genetic risk assessment and MMR gene variant testing in individuals with newly diagnosed CRC can lead to improved outcomes for the patient and at-risk family members. Dating back to 2009, the Evaluation of Genomic Applications in Practice and Prevention (EGAPP), a project developed by the Office of Public Health Genomics at the Centers for Disease Control and Prevention (CDC), reported that there was sufficient evidence to recommend offering tumor screening for Lynch syndrome to individuals with newly diagnosed CRC to reduce morbidity and mortality in relatives.[
Several studies have demonstrated the feasibility of universal screening for Lynch syndrome. Initial experience from one institution found that among 1,566 patients screened using MSI and IHC, 44 patients (2.8%) had Lynch syndrome. For each proband, an average of three additional family members were subsequently diagnosed with Lynch syndrome.[
The consideration to further stratify the recommendation for molecular tumor testing by age (i.e., 70 y) warrants attention as it influences the cost-effectiveness of universal screening strategy.
Loss of MLH1 and PMS2 due to somatic hypermethylation is not uncommon, and is more frequently detected with increasing age at CRC diagnosis.[
Screening individuals with CRC for Lynch syndrome is most often performed in a stepwise fashion based on IHC tumor testing results that evaluate protein expression for the four MMR genes related to Lynch syndrome. One proposed strategy is summarized in
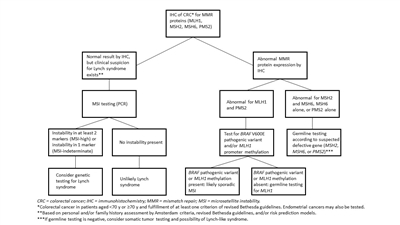
Figure 6. A proposed strategy to evaluate individuals with colorectal cancer for Lynch syndrome based on immunohistochemical tumor testing results. Adapted from Geiersbach KB, Samowitz WS. Microsatellite instability and cancer. Arch Pathol Lab Med 135(10):1269-77, 2011.
Clinicians are increasingly utilizing tumor sequencing to advance therapeutic decisions in a more personalized approach, particularly in patients with metastatic disease. The performance of next-generation tumor sequencing (NGS) of CRCs for the detection of Lynch syndrome was compared with existing screening protocols that include MSI testing and IHC staining (with BRAF p.V600E testing) in 419 CRC cases recruited in a multicenter, population-based study.[
A 2019 retrospective study using data from a large, community-based, integrated U.S. health care system compared the diagnostic performance of age-restricted screening strategies for Lynch syndrome by reflex MMR IHC of all CRCs versus a universal screening strategy without an upper age limit.[
Cost-effectiveness of universal tumor screening for Lynch syndrome
Results are available from a Markov model that incorporated the risks of colorectal, endometrial, and ovarian cancers to estimate the effectiveness and cost-effectiveness of strategies to identify Lynch syndrome among individuals aged 70 years or younger with newly diagnosed CRC.[
NCCN 2024 guidelines support using universal screening on all CRCs to help identify individuals who may have Lynch syndrome. Universal screening can include the following testing methods: IHC testing, MSI testing, and comprehensive tumor NGS panel testing.[
However, it is important to note that the conclusions from this study were contingent upon the number of at-risk relatives who underwent germline testing (through a process known as cascade screening) based on the identification of a germline MMR gene variant in the index case of CRC in the family. In their model, to meet the accepted $50,000 cost-effective threshold, testing a minimum of three to four relatives was necessary.[
Another study addressed the cost-effectiveness of testing for pathogenic variants in the Lynch syndrome–associated genes and evaluated 21 screening strategies, including clinical criteria, use of clinical Lynch syndrome prediction models, and molecular tumor testing.[
Establishment of an upper age limit for universal tumor testing remains controversial. Some experts have endorsed testing only individuals with CRC who are younger than 70 years (reserving testing in individuals ≥70 y for only those meeting the revised Bethesda criteria; with this strategy, 5% of carriers would be missed).[
Another cost-effectiveness analysis was performed using data from 179 consecutive endometrial cancer patients diagnosed at or before age 70 years and screened with MMR IHC and reflex MLH1 promoter hypermethylation, among whom seven Lynch syndrome carriers (3.9%) were identified.[
The cost-effectiveness of universal tumor testing in both CRC and endometrial cancer is largely driven by the assumption of cascade screening through which other at-risk family members will be identified, tested, and subsequently pursue their own cancer risk reduction.[
The cost of germline genetic testing continues to decrease with advancements in DNA analyses, including simultaneous testing of multiple germline variants associated with malignancy, through multigene (panel) tests. As a result, additional cost-effective analyses using more updated data related to germline testing will need to be conducted. Multigene (panel) testing may become a more favorable and cost-effective approach in the future.
Considerations and limitations related to universal tumor testing for Lynch syndrome
While universal screening continues to be adopted nationally, there is significant variability in the uptake and approach to molecular testing. A 2011 survey of the National Society of Genetic Counselors revealed that more than 25% of respondents had some form of universal screening implemented at their center. Tumor screening methods varied; 34 (64.2%) of 53 centers started with IHC, 11 (20.8%) of 53 centers started with MSI testing, and 8 (15.1%) of 53 centers performed both tests on newly diagnosed colorectal tumors.[
Because adherence to universal screening for Lynch syndrome may be poor (many patients are not referred for genetic evaluation and testing), a prospective quality improvement study utilizing the Six Sigma conceptual framework was conducted to improve the implementation of universal genetic screening among young patients with CRC.[
Studies reporting uptake of genetic testing for Lynch syndrome have largely focused on individuals and families who were selected for potential risk of Lynch syndrome based on family history or clinical characteristics. While universal tumor screening is increasingly being adopted to identify newly diagnosed patients who may have a germline variant, few studies have examined the uptake of genetic testing after universal tumor testing. An important implication of universal screening for Lynch syndrome is that it does not result in automatic germline testing in appropriate individuals. In the clinical setting, more follow-up by health care teams to facilitate referral to genetic counseling for patients with abnormal tumor screening results may improve completion of genetic testing.[
Subsequent genetic counseling requires coordination between the pathologist, the referring surgeon or oncologist, and a cancer genetics service. As an illustration, a population-based screening study found that only 54% of patients with an IHC-deficient tumor (that was BRAF pathogenic variant negative) ultimately consented to and proceeded with germline MMR testing.[
In contrast to tumor testing, which is commonly performed without a patient's prior knowledge, germline genetic testing, such as germline testing for MMR pathogenic variants, generally includes genetic counseling and requires patient permission before it is performed. A cross-sectional survey of U.S. cancer programs (20 NCI–designated Comprehensive Cancer Centers and 49 community hospital cancer programs) found that, of those that performed MSI and/or IHC testing as part of standard pathological evaluation at the time of colon cancer diagnosis in all or select cases, none required written informed consent before tumor testing.[
Diagnostic strategies for all individuals diagnosed with endometrial cancer
Given the increased prevalence of endometrial cancer among carriers of MMR pathogenic variants, there is a growing consensus to screen patients with endometrial cancer for Lynch syndrome.
In a study that examined the feasibility and desirability of performing tumor screening of all endometrial cancers, regardless of age at diagnosis or family history of cancer, at least 2.3% (95% CI, 1.3%–4.0%) of newly diagnosed patients had Lynch syndrome.[
Another smaller study of 242 consecutive endometrial cases demonstrated a 4.5% (11/242) prevalence of MMR-deficient cases lacking somatic MLH1 promoter hypermethylation, including four cases (1.7%) with germline MMR variants, four cases (1.7%) with two somatic MMR alterations on NGS, and two cases (0.8%) with otherwise unexplained MMR-deficiency.[
Another study prospectively evaluated universal IHC-based screening of both CRC and endometrial cancer cases, irrespective of age at diagnosis.[
The cost-effectiveness of tumor testing of women diagnosed with endometrial cancer was examined in a model-based simulation study and included IHC testing in the following scenarios: (1) diagnosis before age 50 years; (2) diagnosis before age 60 years; (3) any age at diagnosis with the presence of an FDR with any Lynch syndrome–associated cancer; and (4) all cases irrespective of diagnosis age and family history. Women fulfilling Amsterdam II criteria or those diagnosed before age 50 years with at least one FDR with any Lynch syndrome–associated cancer were directly referred for genetic counseling and genetic testing without IHC testing. A strategy of IHC testing for MMR protein expression in all patients with endometrial cancer and an FDR with any Lynch syndrome–associated cancer was reported to be cost-effective in the detection of Lynch syndrome.[
(Refer to the PDQ summary on
MSI in all cancers
Use of MSI testing across all tumor types has become an important screening tool to select cases that may have a favorable response to immune checkpoint inhibitor therapy. These results may potentially be used to screen for Lynch syndrome in tumors other than CRC. A study evaluated MSI across a wide variety of malignancies and evaluated its use as a potential means to identify Lynch syndrome, regardless of tumor type.[
A single-institution study of 100 histologically confirmed small bowel adenocarcinomas with MSI/MMR tumor studies identified 26 cases with MMR deficiency, 10 of which were due to Lynch syndrome. Most cancers were located in the duodenum, followed by the jejunum and the ileum. The authors recommended that individuals with small bowel adenocarcinomas be screened for Lynch syndrome, given the implications of MMR deficiency on treatment and clinical management.[
Germline genetic testing
Genetic testing for germline pathogenic variants in MLH1, MSH2, MSH6, PMS2, and EPCAM can help formulate appropriate intervention strategies for the affected variant-positive individual and at-risk family members, many of whom may be unaffected by cancer.
If a pathogenic variant is identified in an affected person, then testing for that same pathogenic variant should be offered to all at-risk family members. At-risk relatives who test negative for the identified pathogenic variant in the family are not at increased risk of CRC or other Lynch syndrome–associated malignancies and can follow surveillance recommendations applicable to the general population. Family members who carry the familial pathogenic variant are referred to surveillance and management guidelines for Lynch syndrome. (Refer to the
If no pathogenic variant is identified in the affected family member, then testing is considered negative for Lynch syndrome in that individual. With advances made in DNA sequencing technologies, it is unlikely that current gene testing is not sensitive enough to detect a pathogenic variant in the genes tested. Advances in testing, including the common use of NGS by most commercial testing laboratories have improved upon the detection of certain alterations such as large deletions or genomic rearrangements as well as the presence of a pseudogene PMSCL in PMS2.
Possible reasons why a pathogenic variant may not be detected include the following:
- The family could have a variant in a yet-unidentified gene that causes Lynch syndrome or a predisposition to colon cancer.
- The individual tested in the family may have developed colon cancer through a nongenetic mechanism (i.e., it is a sporadic case also known as a phenocopy), while the other cases in the family are really the result of a germline variant. If this scenario is suspected, testing another affected individual who has had a Lynch syndrome–associated cancer is recommended.
- In cases in which a CRC tumor displayed MSI and/or abnormal IHC, but no germline pathogenic variant was detected, biallelic somatic variants may be the resulting etiology. These are sometimes, confusingly, called Lynch-like syndrome cases, even though they are typically not considered to be familial.
Failure to detect a pathogenic variant could mean that the family truly is not at genetic risk despite a clinical presentation that suggests a genetic basis (e.g., the patient may have biallelic somatic variants in an MMR gene). If no variant can be identified in an affected family member, testing should not be offered to at-risk members because results would be uninformative for the relatives. They would remain at increased risk of CRC by virtue of their family history and should continue with recommended intensive screening.
(Refer to the
Multigene (panel) testing
Germline variant analysis of MLH1, MSH2 (including EPCAM), MSH6, and PMS2 may be considered in instances in which tumor tissue is not available from individuals to test for MSI and/or MMR protein IHC. This approach has become less expensive with the advent of multigene (panel) testing, which is now offered by several clinical laboratories at a cost that may be comparable to single-gene testing. The cost of multigene testing may also approach the cost of tumor screening and may prove to be a cost-effective approach in individuals affected by CRC. At present, multigene tests are not routinely recommended for universal screening for Lynch syndrome among all newly diagnosed CRC patients, but they may be very useful in select populations, such as those with early-onset CRC [
Individuals with early-onset CRC have been shown to have a high frequency and wide spectrum of germline pathogenic variants, indicating that panel testing in this population may be beneficial. In a study of 450 patients with early-onset CRC (mean age at diagnosis, 42.5 y) and a family history including at least one FDR with colon, endometrial, breast, ovarian, and/or pancreatic cancer, 75 germline pathogenic or likely pathogenic variants were identified in 72 patients (16%).[
Multigene testing has also been examined in a larger study of 1,058 individuals with CRC who were unselected for age at diagnosis, personal or family history, or MSI/MMR test results.[
A 2017 study examined the frequency of pathogenic Lynch syndrome–associated gene variants in individuals undergoing multigene testing at a single commercial United States laboratory between 2012 and 2015, and reported on the characteristics of those carriers identified with Lynch syndrome.[
The study reports on genotype-phenotype correlations on 528 Lynch syndrome carriers, the majority of whom had CRC (186, 35.2%) and endometrial cancer (136, 25.8%), followed by breast cancer (124, 23.5%) and ovarian cancer (74, 14%).[
Clinical criteria for the identification of Lynch syndrome, including the Amsterdam criteria, revised Bethesda guidelines, or the PREMM(1,2,6) risk prediction model, would have failed to identify 27.3% of Lynch syndrome carriers in this study.[
Lastly, germline MMR genes have been detected unexpectedly among individuals undergoing multigene testing for cancers not commonly associated with Lynch syndrome, such as breast and prostate cancer. As a result, the cancer spectrum associated with Lynch syndrome may be wider than previously appreciated. For more information, see the
(Refer to the
Cost-effectiveness of multigene (panel) testing
As genetic testing becomes routine rather than the exception, questions regarding the cost of testing are inevitable. Historically, a cost-effectiveness ratio of $50,000 per quality-adjusted life-year (QALY) has been used as the benchmark for good value for care.[
A 2015 study evaluated the cost-effectiveness of multigene testing for CRC and polyposis syndromes in patients referred to a cancer genetics clinic.[
The cost of germline genetic testing continues to decrease with advancements in technology since the time this model analysis was conducted; additional studies are needed to continue to assess the cost-effectiveness of this testing approach.
Prevalence, clinical manifestations, and cancer risks associated with Lynch syndrome
Lynch syndrome is an autosomal dominant syndrome characterized by an early age of onset of CRC, excess synchronous and metachronous colorectal neoplasms, right-sided predominance, and extracolonic tumors, notably endometrial cancer. Lynch syndrome is caused by pathogenic variants in the DNA MMR genes, namely MLH1 on chromosome 3p21;[
Lynch syndrome accounts for about 3% of all newly diagnosed cases of CRC.[
Original reports related to overall and gene-specific prevalence estimates in Lynch syndrome relied heavily on retrospective data from familial cancer registries worldwide. Earlier risk estimates of CRC (and endometrial cancer) reported in Lynch syndrome were subject to ascertainment bias and overestimation, given that data were derived largely from familial cancer registries and cases were often ascertained based on young-onset CRC or an increased number of CRC cases among relatives. Correction of these cancer risk estimates has been made possible through modified segregation analyses, where statistical methodology provides more accurate estimates and adjusts for ascertainment bias. Conversely, risk estimates related to extracolonic malignancies, with the exception of endometrial cancer, may be prone to underestimation because many families may have underreported these cancers in relatives, and Lynch syndrome–related tumors may have occurred later in life.
In a large population-based study of 5,744 CRC cases who were recruited irrespective of family cancer history from the United States, Australia, and Canada, it was estimated that 1 in 279 individuals in the population carry an MMR pathogenic variant associated with Lynch syndrome.[
In another population-based study of 450 individuals with CRC but limited to young onset with diagnoses occurring before age 50 years, germline pathogenic variants were identified in 72 of 450 individuals (16%), as detected by multigene (panel) testing for inherited cancer susceptibility genes. As expected, the majority of identified variants were in genes known to be associated with CRC, predominantly Lynch syndrome (37 of 72 patients, 51.4%). However, 13 of 72 patients (18.1%) had pathogenic variants in genes not traditionally associated with CRC, including but not limited to BRCA1/BRCA2, which accounted for 8% of the identified variants. Because of the high frequency and wide variety of pathogenic variants identified, the authors suggested consideration of multigene testing for all individuals with early-onset CRC.[
Gene-specific considerations and associated CRC risk
The MLH1 and MSH2 genes were originally thought to account for most pathogenic variants of the MMR genes found in Lynch syndrome. However, the prevalence of MSH6 and PMS2 pathogenic variants has been increasing with improved DNA analyses and universal tumor screening of all CRCs.[
MLH1
In early studies, the prevalence of MLH1 pathogenic variants in individuals with Lynch syndrome was reported to be between 41.7% [
MLH1 pathogenic variants are associated with the entire spectrum of malignancies associated with Lynch syndrome.[
Unlike the APC gene of FAP, in which several phenotypes of differing severity and spectrum of disease occur, genotype-phenotype relationships have been elusive in the MMR genes. In a large series of MLH1 pathogenic variant carriers, women with truncating MLH1 pathogenic variants had significantly later onset of endometrial cancer than did those with nontruncating variants.[
MSH2
The prevalence of MSH2 pathogenic variants in individuals or families with Lynch syndrome has varied across studies. MSH2 pathogenic variants were reported in 38% to 54% of Lynch syndrome families in studies including large cancer registries and among cohorts of early-onset CRC (younger than age 55 y).[
The risk of any Lynch syndrome–associated cancer by age 70 years has been found to range between 57% to nearly 80% in MSH2 pathogenic variant carriers.[
The mean age at diagnosis of CRC in MSH2 carriers has been comparable to MLH1 carriers. One study that included 143 affected individuals with MSH2 pathogenic variants found a mean age at CRC diagnosis of 43.9 years (range, 16–90 y). The same study reported a mean age at CRC diagnosis of 42.8 years (range, 16–81 y) in 137 MLH1 pathogenic variant carriers.[
MSH6
Most series have reported a prevalence of germline MSH6 pathogenic variants in approximately 10% of Lynch syndrome families from high-risk clinics and a higher proportion of unselected CRC patients, at approximately 50%.[
The lifetime risk of any Lynch syndrome–associated cancer among MSH6 pathogenic variant carriers is approximately 25% [
The largest series of carriers of MSH6 pathogenic variants reported to date includes 113 families from five countries who were ascertained through family cancer clinics and population-based cancer registries.[
In a more recent prospective study using pooled European registry data of 305 MSH6 carriers without cancer, the cumulative CRC incidence was 20% at age 70 years despite colonoscopic surveillance.[
PMS2
PMS2 was the last of the genes in the MMR family of genes to be identified. This was because lower penetrance among families made it more difficult to identify [
In earlier studies of individuals with CRC and suspected Lynch syndrome, the prevalence of PMS2 pathogenic variants was variable from 2.2% to 5%,[
The lifetime risk of any cancer has been found to range between 25% and 32% for heterozygous PMS2 pathogenic variant carriers.[
The PLSD is a major ongoing initiative to assess cancer risks in Lynch syndrome. Although it lacks specific details regarding screening practices, it includes outcome data from many European programs, classified by age, gender, and MMR gene.[
It is important to note that a more severe phenotype is seen among carriers of biallelic PMS2 pathogenic variants. For more information, see the
The lifetime risk of CRC and endometrial cancer in carriers of these pathogenic variants is summarized in
| Gene | Lifetime Risk of Colorectal Cancer (%) | Lifetime Risk of Endometrial Cancer (%) | References |
|---|---|---|---|
| MLH1 | 41–50 | 34–54 | [ |
| MSH2 | 35–56 | 21–51 | [ |
| MSH6 | 10–22 | 16–49 | [ |
| PMS2 | 10 | 24 | [ |
EPCAM
A subset of individuals with Lynch syndrome (approximately 1%) have a pathogenic variant in EPCAM, which leads to hypermethylation and inactivation of the MSH2 promoter.[
One study of two families with the same EPCAM deletion limited to the 3' end of the gene and not extending into the promoter of MSH2 found few extracolonic cancers and no endometrial cancers.[
Intragenic variability in CRC penetrance among carriers of MMR pathogenic variants
While it is known that MMR pathogenic variant carriers have high lifetime risks of CRC, estimates of these risks vary considerably between studies. Additional factors, including ascertainment bias, heritable modifiers of risk, and nonheritable risk factors may contribute substantially to variation in lifetime risk estimates.
A study from the Colon Cancer Family Registry reported that, depending on the gene that has the pathogenic variant and an individual's sex, 16% to 23% of MLH1 and MSH2 carriers had lifetime CRC risks of less than 10% (i.e., their risks approached average risk for the general population).[
An international registry–based study used retrospective data to examine variation in CRC risk in 5,255 families with Lynch syndrome.[
CMMRD
As described above, patients may carry MMR gene variants in both parental alleles, in a condition known as CMMRD. For more information, see the
The occurrence of such biallelic variants is associated with a characteristic but not diagnostic clinical phenotype. Clinical features include hematologic malignancies and brain tumors in children. When GI tumors occur, the age of onset is strikingly low, sometimes before age 20 years. Café au lait spots and features otherwise suggesting neurofibromatosis are characteristic. Occasionally, patients present with multiple adenomas.
Ethnic variation and founder pathogenic variants in Lynch syndrome
The frequency of MMR variants does not differ markedly from population to population, with similar frequencies identified in a host of different countries. As with hereditary breast and ovarian cancer (HBOC), there are certain variants that occur at higher frequencies within a particular ethnic group. Notable in HBOC are the commonly recurring Ashkenazi Jewish variants, so common that direct-to-consumer testing is offered for these common variants. (Refer to the
Among the first population findings regarding the MMR genes of Lynch syndrome was the recognition of two very common MLH1 variants in Finland, accounting for most cases of Lynch syndrome in this country.[
In the United States, a deletion in exons 1–6 of the MSH2 gene has been estimated to account for as much as 20% of variants in that gene. This so-called American Founder Mutation has been determined by haplotype analysis to date back about 500 years.[
A South American study combining data from Uruguay, Colombia, Brazil, Argentina, and Chile also selected cases of interest according to Amsterdam and Bethesda features, yielding a 60% frequency of MLH1 and 40% frequency of MSH2. MSH6 and PMS2 were not evaluated. Selection bias likely influenced the frequency of variants and perhaps the relative contributions by MLH1 and MSH2. A possible founder variant in Colombia was noted.[
Although testing for commonly recurring founder variants in a given ethnic/geographic area has been considered to be a cost-effective first step when a stepwise strategy is employed, it is likely not necessary when the increasingly common approach of broad panel testing is undertaken as a basic strategy.
Some ethnic groups have increased rates of consanguinity and a subsequent risk of CMMRD. For more information, see the
Ethnic variation in the United States
In this section, the data exploring the distribution of MMR gene variants amongst differing ethnic groups in the United States are presented. The interpretation of these studies is challenging given the presence of selection and ascertainment bias. In addition, even population-based studies are limited by small sample sizes for many ethnic groups and self-reporting of ethnicity/race.
There are few data suggesting the presence of much variation in Lynch syndrome frequency according to geography or ethnicity. Within a small and/or homogeneous ethnic group the presence of founder variants may seem to increase the prevalence of variants in that particular gene. Slight differences in the proportion of MLH1 and MSH2 variants exist from one population to another. MSH6 and PMS2 have been insufficiently studied at the population level as to enable inferences about their relative frequencies.
The most representative population-based studies in the United States, such as that in Columbus, Ohio, have been overrepresented by White individuals, in accordance with their greater overall numbers. Consequently, data on racial and ethnic minorities such as Hispanic and African American individuals suffer from smaller and less rigorously representative samples.
A study conducted in Puerto Rico considered variants in 89 Caribbean Hispanic patients with Lynch syndrome suspected on the grounds of Amsterdam criteria or Bethesda guidelines.[
Clinic-based series from California, Texas, and Puerto Rico yielded an overall variant prevalence similar to those described, with somewhat more MLH1 than MSH2, but also including MSH6 and PMS2. Presence of potential founder variants traceable back to Spain and Europe were noted.[
The closest population-based information on Lynch syndrome in Hispanic individuals is a Southern California study based on the California Tumor Registry, in which 265 patients were identified.[
The problem of small numbers is highlighted by the findings from the more truly population-based studies that have been done in the United States. In a study from Columbus, Ohio, only 8% of the consecutive series patients were African American and the proportion of Hispanic individuals as a subset of White individuals was not stated.[
Lynch syndrome in African Americans
The issues in evaluating prevalence of Lynch syndrome and cancer risks associated with MMR variants in African American individuals are similar to those in Hispanic individuals: a heterogeneous population that has been understudied. A study of clinic-based data from 13 referral centers in the United States identified 51 families with Lynch syndrome with frequencies of MMR gene variants as follows: 61% MLH1, 21% MSH2, 6% MSH6, and 12% PMS2. Age of cancer onset distribution curves were very similar to those seen in White populations.[
Risk of metachronous CRC
A hallmark feature of Lynch syndrome is that carriers of pathogenic MMR gene variants have an increased risk of development of synchronous and metachronous colorectal neoplasms.[
Risk of extracolonic malignancies associated with Lynch syndrome
Patients with Lynch syndrome are at an increased risk of other cancers, especially those of the endometrium. The cumulative risk of extracolonic cancer has been estimated to be 20% by age 70 years in 1,018 women in 86 families, compared with 3% in the general population.[
Endometrial cancer
The most common extracolonic malignancy in Lynch syndrome is endometrial adenocarcinoma, which affects at least one female member in about 50% of Lynch syndrome families. In addition, 50% of women with an MMR gene pathogenic variant will present with endometrial cancer as her first malignancy.[
The lifetime risk of endometrial cancer has been estimated to be from 44% in carriers of MLH1 pathogenic variants to 71% in carriers of MSH2 pathogenic variants, although some earlier studies may have overestimated risk due to ascertainment bias.[
A study of 127 women with Lynch syndrome who had endometrial cancer as their index cancer were found to be at significantly increased risk of other cancers. The following elevated risks were reported: CRC, 48% (95% CI, 27.2%–58.3%); kidney, renal pelvis, and ureter cancer, 28% (95% CI, 11.9%–48.6%); urinary bladder cancer, 24.3% (95% CI, 8.56%–42.9%; and breast cancer, 2.51% (95% CI, 1.17%–4.14%).[
In a study of 113 families that carried MSH6 pathogenic variants from the Colon Cancer Family Registry, female MSH6 carriers had a 26-fold increased incidence of endometrial cancer (HR, 25.5; 95% CI, 16.8–38.7) compared with the general population. A sixfold increased incidence of other cancers associated with Lynch syndrome (HR, 6.0; 95% CI, 3.4–10.7) was observed compared with the general population, but not among male MSH6 carriers.[
Lynch syndrome–associated endometrial cancer is not limited to the endometrioid subtype, and the spectrum of uterine tumors in Lynch syndrome may include clear cell carcinoma, uterine papillary serous carcinoma, and malignant mixed Müllerian tumors.[
Cancer risk in Lynch syndrome beyond CRC and endometrial cancer
Multiple studies demonstrate an increased risk of additional malignancies associated with Lynch syndrome, including cancers of the stomach, pancreas, ovary, small intestine, and brain, transitional cell carcinoma of the bladder, ureters, and renal pelvis, and sebaceous adenomas of the skin.[
The largest prospective study to date is of 446 unaffected carriers of pathogenic variants from the Colon Cancer Family Registry.[
A well-described variant of Lynch syndrome whose phenotype includes multiple cutaneous neoplasms (including sebaceous adenomas, sebaceous carcinomas, and keratoacanthomas) and CRC is Muir-Torre syndrome.[
| Cancer Siteb | General Population Risk (%)c | Risk in Individuals With Lynch Syndrome (%)d | References |
|---|---|---|---|
| CNS = central nervous system. | |||
| a Adapted from Syngal et al.[ |
|||
| b Evolving data suggest a potential association between Lynch syndrome and breast and prostate cancers. (Refer to the |
|||
| c Howlader et al.[ |
|||
| d Range of cancer risk estimates vary based on study sample size, subject ascertainment, and statistical methods. | |||
| Stomach | <1 | 0.2–13 | [ |
| Ovary | 1.3 | 3.4–22 | [ |
| Hepatobiliary tract | <1 | 0.02–4 | [ |
| Urinary tract | <1 | 0.2–25.5 | [ |
| Small bowel | <1 | 0.4–12 | [ |
| Brain/CNS | <1 | 1.2–3.7 | [ |
| Sebaceous neoplasms | <1 | 9.0 | [ |
| Pancreas | 1.6 | 0.4–3.7 | [ |
Additional cancers potentially associated with Lynch syndrome
Additional tumors are being considered as part of the spectrum of Lynch syndrome, but this is controversial. Breast and prostate cancers have been raised as possible Lynch syndrome–associated tumors such that MMR genes are now included on multigene (panel) tests for these cancers.
Breast cancer
The issue of breast cancer risk in Lynch syndrome has been controversial.
Retrospective studies have been inconsistent, but several have demonstrated microsatellite instability in a proportion of breast cancers from individuals with Lynch syndrome;[
A number of subsequent studies have suggested the presence of higher breast cancer risks than previously published,[
Prostate cancer
Prostate cancer was found to be associated with Lynch syndrome in a study of 198 families from two U.S. Lynch syndrome registries in which prostate cancer had not originally been part of the family selection criteria. Prostate cancer risk in relatives of carriers of MMR gene pathogenic variants was 6.3% at age 60 years and 30% at age 80 years, versus a population risk of 2.6% at age 60 years and 18% at age 80 years, with an overall HR of 1.99 (95% CI, 1.31–3.03).[
Adrenocortical cancer
In a series of 114 ACC cases, of which 94 patients had a detailed family history assessment and Li-Fraumeni syndrome was excluded, three patients had family histories that were suggestive of Lynch syndrome. The prevalence of MMR gene pathogenic variants in 94 families was 3.2%, similar to the proportion of Lynch syndrome among unselected colorectal and endometrial cancer patients. In a retrospective review of 135 MMR gene pathogenic variant–positive Lynch syndrome families from the same program, two probands were found to have had a history of ACC. Of the four ACCs in which MSI testing could be performed, all were MSS. These data suggest that if Lynch syndrome is otherwise suspected in an ACC index case, an initial evaluation of the ACC using MSI or IHC testing may be misleading.[
Other cancers
Several additional cancers have been found to be associated with Lynch syndrome in some studies, but further investigation is warranted.
Management of Lynch syndrome
Screening and surveillance in Lynch syndrome
Colon cancer screening and surveillance in Lynch syndrome
Several aspects of the biologic behavior of CRC and its precursor lesion, the adenomatous polyp, in individuals with Lynch syndrome support a different approach to CRC screening in this population as compared with those recommendations for average-risk people in the general population. At present, the recommendations for cancer screening and surveillance in Lynch syndrome consider the differences in cancer risks as compared with those in the general population due to the causative germline deficiency in the MMR system. The following biological differences form the basis of the currently implemented screening strategies in Lynch syndrome:
- CRC and adenomas present at a younger age.
CRCs in Lynch syndrome occur earlier in life than do sporadic cancers; however, the age of onset varies based on which of the MMR genes is altered. (Refer to the
Prevalence, clinical manifestations, and cancer risks associated with Lynch syndrome section of this summary for more information about gene-specific age of onset of CRC.)Carriers of Lynch syndrome pathogenic variants have an increased risk of developing colon adenomas and the onset of adenomas appears to occur at a younger age than in pathogenic variant–negative individuals from the same families.[
464 ] The risk of a carrier of MMR pathogenic variants developing adenomas has been reported to be 3.6 times higher than the risk in noncarriers.[464 ] By age 60 years, 70% of the carriers developed adenomas, compared with 20% of noncarriers. Most of the adenomas in carriers had absence of MMR protein expression and were more likely to have dysplastic features, compared with adenomas from control subjects.[464 ]In one study, the mean age at diagnosis of adenoma in carriers was 43.3 years (range, 23–63.2 y), and the mean age at diagnosis of carcinoma was 45.8 years (range, 25.2–57.6 y).[
464 ] - There is a right-sided predominance of colon cancer.
A larger proportion of Lynch syndrome CRCs (60%–70%) occur in the right colon, suggesting that sigmoidoscopy alone is not an appropriate screening strategy and that a colonoscopy provides a more complete structural examination of the colon. Evidence-based reviews of surveillance colonoscopy in Lynch syndrome have been reported.[
125 ,465 ,466 ] The incidence of CRC throughout life is substantially higher in patients with Lynch syndrome, suggesting that the most-sensitive test available should be used. (Refer toTable 13 for available colon surveillance recommendations.) - The adenoma-carcinoma sequence is accelerated.
The progression from normal mucosa to adenoma to cancer is accelerated,[
467 ,468 ] suggesting that screening should be performed at shorter intervals (every 1–2 years) and with colonoscopy.[468 ,469 ,470 ,471 ] It has been demonstrated that carriers of MMR gene pathogenic variants develop detectable adenomas at an earlier age than do noncarriers.[464 ,464 ] It is not known whether this reflects a greater prevalence of adenomas or the presence of larger adenomas with better detection in Lynch syndrome.
Evidence for the use of colonoscopy for CRC screening and surveillance in Lynch syndrome
The risk of CRC in Lynch syndrome has been studied and updated in a Finnish screening trial, which spans from the early 1980s to present.[
A 15-year controlled screening trial conducted in this series demonstrated a reduction in the incidence of CRC, CRC-specific mortality, and overall mortality with colonoscopy in individuals from Lynch syndrome families.[
The series subsequently limited its attention to subjects without prior diagnosis of adenoma or cancer. The eligible 420 carriers of pathogenic variants had a mean age of 36 years and underwent an average of 2.1 colonoscopies, with a median follow-up of 6.7 years. Adenomas were detected in 28% of subjects. Cumulative risk of one or more adenomas by age 60 years was 68.5% in men and 48.3% in women. Notably, risk of detecting cancer in those free of cancer at baseline exam, and thus regarded as interval cancers, by age 60 years was 34.6% in men and 22.1% in women. The combined cumulative risk of adenoma or cancer by age 60 years was 81.8% in men and 62.9% in women. For both adenomas and carcinomas, about one-half were located proximal to the splenic flexure. While the rates for CRC despite colonoscopy surveillance appear high, the recommended short intervals were not regularly adhered to in this nonrandomized series. These authors recommended surveillance at 2-year intervals. This is in line with most consensus guidelines (refer to
Individuals with Lynch syndrome are at an increased risk of developing synchronous CRC. Of 5,304 CRC cases in the Danish HNPCC Register, including 774 with Lynch syndrome, the relative risks of synchronous CRC (>1 CRC) diagnosed within 1 year of primary CRC for Lynch syndrome, familial CRC (cases meeting Amsterdam I or II criteria) and metachronous CRC (1 CRC at age <50 y or >2 CRCs at age >50 y) were 5.6, 3.2, and 1.9, respectively, compared with sporadic CRC. Thus, the increased risk of synchronous CRC in patients with a strong family history of CRC, and especially Lynch syndrome, should be considered in preoperative colonoscopic examinations.[
Given that colonoscopy is the accepted measure for colon cancer surveillance, preliminary data suggest that the use of chromoendoscopy, such as with indigo carmine, may increase the detection of diminutive, histologically advanced adenomas.[
When an adenoma is detected, the question of whether to test the adenoma for MSI/IHC is raised. One study of patients with prior CRC and known MMR pathogenic variants found eight of 12 adenomas to have both MSI and IHC protein loss.[
Level of evidence (colon surveillance): 2ai
Special considerations: The impact of gene-specific variability in cancer risk on CRC screening recommendations in Lynch syndrome
Because of the variability of gene-specific CRC risks, experts in the field have proposed gene-specific screening and surveillance recommendations. For example, a European consortium [
Available recommendations for colon surveillance in individuals with Lynch syndrome are summarized in
| Organization | MLH1 | MSH2 | MSH6 | PMS2 | EPCAM |
|---|---|---|---|---|---|
| CRC = colorectal cancer; EHTG = European Hereditary Tumor Group; ESCP = European Society of Coloproctology; ESMO = European Society for Medical Oncology; MMR = mismatch repair; NCCN = National Comprehensive Cancer Network. | |||||
| a This table summarizes available guidelines from 2014 and later. Other organizations, including the American Cancer Society, have published guidelines before 2014.[ |
|||||
| b U.S. Multi-Society Task Force on Colorectal Cancer includes the following organizations: American Academy of Family Practice, American College of Gastroenterology, American College of Physicians-American Society of Internal Medicine, American College of Radiology, American Gastroenterological Association, American Society of Colorectal Surgeons, and American Society for Gastrointestinal Endoscopy. | |||||
| c Consider later age forMSH6carriers.[ |
|||||
| d Consider repeating colonoscopy every 5 years forPMS2carriers.[ |
|||||
| e Consider starting at age 30 forMSH6carriers and 35 forPMS2carriers. Consider annual colonoscopy for MMR carriers.[ |
|||||
| NCCN (2024)[ |
High-quality colonoscopy at age 20–25 y or 2–5 y prior to earliest CRC in the family if it was diagnosed before age 25 y; repeat colonoscopy every 1–2 y | High-quality colonoscopy at age 20–25 y or 2–5 y prior to earliest CRC in the family if it was diagnosed before age 25 y; repeat colonoscopy every 1–2 y | High-quality colonoscopy at age 30–35 y or 2–5 y prior to earliest CRC in the family if it was diagnosed before age 30 y; repeat colonoscopy every 1–3 y | High-quality colonoscopy at age 30–35 y or 2–5 y prior to earliest CRC in the family if it was diagnosed before age 30 y; repeat colonoscopy every 1–3 y | NCCN recommends thatEPCAMcarriers be managed the same asMSH2carriers. High-quality colonoscopy at age 20–25 y or 2–5 y prior to the earliest CRC in the family if it was diagnosed before age 25 y; repeat colonoscopy every 1–2 y |
| ESMO (2020)[ |
Colonoscopy at age 25 y or 5 y prior to earliest CRC if diagnosed before age 25 y; repeat every 1–2 y | Colonoscopy at age 25 y or 5 y prior to earliest CRC if diagnosed before age 25 y; repeat every 1–2 y | Colonoscopy at age 35 y or 5 y prior to earliest CRC if diagnosed before age 25 yc; repeat every 1–2 y | Colonoscopy at age 35 y or 5 y prior to earliest CRC if diagnosed before age 25 y; repeat every 1–2 y | Not addressed |
| British Society of Gastroenterology (BSG)/ Association of Coloproctology of Great Britain and Ireland (ACPGBI)/ United Kingdom Cancer Genetics Group (UKCGG) (2020)[ |
Colonoscopy at age 25 y; repeat every 2 y until age 75 y | Colonoscopy at age 25 y; repeat every 2 y until age 75 y | Colonoscopy at age 35 y; repeat every 2 y until age 75 y | Colonoscopy at age 35 y; repeat every 2 y until age 75 y | EPCAMcarriers should be managed as those withMSH2pathogenic variants |
| European guidelines from the EHTG and ESCP; updated Mallorca group guidelines (2021)[ |
Colonoscopy at age 25 y; repeat every 2–3 y | Colonoscopy at age 25 y; repeat every 2–3 y | Colonoscopy at age 35 y; repeat every 2–3 y | Colonoscopy at age 35 y; repeat every 2–3 yd | Not addressed |
| U.S. Multi-Society Task Force on Colorectal Cancer (2014)b[ |
Colonoscopy beginning at age 20–25 y for 2–5 y prior to earliest CRC if before age 25 y; repeat every 1–2 ye | ||||
Extracolonic cancer screening in Lynch syndrome
Endometrial cancer screening in Lynch syndrome
Note: A separate PDQ summary on
Cancer of the endometrium is the most common extracolonic cancer observed in Lynch syndrome families, affecting at least one female in about 50% of Lynch syndrome families. (Refer to the
In the general population, the diagnosis of endometrial cancer is generally made when women present with symptoms like abnormal or postmenopausal bleeding. Endometrial sampling is performed to provide a histological specimen for diagnosis. Eighty percent of women with endometrial cancer present with stage I disease, and there are no data to suggest that the clinical presentation in women with Lynch syndrome differs from that in the general population.
Given their substantial increased risk of endometrial cancer, endometrial cancer screening has been suggested for women with Lynch syndrome who have not had risk-reducing hysterectomies. Proposed screening methods include transvaginal ultrasound (TVUS) (not recommended in premenopausal patients) and/or endometrial biopsy. However, current NCCN guidelines suggest that these screening methods may not benefit women with Lynch syndrome. Screening via endometrial biopsy can be considered in patients with Lynch syndrome, due to its high levels of sensitivity and specificity. Screening may begin at age 30 to 35 years and can be repeated every 1 to 2 years. TVUS, on the other hand, is not sensitive or specific at detecting endometrial cancer. However, this screening method can be considered in women with Lynch syndrome based on a provider's judgment.[
Two studies have examined the use of TVUS in endometrial screening for women with Lynch syndrome.[
A study of 175 women with Lynch syndrome, which included both endometrial sampling and TVUS, showed that endometrial sampling improved sensitivity when compared with TVUS. Endometrial sampling found 11 of the 14 cases of endometrial cancer. Two of these cases were interval cancers that developed in symptomatic women, and one case was an occult endometrial cancer found at the time of hysterectomy. Endometrial sampling also identified 14 additional cases of endometrial hyperplasia. Among the group of 14 women with endometrial cancer, ten also had TVUS screening with endometrial sampling. Four of the ten women had abnormal TVUS, while six women had normal TVUS.[
Some studies suggest that women with a clinical or genetic diagnosis of Lynch syndrome do not participate in intensive gynecologic screening.[
Level of evidence: 5
Ovarian cancer screening in Lynch syndrome
Estimates of the cumulative lifetime risk of ovarian cancer in Lynch syndrome patients range from 3.4% to 22%.[
Level of evidence: None assigned
Risk-reducing surgeries for the prevention of gynecologic cancers in Lynch syndrome
Risk-reducing surgery is an effective strategy for preventing endometrial and ovarian cancers in Lynch syndrome families. A retrospective study of 315 women with pathogenic MMR gene variants compared the rate of endometrial and ovarian cancers among women who did and did not have hysterectomies and oophorectomies. The mean follow-up periods for endometrial cancer were 13.3 years in the surgical group and 7.4 years in the nonsurgical group. The mean follow-up periods for ovarian cancer were 11.2 years in the surgical group and 10.6 years in the nonsurgical group. In the surgical group, no cancers were diagnosed. In contrast, 33% of women were diagnosed with endometrial cancer, and 5.5% of women were diagnosed with ovarian cancer in the nonsurgical group.[
Level of evidence: 3aii
Additional extracolonic cancer screening in Lynch syndrome
The decision to screen for other Lynch syndrome–associated cancers is done on an individual basis and relies on the cancers reported among FDRs and SDRs with Lynch syndrome.
Gastric and small bowel cancers
The lifetime risk of gastric cancer is approximately 8% for male Lynch syndrome carriers and 5% for female Lynch syndrome carriers.[
There are variable reports on the lifetime risk of small bowel cancer associated with Lynch syndrome, ranging from less than 1% to 12%.[
In a single-center retrospective study, researchers analyzed the prevalence of clinically actionable endoscopic findings in 323 asymptomatic Lynch syndrome carriers undergoing EGD for upper GI cancer surveillance.[
Esophageal findings in this study included Barrett's esophagus, which was detected in 6.5% of participants. This percentage is similar to the prevalence of Barrett's esophagus in North American individuals with gastroesophageal reflux disease.[
In addition, 261 subjects underwent gastric biopsy sampling, and the following findings were observed:
- Ten patients had H. pylori (3.8%).
- Fifteen patients had gastric intestinal metaplasia (5.7%).
- Five patients had duodenal adenomas (1.5%) at a mean age of 55.8 years (range, 27–75 y). One patient had an adenoma with high-grade dysplasia, and one patient had an adenoma with tubulovillous histology. Four adenomas were found in the third part of the duodenum, and one adenoma was found in the second part of the duodenum.
- Two patients had gastric adenomas (0.6%). One of these patients had an adenoma with high-grade dysplasia. Patients had gastric adenomas at a mean age of 71.6 years (range, 70–73 y).
EGD surveillance is strongly recommended in patients with Lynch syndrome, given the high prevalence and high incidence of clinically actionable findings on EGD and the favorable benefit of identifying these findings at an early stage.
Level of evidence: 5
Urinary tract cancer
Urinary tract malignancies include those of the transitional cell type of the renal pelvis and ureters, and the bladder. The associated lifetime risk of these malignancies is variable, ranging from less than 1% to as high as 25%, with higher estimates related to pooling the cancers found in different locations within the urinary tract and including the bladder.[
Level of evidence: 5
Pancreatic cancer
An elevated risk of pancreatic cancer among Lynch syndrome carriers has been supported by two cohort studies that adjust for ascertainment bias. One study reported a cumulative risk of pancreatic cancer of 3.7% by age 70 years and an 8.6-fold increase compared with the general population. [
Pancreatic cancer screening may be considered in individuals with MLH1, MSH2, or MSH6 pathogenic variants if they have one or more FDRs or SDRs with exocrine pancreatic cancer (if these family members are on the same side of the family as the individual with Lynch syndrome). Pancreatic cancer screening can begin at age 50 years or 10 years before the youngest pancreatic cancer diagnosis in the family. Screening typically consists of annual contrast-enhanced magnetic resonance imaging/magnetic resonance cholangiopancreatography (MRCP) and/or endoscopic ultrasound (EUS). However, screening can be done more frequently if abnormal findings are found on MRCP/EUS. NCCN recommends that MRCP/EUS occur at a high-volume center that has experience screening individuals with Lynch syndrome. Health care providers are encouraged to have a discussion with patients about pancreatic screening limitations, including the following: the cost of annual pancreatic cancer screening, the high occurrence of benign and indeterminate pancreatic lesions, and the uncertainty regarding the effectiveness of pancreatic cancer screening.[
Level of evidence: 5
Chemoprevention in Lynch syndrome
The Colorectal Adenoma/Carcinoma Prevention Programme (CAPP2) was a double-blind, placebo-controlled, randomized trial to determine the role of aspirin in preventing CRC in patients with Lynch syndrome who were in surveillance programs at a number of international centers.[
In 2020, long-term follow-up data with all participants having surpassed 10 years of follow-up demonstrated a significant reduction in CRC incidence for participants randomly assigned to receive aspirin both by per-protocol analysis (HR, 0.56; 95% CI, 0.34–0.91) and intention-to-treat analysis (HR, 0.65; 95% CI, 0.43–0.97).[
To date, there has been no significant preventive benefit identified in CAPP2 participants randomly assigned to receive resistant starch versus starch-placebo (HR for incident CRC, 0.92; 95% CI, 0.62–1.34; P = .63).[
Experts have speculated that certain Lynch syndrome carriers with lower risks of future incident CRCs (e.g., those with germline PMS2 pathogenic variants, those with prior colectomy, or older individuals) may be less likely to derive benefit from aspirin chemoprevention and may be appropriate for lower dosing.[
The CAPP3 trial, which is evaluating the effect of lower doses of aspirin (blinded 100 mg, 300 mg, and 600 mg enteric-coated aspirin) completed accrual of 1,882 Lynch syndrome carriers in 2019 and data are not expected until at least 5 years of follow-up is complete.[
Because of the level 1 evidence in support of aspirin chemoprevention, clinical practice guidelines consistently recommend that individuals with Lynch syndrome consider taking aspirin daily. Optimal aspirin dosage can be determined by the patient's provider, after having a discussion with the patient about his/her personal risk factors (including pregnancy, in which aspirin use may be contraindicated). NCCN also recommends that providers explain potential advantages and disadvantages of aspirin use to the patient.[
For Lynch syndrome carriers unable to take aspirin, it is unclear whether NSAIDs may have a comparable chemopreventive benefit. A 2015 survey of 1,858 participants in the Colon Cancer Family Registry suggested that aspirin and ibuprofen might both reduce incident CRC in Lynch syndrome carriers.[
Level of evidence: 1aii
Management of Lynch syndrome-associated CRC
Surgical management of CRC in Lynch syndrome
One of the hallmark features of Lynch syndrome is the presence of synchronous and metachronous CRCs. The incidence of metachronous CRCs has been reported to be 16% at 10 years, 41% at 20 years, and 63% at 30 years after segmental colectomy.[
Two studies have shown that patients who undergo extended procedures have fewer metachronous CRCs and additional surgical procedures related to CRC than do patients who undergo segmental resections.[
A retrospective study from the Creighton University Hereditary Cancer Center evaluated the incidence of metachronous CRC and survival in 64 Lynch syndrome pathogenic variant carriers with right-sided colon cancer undergoing either proximal colectomy or total or subtotal colectomy.[
When considering surgical options, it is important to recognize that a subtotal or total colectomy will not eliminate the rectal cancer risk. The lifetime risk of developing cancer in the rectal remnant after an abdominal colectomy has been reported to be 12% at 12 years post-colectomy.[
In patients with Lynch syndrome and rectal cancer, similar surgical options (extended vs. segmental resection) and considerations must be given. Extended procedures include restorative proctocolectomy and IPAA if the sphincter can be saved, or proctocolectomy with loop ileostomy if the sphincter cannot be saved. The risk of metachronous colon cancer after segmental resection for an index rectal cancer has been reported to be between 15% and 27%.[
There are no data about fertility after surgery in Lynch syndrome patients. In female FAP patients, no difference in fecundity after abdominal colectomy and IRA has been reported, whereas there is a 54% decrease in fecundity in patients who undergo restorative proctocolectomy with IPAA compared with the general population.[
In a large Danish registry study, the incidence rate for metachronous CRC was fivefold higher in Lynch syndrome patients, but not significantly higher in familial CRC and moderate familial risk CRC cases when compared with sporadic CRC, demonstrating that the risk of metachronous CRC occurred almost exclusively in Lynch syndrome cases.[
Most clinicians who treat patients with Lynch syndrome will favor an extended procedure at the time of CRC diagnosis. However, as stated above, the choice of surgery must be made on an individual basis by the surgeon and the patient.[
Level of Evidence: 4
Prognostic and therapeutic implications of MSI
As discussed in previous sections, MSI is not only a molecular feature of Lynch syndrome but is also present in 10% to 15% of sporadic cases of CRC (largely due to MLH1 hypermethylation or biallelic somatic variants in an MMR gene). Although MSI testing was initially used to screen patients who might harbor pathogenic MMR gene variants, it has been increasingly recognized that MSI has important prognostic and therapeutic implications. The utility of MSI testing beyond identifying Lynch syndrome has made the case for universal MSI screening more compelling and has contributed to its widespread adoption. Several studies have suggested that stage-specific survival is better for MSI-H CRC compared with MSS cancers. Additionally, the chemotherapeutic agent fluorouracil (5-FU) appears ineffective in the adjuvant treatment of resected MSI-H CRC, in contrast to MSS CRC in which this agent is widely utilized for this purpose. Finally, immunomodulation with agents such as checkpoint inhibitors appears effective in the treatment of advanced MSI-H CRC based on early phase 1 and phase 2 studies, while these agents, at least when utilized as monotherapy, show little activity in MSS CRC.
Prognosis of MSI
Although MSI-H tumors account for 15% of all sporadic CRC, they appear to be more frequent in stage II compared with stage III CRC,[
Several studies subsequently confirmed the improved survival of stage II MSI-H CRC compared with MSS cases. A meta-analysis of 32 studies of 7,642 cases, including 1,277 with MSI-H, showed a combined HR estimate for overall survival (OS) associated with MSI of 0.65 (95% CI, 0.59–0.71; heterogeneity P = .16; I2 [a measure of the percentage of variation across studies that is due to heterogeneity rather than chance] = 20%).[
Consistent with other prior data, clinicopathologic analysis of 85 Lynch syndrome–associated CRCs and 67 sporadic dMMR CRCs demonstrated a significantly superior survival among patients with Lynch syndrome, as well as younger ages at diagnosis and higher numbers of tumor-infiltrating lymphocytes (TILs).[
Given the predilection for MSI-H tumors to involve the right side of the colon, there is a paucity of data on the outcome and prognosis of MSI-H tumors involving the rectum. One study suggested only 2% of rectal cancers are MSI-H.[
The use of adjuvant chemotherapy after surgery for CRC in Lynch syndrome
The finding of MSI in a CRC has been shown in several studies to predict the lack of benefit of adjuvant chemotherapy with 5-FU in resected stage II or stage III colon cancer.[
In 2003, however, the outcomes in a randomized controlled prospective trial of adjuvant chemotherapy in 570 colon cancer patients demonstrated no benefit from adjuvant 5-FU in the group with MSI. Moreover, there were nonsignificant trends towards increased mortality when colon cancers with MSI were treated: twofold for stage III cancers and threefold for stage II cancers.[
Preclinical data suggests the addition of oxaliplatin to 5-FU can overcome the resistance to 5-FU monotherapy seen in MSI-H tumors.[
Level of evidence (against the use of adjuvant therapy): 1ai
Immunotherapy
Tumors that develop via the MSI pathway have more somatic variants than tumors that develop via other pathways. This could imply that dMMR tumors may have more potential antigens (termed neoantigens) and may be more responsive to immune system manipulation than proficient MMR (pMMR) tumors. Microscopically, MSI-H tumors often exhibit abundant tumor-infiltrating lymphocytes, sometimes resulting in a Crohn-like reaction. This histological feature has long suggested the possibility of increased tumor immune surveillance in MSI-H cancers and is one of the main hypotheses for the better stage-specific survival seen in MSI-H compared with MSS cancers.
To test the hypothesis of efficacy of immunomodulation in MSI-H tumors, a phase 2 trial of programmed cell death-1 (PD-1) inhibition was carried out in a small cohort of patients with MSI-H or MSS cancers. Patients with metastatic disease that had failed various chemotherapy regimens were treated with pembrolizumab, an anti–PD-1 immune checkpoint inhibitor.[
A single-arm phase 2 study (CheckMate 142) of another PD-1 inhibitor, nivolumab, was performed in 74 patients with MSI-H/dMMR CRC that had progressed on prior cytotoxic chemotherapy (including 5-FU, irinotecan, and oxaliplatin).[
Based on these data, pembrolizumab 200 mg given intravenously every 3 weeks was approved by the FDA in May 2017 for the treatment of any MSI-H/dMMR metastatic cancer that is refractory to standard therapy and nivolumab 240 mg given intravenously every 2 weeks was granted accelerated approval by the FDA in August 2017 for the treatment of MSI-H/dMMR CRC that is refractory to cytotoxic chemotherapy.
In 2020, treatment-naïve patients with MSI-H/dMMR CRC were enrolled in a phase III trial (KEYNOTE-177) where they were randomized to receive pembrolizumab or chemotherapy. Patients who received pembrolizumab had an increase in PFS when compared with patients who received chemotherapy.[
In another arm of CheckMate 142, 119 individuals with metastatic dMMR CRC were treated with nivolumab plus ipilimumab.[
A retrospective analysis described the pathological responses of 14 patients with MSI-H tumors after treatment with PD-1 inhibitors (with or without CTLA-4 inhibitors). Eight of the patients in this study had Lynch syndrome and all of the patients had unresectable/metastatic CRCs. Patients underwent resection after they completed treatment. The study demonstrated a pathological complete response (PCR) in 13 of the 14 patients, despite radiographic evidence of persistent disease in 12 of these patients. The discordance between imaging and PCR may be related to significant lymphocyte infiltration in patient tumors. The median duration of treatment was 12 months. However, a PCR was demonstrated in a patient who was treated for only 3 months.[
There is debate about when immunotherapeutic agents can be used in patients with non-CRC, MSI-H cancers. Many providers question in which line of therapy immunotherapeutics should be initiated. This question is the subject of multiple ongoing clinical trials. (Use our
Level of evidence: 3b
Vaccines in the treatment or prevention of MSI-related CRC
An alternative approach to immunotherapy in MSI-H CRC involves the use of tumor-directed vaccines. The most promising approaches thus far involve the use of tumor-related neoantigens as epitopes to increase tumor-specific T-cell immunity. Studies are currently under way in the adjuvant treatment of resected stage III CRC (NCT01461148), in patients with metastatic disease (NCT01885702), and in the prevention of CRC in patients with Lynch syndrome (NCT01885702).
Lynch syndrome–related syndromes
Lynch-like or HNPCC-like syndrome
Somatic biallelic MMR gene inactivation is now recognized as a common cause of sporadic MMR deficiency. The literature variably labels these cases as Lynch-like syndrome (LLS). However, this nomenclature can be confusing, since this is a sporadic form of MMR-deficiency, rather than a stand-alone genetic syndrome. For more information, see the
Hamartomatous Polyposis Syndromes
Hamartomatous polyposis syndromes are a distinct and rare group of polyposis diseases characterized by the presence of hamartomatous growths throughout the GI tract. Each syndrome manifests differently, depending on histological type, the number of GI polyps, the number of benign growths, GI cancer risks, and extra-GI cancer risks. Hamartomatous polyposis syndromes include the following:
-
PTEN hamartoma tumor syndromes (PHTS) (including Cowden syndrome) . -
Peutz-Jeghers syndrome (PJS). -
Juvenile polyposis syndrome (JPS).
PTENhamartoma tumor syndromes (including Cowden syndrome)
Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome (BRRS) are part of a spectrum of conditions known collectively as PTEN hamartoma tumor syndromes (PHTS). Approximately 85% of patients diagnosed with Cowden syndrome, and approximately 60% of patients with BRRS have an identifiable PTEN pathogenic variant.[
PTEN functions as a dual-specificity phosphatase that removes phosphate groups from tyrosine, serine, and threonine. PTEN pathogenic variants are diverse and can present as nonsense, missense, frameshift, or splice-site variants. Approximately 40% of variants are found in exon 5, which encodes the phosphatase core motif; several recurrent pathogenic variants have been observed at this location.[
Operational criteria for the diagnosis of Cowden syndrome have been published and subsequently updated.[
Over a 10-year period, the International Cowden Consortium (ICC) prospectively recruited a consecutive series of adult and pediatric patients meeting relaxed ICC criteria for PTEN testing in the United States, Europe, and Asia.[
The age-adjusted risk of CRC was increased in individuals with PTEN pathogenic variants in both studies (SIR, 5.7–10.3).[
| Cancer | Age-Adjusted SIR (95% CI) | Age-Related Penetrance Estimates |
|---|---|---|
| CI = confidence interval; SIR = standardized incidence ratio. | ||
| a Adapted from Tan et al.[ |
||
| b Other historical studies have suggested a lower lifetime risk of breast cancer, in the range of 25%–50%.[ |
||
| Breast | 25.4 (19.8–32.0) | 85% starting around age 30 yb |
| Colorectal | 10.3 (5.6–17.4) | 9% starting around age 40 y |
| Endometrial | 42.9 (28.1–62.8) | 28% starting around age 25 y |
| Kidney | 30.6 (17.8–49.4) | 34% starting around age 40 y |
| Melanoma | 8.5 (4.1–15.6) | 6% with risk beginning in infancy |
| Thyroid | 51.1 (38.1–67.1) | 35% with risk beginning in infancy |
Peutz-Jeghers syndrome
PJS is an early-onset autosomal dominant disorder characterized by melanocytic macules on the lips, the perioral region, and buccal region; and multiple GI polyps, both hamartomatous and adenomatous.[
Females with PJS are also predisposed to the development of cervical adenoma malignum, a rare and very aggressive adenocarcinoma of the cervix.[
Although the risk of malignancy appears to be exceedingly high in individuals with PJS based on the published literature, the possibility that selection and referral biases have resulted in overestimates of these risks should be considered.
| Site | Age (y) | Cumulative Risk (%)b | Reference(s) |
|---|---|---|---|
| GI = gastrointestinal. | |||
| a Reprinted with permission from Macmillan Publishers Ltd: Gastroenterology[ |
|||
| b All cumulative risks were increased compared with the general population (P< .05), with the exception of cervix and testes. | |||
| c GI cancers include colorectal, small intestinal, gastric, esophageal, and pancreatic. | |||
| d Westerman et al.: GI cancer does not include pancreatic cancer.[ |
|||
| e Did not include adenoma malignum of the cervix or Sertoli cell tumors of the testes. | |||
| Any cancer | 60–70 | 37–93 | [ |
| Any GI cancerc,d | 60–70 | 38–66 | [ |
| Gynecological cancer | 60–70 | 13–18 | [ |
| Per origin | |||
| Stomach | 65 | 29 | [ |
| Small bowel | 65 | 13 | [ |
| Colorectum | 65 | 39 | [ |
| Pancreas | 65–70 | 11–36 | [ |
| Lung | 65–70 | 7–17 | [ |
| Breast | 60–70 | 32–54 | [ |
| Uterus | 65 | 9 | [ |
| Ovary | 65 | 21 | [ |
| Cervixe | 65 | 10 | [ |
| Testese | 65 | 9 | [ |
PJS is caused by pathogenic variants in the STK11 (also called LKB1) tumor suppressor gene located on chromosome 19p13.[
Germline variants of the STK11 gene represent a spectrum of nonsense, frameshift, and missense variants, and splice-site variants and large deletions.[
Approximately 85% of variants are localized to regions of the kinase domain of the expressed protein. No strong genotype-phenotype correlations have been identified.[
STK11 has been unequivocally demonstrated to cause PJS. Although earlier estimates using direct DNA sequencing showed a 50% pathogenic variant detection rate in STK11, studies adding techniques to detect large deletions have found pathogenic variants in up to 94% of individuals meeting clinical criteria for PJS.[
Clinical management
NCCN and the U.S. Multi-Society Task Force (USMSTF) on Colorectal Cancer recommend upper endoscopy and high-quality colonoscopy with polypectomy beginning between the ages of 8 to 10 years.[
Management of small bowel hamartomas is important because patients with PJS have risks of bleeding, intussusception, and malignancy. In PJS, cumulative lifetime risk of small bowel cancer is approximately 13%. NCCN guidelines recommend computed tomography enterography (CTE), magnetic resonance enterography (MRE), or video capsule endoscopy (VCE) beginning between the ages of 8 to 10 years for small bowel surveillance in PJS.[
Juvenile polyposis syndrome
JPS is a genetically heterogeneous, rare, childhood- to early adult–onset, autosomal dominant disease that presents characteristically as hamartomatous polyposis throughout the GI tract, although colorectal polyps predominate.[
JPS can be diagnosed clinically (based on fulfillment of specific clinical criteria) or genetically (based on the presence of germline pathogenic variants in SMAD4 or BMPR1A).
A clinical diagnosis of JPS is met by individuals fulfilling one or more of the following criteria:[
- More than five juvenile polyps of the colon or rectum.
- Juvenile polyps in other parts of the GI tract.
- Any number of juvenile polyps and a positive family history of JPS.
Approximately 15% to 60% of JPS cases have germline pathogenic variants in the SMAD4 gene, and 25% to 40% of cases have germline pathogenic variants in the BMPR1A gene.[
Individuals with germline SMAD4 pathogenic variants may have a greater risk to develop severe gastric polyposis [
Individuals with JPS and germline pathogenic variants in SMAD4/BMPR1A have a 38% to 68% lifetime risk of developing CRC by age 60 years, with an average diagnosis occurring between the ages of 34 and 44 years.[
Roughly 22% of JPS patients with germline SMAD4 pathogenic variants also have manifestations of HHT, such as arteriovenous malformations, mucocutaneous telangiectasias, digital clubbing, osteoarthropathy, hepatic arteriovenous malformations, and cerebellar cavernous hemangioma, suggesting that the two syndromes overlap.[
NCCN and the USMSTF on Colorectal Cancer recommend that patients with JPS undergo both high-quality colonoscopy with polypectomy and upper endoscopy with polypectomy beginning between ages 12 and 15 years.[
A severe form of JPS, in which polyposis develops in the first few years of life, is referred to as JPS of infancy. JPS of infancy is often caused by microdeletions of chromosome 10q22-23, a region that includes BMPR1A and PTEN. For more information about PTEN, see the
Oligopolyposis
Oligopolyposis (derived from the Greek prefix, oligo, meaning few) describes polyp counts that are greater than anticipated in average-risk patients but are fewer than 100 polyps. Here, oligopolyposis is used to describe situations in which an individual's polyp count is large enough to suspect a hereditary polyposis syndrome (generally between 10–100 polyps), regardless of the presence or lack of a family history of colorectal polyps/CRC. Since oligopolyposis specifically refers to polyp count, multiple syndromes can present with an oligopolyposis phenotype. It is recommended that most individuals with oligopolyposis have germline genetic testing.[
Multiple syndromes can present with oligopolyposis. These syndromes vary based on polyp histology and the pathogenic variant that is identified. FAP is the most well-described adenomatous polyposis syndrome that occasionally presents with fewer than 100 tubular adenomas. Since the APC gene was discovered in 1992 (pathogenic variants in APC cause FAP), other rare genes have also been implicated in oligopolyposis and cancer. These genes include
In a cross-sectional study of 3,789 patients with 10 or more colorectal polyps, patients underwent multigene panel testing for the following genes: APC, BMPR1A, CDH1, CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, PMS2, PTEN, SMAD4, STK11, and TP53. In this study, pathogenic variants were found in 5% of participants, regardless of the participant's age or polyp cohort.[
NTHL1, POLE, POLD1, and GREM1 pathogenic variant testing is being incorporated into the multigene panel tests for CRC susceptibility (offered commercially with APC and MUTYH testing so a polyposis panel can be ordered up front for patients with oligopolyposis). There are minimal data on the optimal surveillance approach for individuals with germline pathogenic variants in NTHL1 (biallelic carriers only), POLE, or POLD1. It is presumed that CRC risk for these pathogenic variants is comparable to that seen in Lynch syndrome. Hence, some guidelines are endorsing early and frequent colonoscopic screening, similar to that which is conducted in Lynch syndrome patients.
Lastly, oligopolyposis with varying polyp histologies (e.g., adenomas, serrated, inflammatory, and hamartomatous polyps) has been reported in two small case series of individuals previously treated with chemotherapy and radiation therapy for a prior childhood malignancy.[
Polymerase Proofreading–Associated Polyposis (POLEandPOLD1Genes)
Pathogenic variants in related DNA polymerase genes, POLE and POLD1, have been described in families with oligopolyposis, CRC, and endometrial cancer. This condition is called polymerase proofreading–associated polyposis (PPAP).[
The prevalence of POLE and POLD1 pathogenic variants was determined in a prospective cohort of 2,309 unrelated patients with hereditary cancer. These patients all underwent genetic testing with a multigene hereditary cancer panel, in which PPAP was responsible for 0.1% to 0.4% of familial cancer cases (and reaching 0.3% to 0.7% when only CRC and polyposis were considered).[
While PPAP is rare, the age-specific cumulative risks (penetrance) of CRC for POLE and POLD carriers is important to quantify and update, in order to better guide and optimize clinical management. In a retrospective study in which families with POLE or POLD1 pathogenic variants were identified via a PubMed search of relevant studies (prior to 2016) and genotypic data from 669 population-based CRC cases diagnosed in patients under 60 (from the Australasian Colorectal Cancer Family Registry), age-specific cumulative risks were estimated using modified segregation analyses.[
Lifetime risk estimates of CRC in POLE carriers were higher than expected in a subsequent study of 354 individuals with early-onset/familial CRC (n = 218) and/or attenuated adenomatous polyposis (n = 136) at a single center between 2014 and 2017.[
In POLD1 and POLE carriers, NCCN recommends high-quality colonoscopy starting at age 25 to 30 years. Colonoscopy screening can also begin 2 to 5 years before the earliest CRC in the family, if the CRC was diagnosed before age 25 years. Afterwards, colonoscopy is repeated every 1 to 2 years if polyps are identified or every 2 to 3 years if polyps are not identified.[
NTHL1-Associated Polyposis
NTHL1-associated polyposis (or NTHL1 tumor syndrome) was first described in 2015. A study used whole-exome sequencing in 51 individuals (from 48 families) with multiple colonic adenomas.[
A subsequent study of 863 families with CRC and 1,600 families without CRC confirmed an association between biallelic NTHL1 pathogenic variants and inherited CRC risk.[
In a systematic review of 21 papers, the phenotypic and genotypic spectrum of NTHL1 tumor syndrome, including the occurrence of both benign and malignant tumors, were reported in both biallelic and monoallelic carriers.[
- In 47 patients with biallelic pathogenic variants (from 32 families), 23 (49%) were diagnosed with CRC (mean age, 55 y; range, 31–73 y). In addition, 12 of the 22 female patients (55%) were diagnosed with breast cancer (mean age, 49 y; range, 36–63 y). In those who underwent colonoscopy, 93% had colonic adenomas ranging from 2 to 150 polyps, and three patients (6%) had duodenal adenomatosis. Overall, 37 carriers (79%) had at least one primary cancer (cancers occurred in different organs). All ten patients without cancer diagnoses had colonic adenomas. Six female patients (27%) also had gynecological cancers (four patients had endometrial cancer, one patient had ovarian cancer, and one patient had cervical cancer).
- In 158 patients with monoallelic germline NTHL1 variants, 29 individuals (18%) were diagnosed with CRC (mean age, 56 y; range, 36–75 y). Twenty-six out of 68 (38%) monoallelic carriers who underwent colonoscopy had colon polyps or adenomas (1–70 polyps or adenomas). Six patients had more than ten adenomas or polyps. Breast cancer was diagnosed in 59 out of 120 (49%) female monoallelic carriers (mean age, 50 y; range, 24–79 y).
- Limitations of this review included the following: (1) CRC and breast cancer may have been overrepresented due to ascertainment bias; (2) there may have been overlap with other germline pathogenic variants (one Lynch syndrome case and three BRCA2 cases were included in analyses of monoallelic NTHL1 carriers with breast cancer); (3) available data were inconsistent in studies involving monoallelic NTHL1 carriers.
Based on these results, the authors concluded that early endoscopic CRC surveillance can start before age 30 years in biallelic NTHL1 carriers, similar to the surveillance used in MUTYH carriers.[
Consistent with prior reports, monoallelic NTHL1 carriers do not have significantly increased cancer risk. It is not recommended, that these individuals undergo intensive cancer surveillance for GI, breast, or gynecologic cancers.
AXIN2-Related Polyposis
Pathogenic variants in AXIN2, a gene encoding a protein that forms part of the beta-catenin destruction complex, have been associated with oligodontia, ectodermal dysplasia, colorectal polyposis, and CRC in populations enriched with polyposis and CRC.[
The prevalence of AXIN2 pathogenic variants and their contribution to both colorectal polyposis and CRC remains unclear. An AXIN2 pathogenic variant was first implicated in a Finnish family (with multiple members) who had oligodontia (absence of >6 nonwisdom teeth in family members), CRC, and polyposis with autosomal dominant inheritance.[
In AXIN2 carriers, NCCN recommends the initiation of high-quality colonoscopy at age 25 to 30 years. Colonoscopy can be repeated every 2 to 3 years if polyps are not identified or every 1 to 2 years if polyps are found. Surgery can be considered in cases with unmanageable polyposis.[
Hereditary Mixed Polyposis Syndrome (GREM1Gene)
HMPS is a rare hereditary cancer syndrome characterized by the development of various types of colon polyps including serrated adenomas, atypical juvenile polyps, adenomas, and colonic adenocarcinoma. A small number of individuals/families with HMPS harbor a large, 40-kb, single-copy germline duplication upstream of gremlin 1 (GREM1)—the gene that is thought to cause the HMPS phenotype and increased CRC risk.[
Germline GREM1 pathogenic duplications have been observed primarily in individuals/families of Ashkenazi Jewish descent, with varying clinical presentations. Although polyposis appears to be a unifying feature in most families with this duplication, there is a high degree of variability with respect to polyp number, histology, and age of onset. In addition, extracolonic malignancies have been described in several GREM1 pathogenic variant carriers, although the small number of affected individuals limits the ability to definitively demonstrate a causal link to the GREM1 pathogenic variant.
Evidence-based strategies for managing individuals with germline GREM1 pathogenic duplications do not yet exist. However, NCCN guidelines recommend that GREM1 carriers initiate colonoscopic surveillance between ages 25 and 30 years.[
CHEK2
Several studies have suggested that germline CHEK2 pathogenic variants may confer increased risks of colorectal cancer.[
NCCN guidelines recommend that individuals with germline CHEK2 pathogenic variants (and no other CRC risk factors) participate in general population screening for CRC.[
For more information, see the
Serrated Polyposis Syndrome
The term serrated polyposis syndrome (SPS) has been used to characterize individuals who have mostly sessile serrated polyps/lesions (SSPs/SSLs) in the large intestine. Although the term syndrome is used in SPS, most cases appear to be nonfamilial, and the underlying etiology of these polyps is typically unidentifiable.
Germline evaluation of individuals with serrated polyposis is typically unrevealing.[
The WHO states that an individual meets SPS diagnostic criteria if either of the following criteria are fulfilled:[
- A personal history of at least five histologically diagnosed serrated lesions (this includes hyperplastic polyps, SSPs, or SSLs) that are all 5 mm or larger, occurring proximal to the rectum. At least two of these polyps/lesions must be 10 mm or larger.
- A personal history of 20 or more serrated lesions (of any size) in the colon/rectum, with 5 or more lesions occurring proximal to the rectum.
Studies have shown that colorectal adenocarcinoma incidence in patients who met formally defined diagnostic criteria for SPS range from 19.9% to greater than 50%.[
In some individuals with therapy-associated polyposis (TAP), an acquired, mixed-histology form of colorectal and GI tract oligopolyposis, polyps can be seen in childhood/young-adult cancer survivors who were treated with chemotherapy and/or radiotherapy. Some of these individuals will have clinical histories that fulfill SPS diagnostic criteria.[
Evidence-based management guidelines for individuals with SPS do not yet exist. NCCN guidelines recommend undergoing high-quality colonoscopy to clear all SSPs/SSLs that are 5 mm or larger (after the SPS phenotype is clinically identified in an individual).[
MBD4
MBD4-associated neoplasia syndrome is associated with an increased risk of GI polyposis (comparable to AFAP). For more information, see
Advances in Endoscopic Imaging in Hereditary CRC
Performance of endoscopic therapies for adenomas in FAP and Lynch syndrome, and decision-making regarding surgical referral and planning, require accurate estimates of the presence of adenomas. In both AFAP and Lynch syndrome the presence of very subtle adenomas poses special challenges—microadenomas in the case of AFAP and flat, though sometimes large, adenomas in Lynch syndrome.
Chromoendoscopy
The need for sensitive means to endoscopically detect subtle polyps has increased with the recognition of flat adenomas and sessile serrated polyps in otherwise average-risk subjects, very attenuated adenoma phenotypes in AFAP, and subtle flat adenomas in Lynch syndrome. Modern high-resolution endoscopes improve adenoma detection yield, but the use of various vital dyes, especially indigo carmine dye-spray, has further improved detection. Several studies have shown that the improved mucosal contrast achieved with the use of indigo carmine can improve the adenoma detection rate. Whether family history is significant or not, careful clinical evaluation consisting of dye-spray colonoscopy (indigo carmine or methylene blue),[
In various large series of average-risk populations, subtle flat lesions were detected in about 5% to 10% of cases, including adenomas with high-grade dysplasia and invasive adenocarcinoma.[
In a randomized trial of subjects with Lynch syndrome,[
In a German study,[
Fewer evaluations of chromoendoscopy have been performed in AFAP than in Lynch syndrome. One study examined four patients with presumed AFAP and fewer than 20 adenomas upon white-light examination.[
A similar role for chromoendoscopy has been suggested to evaluate the duodenum in FAP. One study from Holland that used indigo carmine dye-spray to detect duodenal adenomas showed an increase in the number and size of adenomas, including some large ones. Overall Spigelman score was not significantly affected.[
Small bowel imaging
Patients with PJS and JPS are at greater risk of disease-related complications in the small bowel (e.g., bleeding, obstruction, intussusception, or cancer). FAP patients, although at great risk of duodenal neoplasia, have a relatively low risk of jejunoileal involvement. The RR of small bowel malignancy is very high in Lynch syndrome, but absolute risk is less than 10%. Although the risks of small bowel neoplasia are high enough to warrant consideration of surveillance in each disease, the technical challenges of doing so have been daunting. Because of the technical challenges and relatively low prevalences, there is virtually no evidence base for small-bowel screening in Lynch syndrome.
Historically, the relative endoscopic inaccessibility of the mid and distal small bowel required radiographic measures for its evaluation, including the barium small bowel series or a variant called tube enteroclysis, in which a nasogastroduodenal tube is placed so that all of the contrast goes into the small intestine quickly and undiluted by gastric juice for more precise imaging. None of these measures were sensitive for small lesions. Previously, therapeutic removal of lesions required laparotomy. However, multiple novel endoscopic approaches have been developed to overcome the technical limitations of small bowel endoscopy, which has enabled jejunal and ileal access for purposes of polypectomy.
For patients with PJS, double-balloon endoscopy or other forms of deep enteroscopy (single-balloon overtube or spiral overtube) are the preferred methods for evaluation of the small bowel.[
In FAP, data from capsule endoscopy [
Capsule endoscopy in the small series of PJS patients described above [
References:
- Bussey HJ: Familial Polyposis Coli: Family Studies, Histopathology, Differential Diagnosis, and Results of Treatment. The Johns Hopkins University Press, 1975.
- Burt RW, Leppert MF, Slattery ML, et al.: Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology 127 (2): 444-51, 2004.
- Choi YH, Cotterchio M, McKeown-Eyssen G, et al.: Penetrance of colorectal cancer among MLH1/MSH2 carriers participating in the colorectal cancer familial registry in Ontario. Hered Cancer Clin Pract 7 (1): 14, 2009.
- Bonadona V, Bonaïti B, Olschwang S, et al.: Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 305 (22): 2304-10, 2011.
- Møller P, Seppälä T, Bernstein I, et al.: Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut 66 (3): 464-472, 2017.
- Baglietto L, Lindor NM, Dowty JG, et al.: Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst 102 (3): 193-201, 2010.
- Aretz S, Uhlhaas S, Goergens H, et al.: MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 119 (4): 807-14, 2006.
- Hearle N, Schumacher V, Menko FH, et al.: Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 12 (10): 3209-15, 2006.
- Coburn MC, Pricolo VE, DeLuca FG, et al.: Malignant potential in intestinal juvenile polyposis syndromes. Ann Surg Oncol 2 (5): 386-91, 1995.
- Desai DC, Neale KF, Talbot IC, et al.: Juvenile polyposis. Br J Surg 82 (1): 14-7, 1995.
- Bülow S, Berk T, Neale K: The history of familial adenomatous polyposis. Fam Cancer 5 (3): 213-20, 2006.
- Herrera L, ed.: Familial Adenomatous Polyposis. Alan R. Liss Inc, 1990.
- Bülow S: Familial polyposis coli. Dan Med Bull 34 (1): 1-15, 1987.
- Campbell WJ, Spence RA, Parks TG: Familial adenomatous polyposis. Br J Surg 81 (12): 1722-33, 1994.
- Inra JA, Steyerberg EW, Grover S, et al.: Racial variation in frequency and phenotypes of APC and MUTYH mutations in 6,169 individuals undergoing genetic testing. Genet Med 17 (10): 815-21, 2015.
- Giardiello FM, Offerhaus JG: Phenotype and cancer risk of various polyposis syndromes. Eur J Cancer 31A (7-8): 1085-7, 1995 Jul-Aug.
- Jagelman DG, DeCosse JJ, Bussey HJ: Upper gastrointestinal cancer in familial adenomatous polyposis. Lancet 1 (8595): 1149-51, 1988.
- Sturt NJ, Gallagher MC, Bassett P, et al.: Evidence for genetic predisposition to desmoid tumours in familial adenomatous polyposis independent of the germline APC mutation. Gut 53 (12): 1832-6, 2004.
- Lynch HT, Fitzgibbons R: Surgery, desmoid tumors, and familial adenomatous polyposis: case report and literature review. Am J Gastroenterol 91 (12): 2598-601, 1996.
- Bülow S, Björk J, Christensen IJ, et al.: Duodenal adenomatosis in familial adenomatous polyposis. Gut 53 (3): 381-6, 2004.
- Burt RW: Colon cancer screening. Gastroenterology 119 (3): 837-53, 2000.
- Galiatsatos P, Foulkes WD: Familial adenomatous polyposis. Am J Gastroenterol 101 (2): 385-98, 2006.
- Bisgaard ML, Bülow S: Familial adenomatous polyposis (FAP): genotype correlation to FAP phenotype with osteomas and sebaceous cysts. Am J Med Genet A 140 (3): 200-4, 2006.
- Petersen GM, Slack J, Nakamura Y: Screening guidelines and premorbid diagnosis of familial adenomatous polyposis using linkage. Gastroenterology 100 (6): 1658-64, 1991.
- Berk T, Cohen Z, Bapat B, et al.: Negative genetic test result in familial adenomatous polyposis: clinical screening implications. Dis Colon Rectum 42 (3): 307-10; discussion 310-2, 1999.
- Jagelman DG: Clinical management of familial adenomatous polyposis. Cancer Surv 8 (1): 159-67, 1989.
- Neale K, Ritchie S, Thomson JP: Screening of offspring of patients with familial adenomatous polyposis: the St. Mark's Hospital polyposis register experience. In: Herrera L, ed.: Familial Adenomatous Polyposis. Alan R. Liss Inc, 1990, pp 61-66.
- Nusliha A, Dalpatadu U, Amarasinghe B, et al.: Congenital hypertrophy of retinal pigment epithelium (CHRPE) in patients with familial adenomatous polyposis (FAP); a polyposis registry experience. BMC Res Notes 7: 734, 2014.
- Chen CS, Phillips KD, Grist S, et al.: Congenital hypertrophy of the retinal pigment epithelium (CHRPE) in familial colorectal cancer. Fam Cancer 5 (4): 397-404, 2006.
- Coleman P, Barnard NA: Congenital hypertrophy of the retinal pigment epithelium: prevalence and ocular features in the optometric population. Ophthalmic Physiol Opt 27 (6): 547-55, 2007.
- Traboulsi EI, Apostolides J, Giardiello FM, et al.: Pigmented ocular fundus lesions and APC mutations in familial adenomatous polyposis. Ophthalmic Genet 17 (4): 167-74, 1996.
- Anthony T, Rodriguez-Bigas MA, Weber TK, et al.: Desmoid tumors. J Am Coll Surg 182 (4): 369-77, 1996.
- Eccles DM, van der Luijt R, Breukel C, et al.: Hereditary desmoid disease due to a frameshift mutation at codon 1924 of the APC gene. Am J Hum Genet 59 (6): 1193-201, 1996.
- Bertario L, Russo A, Sala P, et al.: Genotype and phenotype factors as determinants of desmoid tumors in patients with familial adenomatous polyposis. Int J Cancer 95 (2): 102-7, 2001.
- Lynch HT: Desmoid tumors: genotype-phenotype differences in familial adenomatous polyposis--a nosological dilemma. Am J Hum Genet 59 (6): 1184-5, 1996.
- Scott RJ, Froggatt NJ, Trembath RC, et al.: Familial infiltrative fibromatosis (desmoid tumours) (MIM135290) caused by a recurrent 3' APC gene mutation. Hum Mol Genet 5 (12): 1921-4, 1996.
- Caspari R, Olschwang S, Friedl W, et al.: Familial adenomatous polyposis: desmoid tumours and lack of ophthalmic lesions (CHRPE) associated with APC mutations beyond codon 1444. Hum Mol Genet 4 (3): 337-40, 1995.
- Davies DR, Armstrong JG, Thakker N, et al.: Severe Gardner syndrome in families with mutations restricted to a specific region of the APC gene. Am J Hum Genet 57 (5): 1151-8, 1995.
- Bertario L, Russo A, Sala P, et al.: Multiple approach to the exploration of genotype-phenotype correlations in familial adenomatous polyposis. J Clin Oncol 21 (9): 1698-707, 2003.
- Elayi E, Manilich E, Church J: Polishing the crystal ball: knowing genotype improves ability to predict desmoid disease in patients with familial adenomatous polyposis. Dis Colon Rectum 52 (10): 1762-6, 2009.
- Nieuwenhuis MH, Lefevre JH, Bülow S, et al.: Family history, surgery, and APC mutation are risk factors for desmoid tumors in familial adenomatous polyposis: an international cohort study. Dis Colon Rectum 54 (10): 1229-34, 2011.
- Clark SK, Smith TG, Katz DE, et al.: Identification and progression of a desmoid precursor lesion in patients with familial adenomatous polyposis. Br J Surg 85 (7): 970-3, 1998.
- Hodgson SV, Maher ER: Gastro-intestinal system. In: Hodgson SV, Maher ER: A Practical Guide to Human Cancer Genetics. 2nd ed. Cambridge University Press, 1999, pp 167-175.
- Rodriguez-Bigas MA, Mahoney MC, Karakousis CP, et al.: Desmoid tumors in patients with familial adenomatous polyposis. Cancer 74 (4): 1270-4, 1994.
- Clark SK, Neale KF, Landgrebe JC, et al.: Desmoid tumours complicating familial adenomatous polyposis. Br J Surg 86 (9): 1185-9, 1999.
- Belchetz LA, Berk T, Bapat BV, et al.: Changing causes of mortality in patients with familial adenomatous polyposis. Dis Colon Rectum 39 (4): 384-7, 1996.
- Iwama T, Tamura K, Morita T, et al.: A clinical overview of familial adenomatous polyposis derived from the database of the Polyposis Registry of Japan. Int J Clin Oncol 9 (4): 308-16, 2004.
- Church J, Berk T, Boman BM, et al.: Staging intra-abdominal desmoid tumors in familial adenomatous polyposis: a search for a uniform approach to a troubling disease. Dis Colon Rectum 48 (8): 1528-34, 2005.
- Parc Y, Piquard A, Dozois RR, et al.: Long-term outcome of familial adenomatous polyposis patients after restorative coloproctectomy. Ann Surg 239 (3): 378-82, 2004.
- Church JM, McGannon E, Hull-Boiner S, et al.: Gastroduodenal polyps in patients with familial adenomatous polyposis. Dis Colon Rectum 35 (12): 1170-3, 1992.
- Sarre RG, Frost AG, Jagelman DG, et al.: Gastric and duodenal polyps in familial adenomatous polyposis: a prospective study of the nature and prevalence of upper gastrointestinal polyps. Gut 28 (3): 306-14, 1987.
- Watanabe H, Enjoji M, Yao T, et al.: Gastric lesions in familial adenomatosis coli: their incidence and histologic analysis. Hum Pathol 9 (3): 269-83, 1978.
- Weston BR, Helper DJ, Rex DK: Positive predictive value of endoscopic features deemed typical of gastric fundic gland polyps. J Clin Gastroenterol 36 (5): 399-402, 2003 May-Jun.
- Abraham SC, Nobukawa B, Giardiello FM, et al.: Fundic gland polyps in familial adenomatous polyposis: neoplasms with frequent somatic adenomatous polyposis coli gene alterations. Am J Pathol 157 (3): 747-54, 2000.
- Odze RD, Marcial MA, Antonioli D: Gastric fundic gland polyps: a morphological study including mucin histochemistry, stereometry, and MIB-1 immunohistochemistry. Hum Pathol 27 (9): 896-903, 1996.
- Wu TT, Kornacki S, Rashid A, et al.: Dysplasia and dysregulation of proliferation in foveolar and surface epithelia of fundic gland polyps from patients with familial adenomatous polyposis. Am J Surg Pathol 22 (3): 293-8, 1998.
- Burt RW: Gastric fundic gland polyps. Gastroenterology 125 (5): 1462-9, 2003.
- Bianchi LK, Burke CA, Bennett AE, et al.: Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin Gastroenterol Hepatol 6 (2): 180-5, 2008.
- Jalving M, Koornstra JJ, Wesseling J, et al.: Increased risk of fundic gland polyps during long-term proton pump inhibitor therapy. Aliment Pharmacol Ther 24 (9): 1341-8, 2006.
- Leggett B: FAP: another indication to treat H pylori. Gut 51 (4): 463-4, 2002.
- Nakamura S, Matsumoto T, Kobori Y, et al.: Impact of Helicobacter pylori infection and mucosal atrophy on gastric lesions in patients with familial adenomatous polyposis. Gut 51 (4): 485-9, 2002.
- Iida M, Yao T, Itoh H, et al.: Natural history of gastric adenomas in patients with familial adenomatosis coli/Gardner's syndrome. Cancer 61 (3): 605-11, 1988.
- Bülow S, Alm T, Fausa O, et al.: Duodenal adenomatosis in familial adenomatous polyposis. DAF Project Group. Int J Colorectal Dis 10 (1): 43-6, 1995.
- Park JG, Park KJ, Ahn YO, et al.: Risk of gastric cancer among Korean familial adenomatous polyposis patients. Report of three cases. Dis Colon Rectum 35 (10): 996-8, 1992.
- Iwama T, Mishima Y, Utsunomiya J: The impact of familial adenomatous polyposis on the tumorigenesis and mortality at the several organs. Its rational treatment. Ann Surg 217 (2): 101-8, 1993.
- Offerhaus GJ, Giardiello FM, Krush AJ, et al.: The risk of upper gastrointestinal cancer in familial adenomatous polyposis. Gastroenterology 102 (6): 1980-2, 1992.
- Brosens LA, Keller JJ, Offerhaus GJ, et al.: Prevention and management of duodenal polyps in familial adenomatous polyposis. Gut 54 (7): 1034-43, 2005.
- Ngamruengphong S, Boardman LA, Heigh RI, et al.: Gastric adenomas in familial adenomatous polyposis are common, but subtle, and have a benign course. Hered Cancer Clin Pract 12 (1): 4, 2014.
- Mankaney G, Leone P, Cruise M, et al.: Gastric cancer in FAP: a concerning rise in incidence. Fam Cancer 16 (3): 371-376, 2017.
- Li J, Woods SL, Healey S, et al.: Point Mutations in Exon 1B of APC Reveal Gastric Adenocarcinoma and Proximal Polyposis of the Stomach as a Familial Adenomatous Polyposis Variant. Am J Hum Genet 98 (5): 830-842, 2016.
- Perzin KH, Bridge MF: Adenomas of the small intestine: a clinicopathologic review of 51 cases and a study of their relationship to carcinoma. Cancer 48 (3): 799-819, 1981.
- Ranzi T, Castagnone D, Velio P, et al.: Gastric and duodenal polyps in familial polyposis coli. Gut 22 (5): 363-7, 1981.
- Burke CA, Santisi J, Church J, et al.: The utility of capsule endoscopy small bowel surveillance in patients with polyposis. Am J Gastroenterol 100 (7): 1498-502, 2005.
- Tescher P, Macrae FA, Speer T, et al.: Surveillance of FAP: a prospective blinded comparison of capsule endoscopy and other GI imaging to detect small bowel polyps. Hered Cancer Clin Pract 8 (1): 3, 2010.
- Eliakim R: Video capsule endoscopy of the small bowel. Curr Opin Gastroenterol 26 (2): 129-33, 2010.
- Taylor SA, Halligan S, Moore L, et al.: Multidetector-row CT duodenography in familial adenomatous polyposis: a pilot study. Clin Radiol 59 (10): 939-45, 2004.
- Vasen HF, Bülow S, Myrhøj T, et al.: Decision analysis in the management of duodenal adenomatosis in familial adenomatous polyposis. Gut 40 (6): 716-9, 1997.
- Groves CJ, Saunders BP, Spigelman AD, et al.: Duodenal cancer in patients with familial adenomatous polyposis (FAP): results of a 10 year prospective study. Gut 50 (5): 636-41, 2002.
- Aparicio T, Henriques J, Manfredi S, et al.: Small bowel adenocarcinoma: Results from a nationwide prospective ARCAD-NADEGE cohort study of 347 patients. Int J Cancer 147 (4): 967-977, 2020.
- Bleau BL, Gostout CJ: Endoscopic treatment of ampullary adenomas in familial adenomatous polyposis. J Clin Gastroenterol 22 (3): 237-41, 1996.
- Norton ID, Gostout CJ: Management of periampullary adenoma. Dig Dis 16 (5): 266-73, 1998 Sep-Oct.
- Norton ID, Gostout CJ, Baron TH, et al.: Safety and outcome of endoscopic snare excision of the major duodenal papilla. Gastrointest Endosc 56 (2): 239-43, 2002.
- Saurin JC, Gutknecht C, Napoleon B, et al.: Surveillance of duodenal adenomas in familial adenomatous polyposis reveals high cumulative risk of advanced disease. J Clin Oncol 22 (3): 493-8, 2004.
- Spigelman AD, Williams CB, Talbot IC, et al.: Upper gastrointestinal cancer in patients with familial adenomatous polyposis. Lancet 2 (8666): 783-5, 1989.
- Thiruvengadam SS, Lopez R, O'Malley M, et al.: Spigelman stage IV duodenal polyposis does not precede most duodenal cancer cases in patients with familial adenomatous polyposis. Gastrointest Endosc 89 (2): 345-354.e2, 2019.
- Cetta F, Montalto G, Gori M, et al.: Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: results from a European cooperative study. J Clin Endocrinol Metab 85 (1): 286-92, 2000.
- Cetta F, Curia MC, Montalto G, et al.: Thyroid carcinoma usually occurs in patients with familial adenomatous polyposis in the absence of biallelic inactivation of the adenomatous polyposis coli gene. J Clin Endocrinol Metab 86 (1): 427-32, 2001.
- Seki M, Tanaka K, Kikuchi-Yanoshita R, et al.: Loss of normal allele of the APC gene in an adrenocortical carcinoma from a patient with familial adenomatous polyposis. Hum Genet 89 (3): 298-300, 1992.
- Marchesa P, Fazio VW, Church JM, et al.: Adrenal masses in patients with familial adenomatous polyposis. Dis Colon Rectum 40 (9): 1023-8, 1997.
- Kallenberg FGJ, Bastiaansen BAJ, Nio CY, et al.: Adrenal Lesions in Patients With (Attenuated) Familial Adenomatous Polyposis and MUTYH-Associated Polyposis. Dis Colon Rectum 60 (10): 1057-1064, 2017.
- Cetta F, Mazzarella L, Bon G, et al.: Genetic alterations in hepatoblastoma and hepatocellular carcinoma associated with familial adenomatous polyposis. Med Pediatr Oncol 41 (5): 496-7, 2003.
- Young J, Barker M, Robertson T, et al.: A case of myoepithelial carcinoma displaying biallelic inactivation of the tumour suppressor gene APC in a patient with familial adenomatous polyposis. J Clin Pathol 55 (3): 230-1, 2002.
- Cetta F, Montalto G, Petracci M: Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut 41 (3): 417, 1997.
- Giardiello FM, Petersen GM, Brensinger JD, et al.: Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut 39 (96): 867-9, 1996.
- Ding SF, Michail NE, Habib NA: Genetic changes in hepatoblastoma. J Hepatol 20 (5): 672-5, 1994.
- Hughes LJ, Michels VV: Risk of hepatoblastoma in familial adenomatous polyposis. Am J Med Genet 43 (6): 1023-5, 1992.
- Bernstein IT, Bülow S, Mauritzen K: Hepatoblastoma in two cousins in a family with adenomatous polyposis. Report of two cases. Dis Colon Rectum 35 (4): 373-4, 1992.
- Giardiello FM, Offerhaus GJ, Krush AJ, et al.: Risk of hepatoblastoma in familial adenomatous polyposis. J Pediatr 119 (5): 766-8, 1991.
- Perilongo G: Link confirmed between FAP and hepatoblastoma. Oncology (Huntingt) 5 (7): 14, 1991.
- Toyama WM, Wagner S: Hepatoblastoma with familial polyposis coli: another case and corrected pedigree. Surgery 108 (1): 123-4, 1990.
- Kurahashi H, Takami K, Oue T, et al.: Biallelic inactivation of the APC gene in hepatoblastoma. Cancer Res 55 (21): 5007-11, 1995.
- Hirschman BA, Pollock BH, Tomlinson GE: The spectrum of APC mutations in children with hepatoblastoma from familial adenomatous polyposis kindreds. J Pediatr 147 (2): 263-6, 2005.
- Hamilton SR, Liu B, Parsons RE, et al.: The molecular basis of Turcot's syndrome. N Engl J Med 332 (13): 839-47, 1995.
- Laurent-Puig P, Béroud C, Soussi T: APC gene: database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res 26 (1): 269-70, 1998.
- Yan H, Dobbie Z, Gruber SB, et al.: Small changes in expression affect predisposition to tumorigenesis. Nat Genet 30 (1): 25-6, 2002.
- Spirio L, Olschwang S, Groden J, et al.: Alleles of the APC gene: an attenuated form of familial polyposis. Cell 75 (5): 951-7, 1993.
- Brensinger JD, Laken SJ, Luce MC, et al.: Variable phenotype of familial adenomatous polyposis in pedigrees with 3' mutation in the APC gene. Gut 43 (4): 548-52, 1998.
- Soravia C, Berk T, Madlensky L, et al.: Genotype-phenotype correlations in attenuated adenomatous polyposis coli. Am J Hum Genet 62 (6): 1290-301, 1998.
- Pedemonte S, Sciallero S, Gismondi V, et al.: Novel germline APC variants in patients with multiple adenomas. Genes Chromosomes Cancer 22 (4): 257-67, 1998.
- Rozen P, Samuel Z, Shomrat R, et al.: Notable intrafamilial phenotypic variability in a kindred with familial adenomatous polyposis and an APC mutation in exon 9. Gut 45 (6): 829-33, 1999.
- Fearnhead NS: Familial adenomatous polyposis and MYH. Lancet 362 (9377): 5-6, 2003.
- Al-Tassan N, Chmiel NH, Maynard J, et al.: Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat Genet 30 (2): 227-32, 2002.
- Bellido F, Pineda M, Aiza G, et al.: POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med 18 (4): 325-32, 2016.
- Spier I, Holzapfel S, Altmüller J, et al.: Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int J Cancer 137 (2): 320-31, 2015.
- Bakry D, Aronson M, Durno C, et al.: Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: report from the constitutional mismatch repair deficiency consortium. Eur J Cancer 50 (5): 987-96, 2014.
- Grover S, Kastrinos F, Steyerberg EW, et al.: Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA 308 (5): 485-92, 2012.
- Sieber OM, Lamlum H, Crabtree MD, et al.: Whole-gene APC deletions cause classical familial adenomatous polyposis, but not attenuated polyposis or "multiple" colorectal adenomas. Proc Natl Acad Sci U S A 99 (5): 2954-8, 2002.
- Michils G, Tejpar S, Thoelen R, et al.: Large deletions of the APC gene in 15% of mutation-negative patients with classical polyposis (FAP): a Belgian study. Hum Mutat 25 (2): 125-34, 2005.
- Kadiyska TK, Todorov TP, Bichev SN, et al.: APC promoter 1B deletion in familial polyposis--implications for mutation-negative families. Clin Genet 85 (5): 452-7, 2014.
- Jasperson KW, Patel SG, Ahnen DJ, et al., eds.: APC-Associated Polyposis Conditions. In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp.
Available online. Last accessed May 24, 2024. - Bisgaard ML, Fenger K, Bülow S, et al.: Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat 3 (2): 121-5, 1994.
- Patenaude AF: Cancer susceptibility testing: risks, benefits, and personal beliefs. In: Clarke A, ed.: The Genetic Testing of Children. BIOS Scientific, 1998, pp 145-156.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, and Gastric. Version 3.2024. Plymouth Meeting, PA: National Comprehensive Cancer Network, 2024.
Available with free registration. Last accessed December 13, 2024. - Nugent KP, Spigelman AD, Phillips RK: Life expectancy after colectomy and ileorectal anastomosis for familial adenomatous polyposis. Dis Colon Rectum 36 (11): 1059-62, 1993.
- Barrow P, Khan M, Lalloo F, et al.: Systematic review of the impact of registration and screening on colorectal cancer incidence and mortality in familial adenomatous polyposis and Lynch syndrome. Br J Surg 100 (13): 1719-31, 2013.
- Koskenvuo L, Pitkäniemi J, Rantanen M, et al.: Impact of Screening on Survival in Familial Adenomatous Polyposis. J Clin Gastroenterol 50 (1): 40-4, 2016.
- Hakulinen T, Seppä K, Lambert PC: Choosing the relative survival method for cancer survival estimation. Eur J Cancer 47 (14): 2202-10, 2011.
- Petersen GM: Genetic testing and counseling in familial adenomatous polyposis. Oncology (Huntingt) 10 (1): 89-94; discussion 97-8, 1996.
- Church J, Burke C, McGannon E, et al.: Risk of rectal cancer in patients after colectomy and ileorectal anastomosis for familial adenomatous polyposis: a function of available surgical options. Dis Colon Rectum 46 (9): 1175-81, 2003.
- Guillem JG, Wood WC, Moley JF, et al.: ASCO/SSO review of current role of risk-reducing surgery in common hereditary cancer syndromes. Ann Surg Oncol 13 (10): 1296-321, 2006.
- Bertario L, Russo A, Radice P, et al.: Genotype and phenotype factors as determinants for rectal stump cancer in patients with familial adenomatous polyposis. Hereditary Colorectal Tumors Registry. Ann Surg 231 (4): 538-43, 2000.
- Heiskanen I, Järvinen HJ: Fate of the rectal stump after colectomy and ileorectal anastomosis for familial adenomatous polyposis. Int J Colorectal Dis 12 (1): 9-13, 1997.
- Bülow S, Bülow C, Vasen H, et al.: Colectomy and ileorectal anastomosis is still an option for selected patients with familial adenomatous polyposis. Dis Colon Rectum 51 (9): 1318-23, 2008.
- De Cosse JJ, Bülow S, Neale K, et al.: Rectal cancer risk in patients treated for familial adenomatous polyposis. The Leeds Castle Polyposis Group. Br J Surg 79 (12): 1372-5, 1992.
- Bess MA, Adson MA, Elveback LR, et al.: Rectal cancer following colectomy for polyposis. Arch Surg 115 (4): 460-7, 1980.
- Iwama T, Mishima Y: Factors affecting the risk of rectal cancer following rectum-preserving surgery in patients with familial adenomatous polyposis. Dis Colon Rectum 37 (10): 1024-6, 1994.
- Bülow S, Højen H, Buntzen S, et al.: Primary and secondary restorative proctocolectomy for familial adenomatous polyposis: complications and long-term bowel function. Colorectal Dis 15 (4): 436-41, 2013.
- Church J, Burke C, McGannon E, et al.: Predicting polyposis severity by proctoscopy: how reliable is it? Dis Colon Rectum 44 (9): 1249-54, 2001.
- Nieuwenhuis MH, Bülow S, Björk J, et al.: Genotype predicting phenotype in familial adenomatous polyposis: a practical application to the choice of surgery. Dis Colon Rectum 52 (7): 1259-63, 2009.
- Vasen HF, van der Luijt RB, Slors JF, et al.: Molecular genetic tests as a guide to surgical management of familial adenomatous polyposis. Lancet 348 (9025): 433-5, 1996.
- Wu JS, Paul P, McGannon EA, et al.: APC genotype, polyp number, and surgical options in familial adenomatous polyposis. Ann Surg 227 (1): 57-62, 1998.
- Nieuwenhuis MH, Mathus-Vliegen LM, Slors FJ, et al.: Genotype-phenotype correlations as a guide in the management of familial adenomatous polyposis. Clin Gastroenterol Hepatol 5 (3): 374-8, 2007.
- Lovegrove RE, Tilney HS, Heriot AG, et al.: A comparison of adverse events and functional outcomes after restorative proctocolectomy for familial adenomatous polyposis and ulcerative colitis. Dis Colon Rectum 49 (9): 1293-306, 2006.
- Parc YR, Olschwang S, Desaint B, et al.: Familial adenomatous polyposis: prevalence of adenomas in the ileal pouch after restorative proctocolectomy. Ann Surg 233 (3): 360-4, 2001.
- Groves CJ, Beveridge G, Swain DJ, et al.: Prevalence and morphology of pouch and ileal adenomas in familial adenomatous polyposis. Dis Colon Rectum 48 (4): 816-23, 2005.
- Lee CHA, Kalady MF, Burke CA, et al.: Incidence and Management of Rectal Cuff and Anal Transitional Zone Neoplasia in Patients With Familial Adenomatous Polyposis. Dis Colon Rectum 64 (8): 977-985, 2021.
- Ooi BS, Remzi FH, Gramlich T, et al.: Anal transitional zone cancer after restorative proctocolectomy and ileoanal anastomosis in familial adenomatous polyposis: report of two cases. Dis Colon Rectum 46 (10): 1418-23; discussion 1422-3, 2003.
- Steinbach G, Lynch PM, Phillips RK, et al.: The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 342 (26): 1946-52, 2000.
- Giardiello FM, Yang VW, Hylind LM, et al.: Primary chemoprevention of familial adenomatous polyposis with sulindac. N Engl J Med 346 (14): 1054-9, 2002.
- Lynch PM, Burke CA, Phillips R, et al.: An international randomised trial of celecoxib versus celecoxib plus difluoromethylornithine in patients with familial adenomatous polyposis. Gut 65 (2): 286-95, 2016.
- Burke CA, Dekker E, Lynch P, et al.: Eflornithine plus Sulindac for Prevention of Progression in Familial Adenomatous Polyposis. N Engl J Med 383 (11): 1028-1039, 2020.
- Balaguer F, Stoffel EM, Burke CA, et al.: Combination of Sulindac and Eflornithine Delays the Need for Lower Gastrointestinal Surgery in Patients With Familial Adenomatous Polyposis: Post Hoc Analysis of a Randomized Clinical Trial. Dis Colon Rectum 65 (4): 536-545, 2022.
- Lynch PM, Ayers GD, Hawk E, et al.: The safety and efficacy of celecoxib in children with familial adenomatous polyposis. Am J Gastroenterol 105 (6): 1437-43, 2010.
- West NJ, Clark SK, Phillips RK, et al.: Eicosapentaenoic acid reduces rectal polyp number and size in familial adenomatous polyposis. Gut 59 (7): 918-25, 2010.
- Phillips RK, Wallace MH, Lynch PM, et al.: A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut 50 (6): 857-60, 2002.
- Nugent KP, Farmer KC, Spigelman AD, et al.: Randomized controlled trial of the effect of sulindac on duodenal and rectal polyposis and cell proliferation in patients with familial adenomatous polyposis. Br J Surg 80 (12): 1618-9, 1993.
- Jacoby RF, Cole CE, Hawk ET, et al.: Ursodeoxycholate/Sulindac combination treatment effectively prevents intestinal adenomas in a mouse model of polyposis. Gastroenterology 127 (3): 838-44, 2004.
- Parc Y, Desaint B, Fléjou JF, et al.: The effect of ursodesoxycholic acid on duodenal adenomas in familial adenomatous polyposis: a prospective randomized placebo-control trial. Colorectal Dis 14 (7): 854-60, 2012.
- van Heumen BW, Roelofs HM, Vink-Börger ME, et al.: Ursodeoxycholic acid counteracts celecoxib in reduction of duodenal polyps in patients with familial adenomatous polyposis: a multicentre, randomized controlled trial. Orphanet J Rare Dis 8: 118, 2013.
- Fitzgerald GA: Coxibs and cardiovascular disease. N Engl J Med 351 (17): 1709-11, 2004.
- Solomon SD, McMurray JJ, Pfeffer MA, et al.: Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med 352 (11): 1071-80, 2005.
- Bresalier RS, Sandler RS, Quan H, et al.: Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med 352 (11): 1092-102, 2005.
- Giardiello FM, Hamilton SR, Krush AJ, et al.: Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med 328 (18): 1313-6, 1993.
- Roberts RB, Min L, Washington MK, et al.: Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc Natl Acad Sci U S A 99 (3): 1521-6, 2002.
- Samadder NJ, Neklason DW, Boucher KM, et al.: Effect of Sulindac and Erlotinib vs Placebo on Duodenal Neoplasia in Familial Adenomatous Polyposis: A Randomized Clinical Trial. JAMA 315 (12): 1266-75, 2016 Mar 22-29.
- Samadder NJ, Kuwada SK, Boucher KM, et al.: Association of Sulindac and Erlotinib vs Placebo With Colorectal Neoplasia in Familial Adenomatous Polyposis: Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol 4 (5): 671-677, 2018.
- Rinella ES, Threadgill DW: Efficacy of EGFR inhibition is modulated by model, sex, genetic background and diet: implications for preclinical cancer prevention and therapy trials. PLoS One 7 (6): e39552, 2012.
- Tonelli F, Ficari F, Valanzano R, et al.: Treatment of desmoids and mesenteric fibromatosis in familial adenomatous polyposis with raloxifene. Tumori 89 (4): 391-6, 2003 Jul-Aug.
- Hansmann A, Adolph C, Vogel T, et al.: High-dose tamoxifen and sulindac as first-line treatment for desmoid tumors. Cancer 100 (3): 612-20, 2004.
- Lindor NM, Dozois R, Nelson H, et al.: Desmoid tumors in familial adenomatous polyposis: a pilot project evaluating efficacy of treatment with pirfenidone. Am J Gastroenterol 98 (8): 1868-74, 2003.
- Mace J, Sybil Biermann J, Sondak V, et al.: Response of extraabdominal desmoid tumors to therapy with imatinib mesylate. Cancer 95 (11): 2373-9, 2002.
- Penel N, Le Cesne A, Bui BN, et al.: Imatinib for progressive and recurrent aggressive fibromatosis (desmoid tumors): an FNCLCC/French Sarcoma Group phase II trial with a long-term follow-up. Ann Oncol 22 (2): 452-7, 2011.
- Kasper B, Gruenwald V, Reichardt P, et al.: Imatinib induces sustained progression arrest in RECIST progressive desmoid tumours: Final results of a phase II study of the German Interdisciplinary Sarcoma Group (GISG). Eur J Cancer 76: 60-67, 2017.
- Gounder MM, Mahoney MR, Van Tine BA, et al.: Sorafenib for Advanced and Refractory Desmoid Tumors. N Engl J Med 379 (25): 2417-2428, 2018.
- Latchford AR, Sturt NJ, Neale K, et al.: A 10-year review of surgery for desmoid disease associated with familial adenomatous polyposis. Br J Surg 93 (10): 1258-64, 2006.
- Heiskanen I, Järvinen HJ: Occurrence of desmoid tumours in familial adenomatous polyposis and results of treatment. Int J Colorectal Dis 11 (4): 157-62, 1996.
- Wood LD, Salaria SN, Cruise MW, et al.: Upper GI tract lesions in familial adenomatous polyposis (FAP): enrichment of pyloric gland adenomas and other gastric and duodenal neoplasms. Am J Surg Pathol 38 (3): 389-93, 2014.
- Nakamura K, Nonaka S, Nakajima T, et al.: Clinical outcomes of gastric polyps and neoplasms in patients with familial adenomatous polyposis. Endosc Int Open 5 (3): E137-E145, 2017.
- Park JS, Choi GS, Kim HJ, et al.: Natural orifice specimen extraction versus conventional laparoscopically assisted right hemicolectomy. Br J Surg 98 (5): 710-5, 2011.
- Johnson MD, Mackey R, Brown N, et al.: Outcome based on management for duodenal adenomas: sporadic versus familial disease. J Gastrointest Surg 14 (2): 229-35, 2010.
- de Vos tot Nederveen Cappel WH, Järvinen HJ, Björk J, et al.: Worldwide survey among polyposis registries of surgical management of severe duodenal adenomatosis in familial adenomatous polyposis. Br J Surg 90 (6): 705-10, 2003.
- Balmaña J, Balaguer F, Cervantes A, et al.: Familial risk-colorectal cancer: ESMO Clinical Practice Guidelines. Ann Oncol 24 (Suppl 6): vi73-80, 2013.
- Bülow S, Christensen IJ, Højen H, et al.: Duodenal surveillance improves the prognosis after duodenal cancer in familial adenomatous polyposis. Colorectal Dis 14 (8): 947-52, 2012.
- Ahmad NA, Kochman ML, Long WB, et al.: Efficacy, safety, and clinical outcomes of endoscopic mucosal resection: a study of 101 cases. Gastrointest Endosc 55 (3): 390-6, 2002.
- Heiskanen I, Kellokumpu I, Järvinen H: Management of duodenal adenomas in 98 patients with familial adenomatous polyposis. Endoscopy 31 (6): 412-6, 1999.
- Penna C, Phillips RK, Tiret E, et al.: Surgical polypectomy of duodenal adenomas in familial adenomatous polyposis: experience of two European centres. Br J Surg 80 (8): 1027-9, 1993.
- Mackey R, Walsh RM, Chung R, et al.: Pancreas-sparing duodenectomy is effective management for familial adenomatous polyposis. J Gastrointest Surg 9 (8): 1088-93; discussion 1093, 2005.
- Collard MK, Lefevre JH, Ahmed O, et al.: Ten-year impact of pancreaticoduodenectomy on bowel function and quality of life of patients with ileal pouch-anal anastomosis for familial adenomatous polyposis. HPB (Oxford) 22 (10): 1402-1410, 2020.
- Lepistö A, Kiviluoto T, Halttunen J, et al.: Surveillance and treatment of duodenal adenomatosis in familial adenomatous polyposis. Endoscopy 41 (6): 504-9, 2009.
- Wallace MH, Phillips RK: Upper gastrointestinal disease in patients with familial adenomatous polyposis. Br J Surg 85 (6): 742-50, 1998.
- Parc Y, Mabrut JY, Shields C, et al.: Surgical management of the duodenal manifestations of familial adenomatous polyposis. Br J Surg 98 (4): 480-4, 2011.
- Penna C, Bataille N, Balladur P, et al.: Surgical treatment of severe duodenal polyposis in familial adenomatous polyposis. Br J Surg 85 (5): 665-8, 1998.
- Hirasawa R, Iishi H, Tatsuta M, et al.: Clinicopathologic features and endoscopic resection of duodenal adenocarcinomas and adenomas with the submucosal saline injection technique. Gastrointest Endosc 46 (6): 507-13, 1997.
- Catalano MF, Linder JD, Chak A, et al.: Endoscopic management of adenoma of the major duodenal papilla. Gastrointest Endosc 59 (2): 225-32, 2004.
- Alarcon FJ, Burke CA, Church JM, et al.: Familial adenomatous polyposis: efficacy of endoscopic and surgical treatment for advanced duodenal adenomas. Dis Colon Rectum 42 (12): 1533-6, 1999.
- Biasco G, Nobili E, Calabrese C, et al.: Impact of surgery on the development of duodenal cancer in patients with familial adenomatous polyposis. Dis Colon Rectum 49 (12): 1860-6, 2006.
- Chung RS, Church JM, vanStolk R: Pancreas-sparing duodenectomy: indications, surgical technique, and results. Surgery 117 (3): 254-9, 1995.
- Tsiotos GG, Sarr MG: Pancreas-preserving total duodenectomy. Dig Surg 15 (5): 398-403, 1998.
- Sarmiento JM, Thompson GB, Nagorney DM, et al.: Pancreas-sparing duodenectomy for duodenal polyposis. Arch Surg 137 (5): 557-62; discussion 562-3, 2002.
- Kalady MF, Clary BM, Tyler DS, et al.: Pancreas-preserving duodenectomy in the management of duodenal familial adenomatous polyposis. J Gastrointest Surg 6 (1): 82-7, 2002 Jan-Feb.
- Eisenberger CF, Knoefel WT, Peiper M, et al.: Pancreas-sparing duodenectomy in duodenal pathology: indications and results. Hepatogastroenterology 51 (57): 727-31, 2004 May-Jun.
- Jasperson KW, Tuohy TM, Neklason DW, et al.: Hereditary and familial colon cancer. Gastroenterology 138 (6): 2044-58, 2010.
- Jarrar AM, Milas M, Mitchell J, et al.: Screening for thyroid cancer in patients with familial adenomatous polyposis. Ann Surg 253 (3): 515-21, 2011.
- Aretz S, Koch A, Uhlhaas S, et al.: Should children at risk for familial adenomatous polyposis be screened for hepatoblastoma and children with apparently sporadic hepatoblastoma be screened for APC germline mutations? Pediatr Blood Cancer 47 (6): 811-8, 2006.
- Leppert M, Burt R, Hughes JP, et al.: Genetic analysis of an inherited predisposition to colon cancer in a family with a variable number of adenomatous polyps. N Engl J Med 322 (13): 904-8, 1990.
- Lynch HT, Smyrk TC: Classification of familial adenomatous polyposis: a diagnostic nightmare. Am J Hum Genet 62 (6): 1288-9, 1998.
- Giardiello FM, Brensinger JD, Luce MC, et al.: Phenotypic expression of disease in families that have mutations in the 5' region of the adenomatous polyposis coli gene. Ann Intern Med 126 (7): 514-9, 1997.
- White S, Bubb VJ, Wyllie AH: Germline APC mutation (Gln1317) in a cancer-prone family that does not result in familial adenomatous polyposis. Genes Chromosomes Cancer 15 (2): 122-8, 1996.
- Gonçalves V, Theisen P, Antunes O, et al.: A missense mutation in the APC tumor suppressor gene disrupts an ASF/SF2 splicing enhancer motif and causes pathogenic skipping of exon 14. Mutat Res 662 (1-2): 33-6, 2009.
- Heppner Goss K, Trzepacz C, Tuohy TM, et al.: Attenuated APC alleles produce functional protein from internal translation initiation. Proc Natl Acad Sci U S A 99 (12): 8161-6, 2002.
- Nieuwenhuis MH, Vasen HF: Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol 61 (2): 153-61, 2007.
- Scott RJ, Meldrum C, Crooks R, et al.: Familial adenomatous polyposis: more evidence for disease diversity and genetic heterogeneity. Gut 48 (4): 508-14, 2001.
- Knudsen AL, Bisgaard ML, Bülow S: Attenuated familial adenomatous polyposis (AFAP). A review of the literature. Fam Cancer 2 (1): 43-55, 2003.
- Vasen HF, Möslein G, Alonso A, et al.: Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 57 (5): 704-13, 2008.
- Nielsen M, Morreau H, Vasen HF, et al.: MUTYH-associated polyposis (MAP). Crit Rev Oncol Hematol 79 (1): 1-16, 2011.
- Nielsen M, Joerink-van de Beld MC, Jones N, et al.: Analysis of MUTYH genotypes and colorectal phenotypes in patients With MUTYH-associated polyposis. Gastroenterology 136 (2): 471-6, 2009.
- Nielsen M, Franken PF, Reinards TH, et al.: Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP). J Med Genet 42 (9): e54, 2005.
- Knopperts AP, Nielsen M, Niessen RC, et al.: Contribution of bi-allelic germline MUTYH mutations to early-onset and familial colorectal cancer and to low number of adenomatous polyps: case-series and literature review. Fam Cancer 12 (1): 43-50, 2013.
- Sampson JR, Dolwani S, Jones S, et al.: Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 362 (9377): 39-41, 2003.
- Dolwani S, Williams GT, West KP, et al.: Analysis of inherited MYH/(MutYH) mutations in British Asian patients with colorectal cancer. Gut 56 (4): 593, 2007.
- Gismondi V, Meta M, Bonelli L, et al.: Prevalence of the Y165C, G382D and 1395delGGA germline mutations of the MYH gene in Italian patients with adenomatous polyposis coli and colorectal adenomas. Int J Cancer 109 (5): 680-4, 2004.
- Ricci MT, Miccoli S, Turchetti D, et al.: Type and frequency of MUTYH variants in Italian patients with suspected MAP: a retrospective multicenter study. J Hum Genet 62 (2): 309-315, 2017.
- Isidro G, Laranjeira F, Pires A, et al.: Germline MUTYH (MYH) mutations in Portuguese individuals with multiple colorectal adenomas. Hum Mutat 24 (4): 353-4, 2004.
- Kim DW, Kim IJ, Kang HC, et al.: Germline mutations of the MYH gene in Korean patients with multiple colorectal adenomas. Int J Colorectal Dis 22 (10): 1173-8, 2007.
- Yanaru-Fujisawa R, Matsumoto T, Ushijima Y, et al.: Genomic and functional analyses of MUTYH in Japanese patients with adenomatous polyposis. Clin Genet 73 (6): 545-53, 2008.
- Kim JC, Ka IH, Lee YM, et al.: MYH, OGG1, MTH1, and APC alterations involved in the colorectal tumorigenesis of Korean patients with multiple adenomas. Virchows Arch 450 (3): 311-9, 2007.
- Hampel H: Genetic testing for hereditary colorectal cancer. Surg Oncol Clin N Am 18 (4): 687-703, 2009.
- Jones N, Vogt S, Nielsen M, et al.: Increased colorectal cancer incidence in obligate carriers of heterozygous mutations in MUTYH. Gastroenterology 137 (2): 489-94, 494.e1; quiz 725-6, 2009.
- Sieber OM, Lipton L, Crabtree M, et al.: Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med 348 (9): 791-9, 2003.
- Nieuwenhuis MH, Vogt S, Jones N, et al.: Evidence for accelerated colorectal adenoma--carcinoma progression in MUTYH-associated polyposis? Gut 61 (5): 734-8, 2012.
- Win AK, Dowty JG, Cleary SP, et al.: Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology 146 (5): 1208-11.e1-5, 2014.
- Morak M, Laner A, Bacher U, et al.: MUTYH-associated polyposis - variability of the clinical phenotype in patients with biallelic and monoallelic MUTYH mutations and report on novel mutations. Clin Genet 78 (4): 353-63, 2010.
- Boparai KS, Dekker E, Van Eeden S, et al.: Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology 135 (6): 2014-8, 2008.
- Nascimbeni R, Pucciarelli S, Di Lorenzo D, et al.: Rectum-sparing surgery may be appropriate for biallelic MutYH-associated polyposis. Dis Colon Rectum 53 (12): 1670-5, 2010.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Colorectal Cancer Screening. Version 1.2024. Plymouth Meeting, PA: National Comprehensive Cancer Network, 2024.
Available with free registration. Last accessed March 3, 2024. - Win AK, Cleary SP, Dowty JG, et al.: Cancer risks for monoallelic MUTYH mutation carriers with a family history of colorectal cancer. Int J Cancer 129 (9): 2256-62, 2011.
- Vogt S, Jones N, Christian D, et al.: Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 137 (6): 1976-85.e1-10, 2009.
- Lefevre JH, Rodrigue CM, Mourra N, et al.: Implication of MYH in colorectal polyposis. Ann Surg 244 (6): 874-9; discussion 879-80, 2006.
- Wasielewski M, Out AA, Vermeulen J, et al.: Increased MUTYH mutation frequency among Dutch families with breast cancer and colorectal cancer. Breast Cancer Res Treat 124 (3): 635-41, 2010.
- Poulsen ML, Bisgaard ML: MUTYH Associated Polyposis (MAP). Curr Genomics 9 (6): 420-35, 2008.
- Goodenberger M, Lindor NM: Lynch syndrome and MYH-associated polyposis: review and testing strategy. J Clin Gastroenterol 45 (6): 488-500, 2011.
- Walton SJ, Kallenberg FG, Clark SK, et al.: Frequency and Features of Duodenal Adenomas in Patients With MUTYH-Associated Polyposis. Clin Gastroenterol Hepatol 14 (7): 986-92, 2016.
- Win AK, Hopper JL, Jenkins MA: Association between monoallelic MUTYH mutation and colorectal cancer risk: a meta-regression analysis. Fam Cancer 10 (1): 1-9, 2011.
- Giráldez MD, Balaguer F, Caldés T, et al.: Association of MUTYH and MSH6 germline mutations in colorectal cancer patients. Fam Cancer 8 (4): 525-31, 2009.
- Steinke V, Rahner N, Morak M, et al.: No association between MUTYH and MSH6 germline mutations in 64 HNPCC patients. Eur J Hum Genet 16 (5): 587-92, 2008.
- Win AK, Reece JC, Buchanan DD, et al.: Risk of colorectal cancer for people with a mutation in both a MUTYH and a DNA mismatch repair gene. Fam Cancer 14 (4): 575-83, 2015.
- Boland CR, Troncale FJ: Familial colonic cancer without antecedent polyposis. Ann Intern Med 100 (5): 700-1, 1984.
- Vasen HF, Mecklin JP, Khan PM, et al.: The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 34 (5): 424-5, 1991.
- Bodmer WF, Bailey CJ, Bodmer J, et al.: Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 328 (6131): 614-6, 1987 Aug 13-19.
- Groden J, Thliveris A, Samowitz W, et al.: Identification and characterization of the familial adenomatous polyposis coli gene. Cell 66 (3): 589-600, 1991.
- Vasen HF, Watson P, Mecklin JP, et al.: New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 116 (6): 1453-6, 1999.
- Lindor NM, Rabe K, Petersen GM, et al.: Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 293 (16): 1979-85, 2005.
- Rodriguez-Bigas MA, Boland CR, Hamilton SR, et al.: A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst 89 (23): 1758-62, 1997.
- Umar A, Boland CR, Terdiman JP, et al.: Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 96 (4): 261-8, 2004.
- Laghi L, Bianchi P, Roncalli M, et al.: Re: Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 96 (18): 1402-3; author reply 1403-4, 2004.
- Hampel H, Frankel WL, Martin E, et al.: Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 26 (35): 5783-8, 2008.
- Grover S, Stoffel EM, Bussone L, et al.: Physician assessment of family cancer history and referral for genetic evaluation in colorectal cancer patients. Clin Gastroenterol Hepatol 2 (9): 813-9, 2004.
- Barnetson RA, Tenesa A, Farrington SM, et al.: Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med 354 (26): 2751-63, 2006.
- Kastrinos F, Steyerberg EW, Mercado R, et al.: The PREMM(1,2,6) model predicts risk of MLH1, MSH2, and MSH6 germline mutations based on cancer history. Gastroenterology 140 (1): 73-81, 2011.
- Chen S, Wang W, Lee S, et al.: Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA 296 (12): 1479-87, 2006.
- Kastrinos F, Uno H, Ukaegbu C, et al.: Development and Validation of the PREMM5 Model for Comprehensive Risk Assessment of Lynch Syndrome. J Clin Oncol 35 (19): 2165-2172, 2017.
- Yurgelun MB, Uno H, Furniss CS, et al.: Development and Validation of the PREMMplus Model for Multigene Hereditary Cancer Risk Assessment. J Clin Oncol 40 (35): 4083-4094, 2022.
- Kastrinos F, Allen JI, Stockwell DH, et al.: Development and validation of a colon cancer risk assessment tool for patients undergoing colonoscopy. Am J Gastroenterol 104 (6): 1508-18, 2009.
- Balaguer F, Balmaña J, Castellví-Bel S, et al.: Validation and extension of the PREMM1,2 model in a population-based cohort of colorectal cancer patients. Gastroenterology 134 (1): 39-46, 2008.
- Balmaña J, Balaguer F, Castellví-Bel S, et al.: Comparison of predictive models, clinical criteria and molecular tumour screening for the identification of patients with Lynch syndrome in a population-based cohort of colorectal cancer patients. J Med Genet 45 (9): 557-63, 2008.
- Green RC, Parfrey PS, Woods MO, et al.: Prediction of Lynch syndrome in consecutive patients with colorectal cancer. J Natl Cancer Inst 101 (5): 331-40, 2009.
- Kastrinos F, Steyerberg EW, Balmaña J, et al.: Comparison of the clinical prediction model PREMM(1,2,6) and molecular testing for the systematic identification of Lynch syndrome in colorectal cancer. Gut 62 (2): 272-9, 2013.
- Khan O, Blanco A, Conrad P, et al.: Performance of Lynch syndrome predictive models in a multi-center US referral population. Am J Gastroenterol 106 (10): 1822-7; quiz 1828, 2011.
- Pouchet CJ, Wong N, Chong G, et al.: A comparison of models used to predict MLH1, MSH2 and MSH6 mutation carriers. Ann Oncol 20 (4): 681-8, 2009.
- Monzon JG, Cremin C, Armstrong L, et al.: Validation of predictive models for germline mutations in DNA mismatch repair genes in colorectal cancer. Int J Cancer 126 (4): 930-9, 2010.
- Kastrinos F, Balmaña J, Syngal S: Prediction models in Lynch syndrome. Fam Cancer 12 (2): 217-28, 2013.
- Balmaña J, Stockwell DH, Steyerberg EW, et al.: Prediction of MLH1 and MSH2 mutations in Lynch syndrome. JAMA 296 (12): 1469-78, 2006.
- Luba DG, DiSario JA, Rock C, et al.: Community Practice Implementation of a Self-administered Version of PREMM1,2,6 to Assess Risk for Lynch Syndrome. Clin Gastroenterol Hepatol 16 (1): 49-58, 2018.
- Kastrinos F, Ojha RP, Leenen C, et al.: Comparison of Prediction Models for Lynch Syndrome Among Individuals With Colorectal Cancer. J Natl Cancer Inst 108 (2): , 2016.
- Weber JL, May PE: Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet 44 (3): 388-96, 1989.
- Vilar E, Gruber SB: Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol 7 (3): 153-62, 2010.
- Haraldsdottir S, Roth R, Pearlman R, et al.: Mismatch repair deficiency concordance between primary colorectal cancer and corresponding metastasis. Fam Cancer 15 (2): 253-60, 2016.
- Grady WM, Carethers JM: Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 135 (4): 1079-99, 2008.
- Greenson JK, Huang SC, Herron C, et al.: Pathologic predictors of microsatellite instability in colorectal cancer. Am J Surg Pathol 33 (1): 126-33, 2009.
- Boland CR, Thibodeau SN, Hamilton SR, et al.: A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 58 (22): 5248-57, 1998.
- Thibodeau SN, French AJ, Roche PC, et al.: Altered expression of hMSH2 and hMLH1 in tumors with microsatellite instability and genetic alterations in mismatch repair genes. Cancer Res 56 (21): 4836-40, 1996.
- Cawkwell L, Gray S, Murgatroyd H, et al.: Choice of management strategy for colorectal cancer based on a diagnostic immunohistochemical test for defective mismatch repair. Gut 45 (3): 409-15, 1999.
- Lindor NM, Burgart LJ, Leontovich O, et al.: Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J Clin Oncol 20 (4): 1043-8, 2002.
- de La Chapelle A: Microsatellite instability phenotype of tumors: genotyping or immunohistochemistry? The jury is still out. J Clin Oncol 20 (4): 897-9, 2002.
- Peltomäki P: Update on Lynch syndrome genomics. Fam Cancer 15 (3): 385-93, 2016.
- Rumilla K, Schowalter KV, Lindor NM, et al.: Frequency of deletions of EPCAM (TACSTD1) in MSH2-associated Lynch syndrome cases. J Mol Diagn 13 (1): 93-9, 2011.
- Kovacs ME, Papp J, Szentirmay Z, et al.: Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat 30 (2): 197-203, 2009.
- Rosty C, Clendenning M, Walsh MD, et al.: Germline mutations in PMS2 and MLH1 in individuals with solitary loss of PMS2 expression in colorectal carcinomas from the Colon Cancer Family Registry Cohort. BMJ Open 6 (2): e010293, 2016.
- Lynch HT, Boland CR, Rodriguez-Bigas MA, et al.: Who should be sent for genetic testing in hereditary colorectal cancer syndromes? J Clin Oncol 25 (23): 3534-42, 2007.
- Cunningham JM, Kim CY, Christensen ER, et al.: The frequency of hereditary defective mismatch repair in a prospective series of unselected colorectal carcinomas. Am J Hum Genet 69 (4): 780-90, 2001.
- Esteller M: Epigenetics in cancer. N Engl J Med 358 (11): 1148-59, 2008.
- Wang L, Cunningham JM, Winters JL, et al.: BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res 63 (17): 5209-12, 2003.
- Domingo E, Espín E, Armengol M, et al.: Activated BRAF targets proximal colon tumors with mismatch repair deficiency and MLH1 inactivation. Genes Chromosomes Cancer 39 (2): 138-42, 2004.
- Deng G, Bell I, Crawley S, et al.: BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res 10 (1 Pt 1): 191-5, 2004.
- Domingo E, Niessen RC, Oliveira C, et al.: BRAF-V600E is not involved in the colorectal tumorigenesis of HNPCC in patients with functional MLH1 and MSH2 genes. Oncogene 24 (24): 3995-8, 2005.
- Haraldsdottir S, Hampel H, Tomsic J, et al.: Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology 147 (6): 1308-1316.e1, 2014.
- Pearlman R, Frankel WL, Swanson BJ, et al.: Prospective Statewide Study of Universal Screening for Hereditary Colorectal Cancer: The Ohio Colorectal Cancer Prevention Initiative. JCO Precis Oncol 5: , 2021.
- Hampel H, Pearlman R, de la Chapelle A, et al.: Double somatic mismatch repair gene pathogenic variants as common as Lynch syndrome among endometrial cancer patients. Gynecol Oncol 160 (1): 161-168, 2021.
- Dixon K, Asrat MJ, Bedard AC, et al.: Integrating Tumor Sequencing Into Clinical Practice for Patients With Mismatch Repair-Deficient Lynch Syndrome Spectrum Cancers. Clin Transl Gastroenterol 12 (8): e00397, 2021.
- Antelo M, Golubicki M, Roca E, et al.: Lynch-like syndrome is as frequent as Lynch syndrome in early-onset nonfamilial nonpolyposis colorectal cancer. Int J Cancer 145 (3): 705-713, 2019.
- Carethers JM, Stoffel EM: Lynch syndrome and Lynch syndrome mimics: The growing complex landscape of hereditary colon cancer. World J Gastroenterol 21 (31): 9253-61, 2015.
- Bittles AH, Black ML: Evolution in health and medicine Sackler colloquium: Consanguinity, human evolution, and complex diseases. Proc Natl Acad Sci U S A 107 (Suppl 1): 1779-86, 2010.
- Hitchins MP: The role of epigenetics in Lynch syndrome. Fam Cancer 12 (2): 189-205, 2013.
- Gazzoli I, Loda M, Garber J, et al.: A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res 62 (14): 3925-8, 2002.
- Gylling A, Ridanpää M, Vierimaa O, et al.: Large genomic rearrangements and germline epimutations in Lynch syndrome. Int J Cancer 124 (10): 2333-40, 2009.
- Hitchins MP, Rapkins RW, Kwok CT, et al.: Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5'UTR. Cancer Cell 20 (2): 200-13, 2011.
- Goel A, Nguyen TP, Leung HC, et al.: De novo constitutional MLH1 epimutations confer early-onset colorectal cancer in two new sporadic Lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. Int J Cancer 128 (4): 869-78, 2011.
- Hitchins MP, Wong JJ, Suthers G, et al.: Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med 356 (7): 697-705, 2007.
- Hampel H, Frankel WL, Martin E, et al.: Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 352 (18): 1851-60, 2005.
- Ladabaum U, Wang G, Terdiman J, et al.: Strategies to identify the Lynch syndrome among patients with colorectal cancer: a cost-effectiveness analysis. Ann Intern Med 155 (2): 69-79, 2011.
- Piñol V, Castells A, Andreu M, et al.: Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA 293 (16): 1986-94, 2005.
- Baudhuin LM, Burgart LJ, Leontovich O, et al.: Use of microsatellite instability and immunohistochemistry testing for the identification of individuals at risk for Lynch syndrome. Fam Cancer 4 (3): 255-65, 2005.
- Lagerstedt Robinson K, Liu T, Vandrovcova J, et al.: Lynch syndrome (hereditary nonpolyposis colorectal cancer) diagnostics. J Natl Cancer Inst 99 (4): 291-9, 2007.
- Schofield L, Watson N, Grieu F, et al.: Population-based detection of Lynch syndrome in young colorectal cancer patients using microsatellite instability as the initial test. Int J Cancer 124 (5): 1097-102, 2009.
- Mills AM, Liou S, Ford JM, et al.: Lynch syndrome screening should be considered for all patients with newly diagnosed endometrial cancer. Am J Surg Pathol 38 (11): 1501-9, 2014.
- Giardiello FM, Allen JI, Axilbund JE, et al.: Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task Force on colorectal cancer. Am J Gastroenterol 109 (8): 1159-79, 2014.
- Rubenstein JH, Enns R, Heidelbaugh J, et al.: American Gastroenterological Association Institute Guideline on the Diagnosis and Management of Lynch Syndrome. Gastroenterology 149 (3): 777-82; quiz e16-7, 2015.
- Committee on Practice Bulletins-Gynecology, Society of Gynecologic Oncology: ACOG Practice Bulletin No. 147: Lynch syndrome. Obstet Gynecol 124 (5): 1042-54, 2014.
- Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group: Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med 11 (1): 35-41, 2009.
- Palomaki GE, McClain MR, Melillo S, et al.: EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med 11 (1): 42-65, 2009.
- Moreira L, Balaguer F, Lindor N, et al.: Identification of Lynch syndrome among patients with colorectal cancer. JAMA 308 (15): 1555-65, 2012.
- Boland CR, Shike M: Report from the Jerusalem workshop on Lynch syndrome-hereditary nonpolyposis colorectal cancer. Gastroenterology 138 (7): 2197.e1-7, 2010.
- Crucianelli F, Tricarico R, Turchetti D, et al.: MLH1 constitutional and somatic methylation in patients with MLH1 negative tumors fulfilling the revised Bethesda criteria. Epigenetics 9 (10): 1431-8, 2014.
- Leenen CH, Goverde A, de Bekker-Grob EW, et al.: Cost-effectiveness of routine screening for Lynch syndrome in colorectal cancer patients up to 70 years of age. Genet Med 18 (10): 966-73, 2016.
- Hampel H, Pearlman R, Beightol M, et al.: Assessment of Tumor Sequencing as a Replacement for Lynch Syndrome Screening and Current Molecular Tests for Patients With Colorectal Cancer. JAMA Oncol 4 (6): 806-813, 2018.
- Li D, Hoodfar E, Jiang SF, et al.: Comparison of Universal Versus Age-Restricted Screening of Colorectal Tumors for Lynch Syndrome Using Mismatch Repair Immunohistochemistry: A Cohort Study. Ann Intern Med 171 (1): 19-26, 2019.
- Barzi A, Sadeghi S, Kattan MW, et al.: Comparative effectiveness of screening strategies for Lynch syndrome. J Natl Cancer Inst 107 (4): , 2015.
- Stoffel EM, Mangu PB, Gruber SB, et al.: Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology Clinical Practice Guidelines. J Clin Oncol 33 (2): 209-17, 2015.
- Goverde A, Spaander MC, van Doorn HC, et al.: Cost-effectiveness of routine screening for Lynch syndrome in endometrial cancer patients up to 70years of age. Gynecol Oncol 143 (3): 453-459, 2016.
- Cohen SA: Current Lynch syndrome tumor screening practices: a survey of genetic counselors. J Genet Couns 23 (1): 38-47, 2014.
- Beamer LC, Grant ML, Espenschied CR, et al.: Reflex immunohistochemistry and microsatellite instability testing of colorectal tumors for Lynch syndrome among US cancer programs and follow-up of abnormal results. J Clin Oncol 30 (10): 1058-63, 2012.
- Dineen S, Lynch PM, Rodriguez-Bigas MA, et al.: A Prospective Six Sigma Quality Improvement Trial to Optimize Universal Screening for Genetic Syndrome Among Patients With Young-Onset Colorectal Cancer. J Natl Compr Canc Netw 13 (7): 865-72, 2015.
- Heald B, Plesec T, Liu X, et al.: Implementation of universal microsatellite instability and immunohistochemistry screening for diagnosing lynch syndrome in a large academic medical center. J Clin Oncol 31 (10): 1336-40, 2013.
- Cragun D, DeBate RD, Vadaparampil ST, et al.: Comparing universal Lynch syndrome tumor-screening programs to evaluate associations between implementation strategies and patient follow-through. Genet Med 16 (10): 773-82, 2014.
- Ward RL, Hicks S, Hawkins NJ: Population-based molecular screening for Lynch syndrome: implications for personalized medicine. J Clin Oncol 31 (20): 2554-62, 2013.
- Hampel H, Frankel W, Panescu J, et al.: Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res 66 (15): 7810-7, 2006.
- Hampel H, Panescu J, Lockman J, et al.: Comment on: Screening for Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer) among Endometrial Cancer Patients. Cancer Res 67 (19): 9603, 2007.
- Watkins JC, Yang EJ, Muto MG, et al.: Universal Screening for Mismatch-Repair Deficiency in Endometrial Cancers to Identify Patients With Lynch Syndrome and Lynch-like Syndrome. Int J Gynecol Pathol 36 (2): 115-127, 2017.
- Adar T, Rodgers LH, Shannon KM, et al.: Universal screening of both endometrial and colon cancers increases the detection of Lynch syndrome. Cancer 124 (15): 3145-3153, 2018.
- Kwon JS, Scott JL, Gilks CB, et al.: Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 29 (16): 2247-52, 2011.
- Latham A, Srinivasan P, Kemel Y, et al.: Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J Clin Oncol 37 (4): 286-295, 2019.
- Middha S, Zhang L, Nafa K, et al.: Reliable Pan-Cancer Microsatellite Instability Assessment by Using Targeted Next-Generation Sequencing Data. JCO Precis Oncol 2017: , 2017.
- Latham A, Shia J, Patel Z, et al.: Characterization and Clinical Outcomes of DNA Mismatch Repair-deficient Small Bowel Adenocarcinoma. Clin Cancer Res 27 (5): 1429-1437, 2021.
- Pearlman R, Frankel WL, Swanson B, et al.: Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol 3 (4): 464-471, 2017.
- Yurgelun MB, Allen B, Kaldate RR, et al.: Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology 149 (3): 604-13.e20, 2015.
- You YN, Borras E, Chang K, et al.: Detection of Pathogenic Germline Variants Among Patients With Advanced Colorectal Cancer Undergoing Tumor Genomic Profiling for Precision Medicine. Dis Colon Rectum 62 (4): 429-437, 2019.
- Yurgelun MB, Kulke MH, Fuchs CS, et al.: Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer. J Clin Oncol 35 (10): 1086-1095, 2017.
- Espenschied CR, LaDuca H, Li S, et al.: Multigene Panel Testing Provides a New Perspective on Lynch Syndrome. J Clin Oncol 35 (22): 2568-2575, 2017.
- Roberts ME, Jackson SA, Susswein LR, et al.: MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet Med 20 (10): 1167-1174, 2018.
- Neumann PJ, Cohen JT, Weinstein MC: Updating cost-effectiveness--the curious resilience of the $50,000-per-QALY threshold. N Engl J Med 371 (9): 796-7, 2014.
- Gallego CJ, Shirts BH, Bennette CS, et al.: Next-Generation Sequencing Panels for the Diagnosis of Colorectal Cancer and Polyposis Syndromes: A Cost-Effectiveness Analysis. J Clin Oncol 33 (18): 2084-91, 2015.
- Bronner CE, Baker SM, Morrison PT, et al.: Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 368 (6468): 258-61, 1994.
- Papadopoulos N, Nicolaides NC, Wei YF, et al.: Mutation of a mutL homolog in hereditary colon cancer. Science 263 (5153): 1625-9, 1994.
- Fishel R, Lescoe MK, Rao MR, et al.: The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 75 (5): 1027-38, 1993.
- Leach FS, Nicolaides NC, Papadopoulos N, et al.: Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 75 (6): 1215-25, 1993.
- Miyaki M, Konishi M, Tanaka K, et al.: Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet 17 (3): 271-2, 1997.
- Nicolaides NC, Papadopoulos N, Liu B, et al.: Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 371 (6492): 75-80, 1994.
- Worthley DL, Walsh MD, Barker M, et al.: Familial mutations in PMS2 can cause autosomal dominant hereditary nonpolyposis colorectal cancer. Gastroenterology 128 (5): 1431-6, 2005.
- Peltomäki P, Aaltonen LA, Sistonen P, et al.: Genetic mapping of a locus predisposing to human colorectal cancer. Science 260 (5109): 810-2, 1993.
- Lindblom A, Tannergård P, Werelius B, et al.: Genetic mapping of a second locus predisposing to hereditary non-polyposis colon cancer. Nat Genet 5 (3): 279-82, 1993.
- Ligtenberg MJ, Kuiper RP, Chan TL, et al.: Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3' exons of TACSTD1. Nat Genet 41 (1): 112-7, 2009.
- Kuiper RP, Vissers LE, Venkatachalam R, et al.: Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum Mutat 32 (4): 407-14, 2011.
- Vasen HF: Clinical description of the Lynch syndrome [hereditary nonpolyposis colorectal cancer (HNPCC)]. Fam Cancer 4 (3): 219-25, 2005.
- National Cancer Institute: SEER Stat Fact Sheets: Colorectal Cancer. Bethesda, Md: National Institutes of Health.
Available online . Last accessed September 5, 2024. - Hampel H, Stephens JA, Pukkala E, et al.: Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology 129 (2): 415-21, 2005.
- Win AK, Jenkins MA, Dowty JG, et al.: Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer Epidemiol Biomarkers Prev 26 (3): 404-412, 2017.
- Marra G, Boland CR: Hereditary nonpolyposis colorectal cancer: the syndrome, the genes, and historical perspectives. J Natl Cancer Inst 87 (15): 1114-25, 1995.
- Peltomäki P, Vasen HF: Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 113 (4): 1146-58, 1997.
- Plazzer JP, Sijmons RH, Woods MO, et al.: The InSiGHT database: utilizing 100 years of insights into Lynch syndrome. Fam Cancer 12 (2): 175-80, 2013.
- Peltomäki P: Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol 21 (6): 1174-9, 2003.
- Vasen HF, Stormorken A, Menko FH, et al.: MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol 19 (20): 4074-80, 2001.
- Ryan NAJ, Morris J, Green K, et al.: Association of Mismatch Repair Mutation With Age at Cancer Onset in Lynch Syndrome: Implications for Stratified Surveillance Strategies. JAMA Oncol 3 (12): 1702-1706, 2017.
- Quehenberger F, Vasen HF, van Houwelingen HC: Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet 42 (6): 491-6, 2005.
- Lin KM, Shashidharan M, Thorson AG, et al.: Cumulative incidence of colorectal and extracolonic cancers in MLH1 and MSH2 mutation carriers of hereditary nonpolyposis colorectal cancer. J Gastrointest Surg 2 (1): 67-71, 1998 Jan-Feb.
- Plaschke J, Engel C, Krüger S, et al.: Lower incidence of colorectal cancer and later age of disease onset in 27 families with pathogenic MSH6 germline mutations compared with families with MLH1 or MSH2 mutations: the German Hereditary Nonpolyposis Colorectal Cancer Consortium. J Clin Oncol 22 (22): 4486-94, 2004.
- Berends MJ, Wu Y, Sijmons RH, et al.: Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am J Hum Genet 70 (1): 26-37, 2002.
- Ramsoekh D, Wagner A, van Leerdam ME, et al.: A high incidence of MSH6 mutations in Amsterdam criteria II-negative families tested in a diagnostic setting. Gut 57 (11): 1539-44, 2008.
- Peltomäki P, Vasen H: Mutations associated with HNPCC predisposition -- Update of ICG-HNPCC/INSiGHT mutation database. Dis Markers 20 (4-5): 269-76, 2004.
- Kolodner RD, Tytell JD, Schmeits JL, et al.: Germ-line msh6 mutations in colorectal cancer families. Cancer Res 59 (20): 5068-74, 1999.
- Peterlongo P, Nafa K, Lerman GS, et al.: MSH6 germline mutations are rare in colorectal cancer families. Int J Cancer 107 (4): 571-9, 2003.
- Hendriks YM, Wagner A, Morreau H, et al.: Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology 127 (1): 17-25, 2004.
- Goodenberger ML, Thomas BC, Riegert-Johnson D, et al.: PMS2 monoallelic mutation carriers: the known unknown. Genet Med 18 (1): 13-9, 2016.
- Hendriks YM, Jagmohan-Changur S, van der Klift HM, et al.: Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Gastroenterology 130 (2): 312-22, 2006.
- Truninger K, Menigatti M, Luz J, et al.: Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology 128 (5): 1160-71, 2005.
- Senter L, Clendenning M, Sotamaa K, et al.: The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 135 (2): 419-28, 2008.
- ten Broeke SW, Brohet RM, Tops CM, et al.: Lynch syndrome caused by germline PMS2 mutations: delineating the cancer risk. J Clin Oncol 33 (4): 319-25, 2015.
- Ten Broeke SW, van der Klift HM, Tops CMJ, et al.: Cancer Risks for PMS2-Associated Lynch Syndrome. J Clin Oncol 36 (29): 2961-2968, 2018.
- Møller P, Seppälä T, Bernstein I, et al.: Incidence of and survival after subsequent cancers in carriers of pathogenic MMR variants with previous cancer: a report from the prospective Lynch syndrome database. Gut 66 (9): 1657-1664, 2017.
- Møller P, Seppälä TT, Bernstein I, et al.: Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut 67 (7): 1306-1316, 2018.
- Engel C, Vasen HF, Seppälä T, et al.: No Difference in Colorectal Cancer Incidence or Stage at Detection by Colonoscopy Among 3 Countries With Different Lynch Syndrome Surveillance Policies. Gastroenterology 155 (5): 1400-1409.e2, 2018.
- Ligtenberg MJ, Kuiper RP, Geurts van Kessel A, et al.: EPCAM deletion carriers constitute a unique subgroup of Lynch syndrome patients. Fam Cancer 12 (2): 169-74, 2013.
- Kempers MJ, Kuiper RP, Ockeloen CW, et al.: Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. Lancet Oncol 12 (1): 49-55, 2011.
- Lynch HT, Riegert-Johnson DL, Snyder C, et al.: Lynch syndrome-associated extracolonic tumors are rare in two extended families with the same EPCAM deletion. Am J Gastroenterol 106 (10): 1829-36, 2011.
- Dowty JG, Win AK, Buchanan DD, et al.: Cancer risks for MLH1 and MSH2 mutation carriers. Hum Mutat 34 (3): 490-7, 2013.
- International Mismatch Repair Consortium: Variation in the risk of colorectal cancer in families with Lynch syndrome: a retrospective cohort study. Lancet Oncol 22 (7): 1014-1022, 2021.
- Desai DC, Lockman JC, Chadwick RB, et al.: Recurrent germline mutation in MSH2 arises frequently de novo. J Med Genet 37 (9): 646-52, 2000.
- Nyström-Lahti M, Kristo P, Nicolaides NC, et al.: Founding mutations and Alu-mediated recombination in hereditary colon cancer. Nat Med 1 (11): 1203-6, 1995.
- Moisio AL, Sistonen P, Weissenbach J, et al.: Age and origin of two common MLH1 mutations predisposing to hereditary colon cancer. Am J Hum Genet 59 (6): 1243-51, 1996.
- Caluseriu O, Di Gregorio C, Lucci-Cordisco E, et al.: A founder MLH1 mutation in families from the districts of Modena and Reggio-Emilia in northern Italy with hereditary non-polyposis colorectal cancer associated with protein elongation and instability. J Med Genet 41 (3): e34, 2004.
- Chan TL, Chan YW, Ho JW, et al.: MSH2 c.1452-1455delAATG is a founder mutation and an important cause of hereditary nonpolyposis colorectal cancer in the southern Chinese population. Am J Hum Genet 74 (5): 1035-42, 2004.
- Clendenning M, Baze ME, Sun S, et al.: Origins and prevalence of the American Founder Mutation of MSH2. Cancer Res 68 (7): 2145-53, 2008.
- Dominguez-Valentin M, Nilbert M, Wernhoff P, et al.: Mutation spectrum in South American Lynch syndrome families. Hered Cancer Clin Pract 11 (1): 18, 2013.
- Cruz-Correa M, Diaz-Algorri Y, Pérez-Mayoral J, et al.: Clinical characterization and mutation spectrum in Caribbean Hispanic families with Lynch syndrome. Fam Cancer 14 (3): 415-25, 2015.
- Sunga AY, Ricker C, Espenschied CR, et al.: Spectrum of mismatch repair gene mutations and clinical presentation of Hispanic individuals with Lynch syndrome. Cancer Genet 212-213: 1-7, 2017.
- Ricker CN, Hanna DL, Peng C, et al.: DNA mismatch repair deficiency and hereditary syndromes in Latino patients with colorectal cancer. Cancer 123 (19): 3732-3743, 2017.
- Guindalini RS, Win AK, Gulden C, et al.: Mutation spectrum and risk of colorectal cancer in African American families with Lynch syndrome. Gastroenterology 149 (6): 1446-53, 2015.
- Parry S, Win AK, Parry B, et al.: Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 60 (7): 950-7, 2011.
- Signoroni S, Piozzi GN, Ricci MT, et al.: Risk factors for metachronous colorectal cancer in Lynch syndrome patients: a registry-based observational mono-institutional study cohort. Int J Clin Oncol 25 (9): 1644-1652, 2020.
- Watson P, Vasen HF, Mecklin JP, et al.: The risk of endometrial cancer in hereditary nonpolyposis colorectal cancer. Am J Med 96 (6): 516-20, 1994.
- Watson P, Lynch HT: Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer 71 (3): 677-85, 1993.
- Voskuil DW, Vasen HF, Kampman E, et al.: Colorectal cancer risk in HNPCC families: development during lifetime and in successive generations. National Collaborative Group on HNPCC. Int J Cancer 72 (2): 205-9, 1997.
- Heinimann K, Müller H, Weber W, et al.: Disease expression in Swiss hereditary non-polyposis colorectal cancer (HNPCC) kindreds. Int J Cancer 74 (3): 281-5, 1997.
- Lu KH, Dinh M, Kohlmann W, et al.: Gynecologic cancer as a "sentinel cancer" for women with hereditary nonpolyposis colorectal cancer syndrome. Obstet Gynecol 105 (3): 569-74, 2005.
- Tan YY, McGaughran J, Ferguson K, et al.: Improving identification of lynch syndrome patients: a comparison of research data with clinical records. Int J Cancer 132 (12): 2876-83, 2013.
- Win AK, Young JP, Lindor NM, et al.: Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol 30 (9): 958-64, 2012.
- Win AK, Lindor NM, Winship I, et al.: Risks of colorectal and other cancers after endometrial cancer for women with Lynch syndrome. J Natl Cancer Inst 105 (4): 274-9, 2013.
- Broaddus RR, Lynch HT, Chen LM, et al.: Pathologic features of endometrial carcinoma associated with HNPCC: a comparison with sporadic endometrial carcinoma. Cancer 106 (1): 87-94, 2006.
- Vasen HF, Offerhaus GJ, den Hartog Jager FC, et al.: The tumour spectrum in hereditary non-polyposis colorectal cancer: a study of 24 kindreds in the Netherlands. Int J Cancer 46 (1): 31-4, 1990.
- Aarnio M, Mecklin JP, Aaltonen LA, et al.: Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer 64 (6): 430-3, 1995.
- Ketabi Z, Bartuma K, Bernstein I, et al.: Ovarian cancer linked to Lynch syndrome typically presents as early-onset, non-serous epithelial tumors. Gynecol Oncol 121 (3): 462-5, 2011.
- Borelli I, Casalis Cavalchini GC, Del Peschio S, et al.: A founder MLH1 mutation in Lynch syndrome families from Piedmont, Italy, is associated with an increased risk of pancreatic tumours and diverse immunohistochemical patterns. Fam Cancer 13 (3): 401-13, 2014.
- Raymond VM, Mukherjee B, Wang F, et al.: Elevated risk of prostate cancer among men with Lynch syndrome. J Clin Oncol 31 (14): 1713-8, 2013.
- Raymond VM, Everett JN, Furtado LV, et al.: Adrenocortical carcinoma is a lynch syndrome-associated cancer. J Clin Oncol 31 (24): 3012-8, 2013.
- Haraldsdottir S, Hampel H, Wei L, et al.: Prostate cancer incidence in males with Lynch syndrome. Genet Med 16 (7): 553-7, 2014.
- Bapat B, Xia L, Madlensky L, et al.: The genetic basis of Muir-Torre syndrome includes the hMLH1 locus. Am J Hum Genet 59 (3): 736-9, 1996.
- Lynch HT, Lynch PM, Pester J, et al.: The cancer family syndrome. Rare cutaneous phenotypic linkage of Torre's syndrome. Arch Intern Med 141 (5): 607-11, 1981.
- Suspiro A, Fidalgo P, Cravo M, et al.: The Muir-Torre syndrome: a rare variant of hereditary nonpolyposis colorectal cancer associated with hMSH2 mutation. Am J Gastroenterol 93 (9): 1572-4, 1998.
- Kruse R, Rütten A, Lamberti C, et al.: Muir-Torre phenotype has a frequency of DNA mismatch-repair-gene mutations similar to that in hereditary nonpolyposis colorectal cancer families defined by the Amsterdam criteria. Am J Hum Genet 63 (1): 63-70, 1998.
- South CD, Hampel H, Comeras I, et al.: The frequency of Muir-Torre syndrome among Lynch syndrome families. J Natl Cancer Inst 100 (4): 277-81, 2008.
- Kacerovska D, Cerna K, Martinek P, et al.: MSH6 mutation in a family affected by Muir-Torre syndrome. Am J Dermatopathol 34 (6): 648-52, 2012.
- Tavakkol Z, Keller JJ, Furmanczyk PS, et al.: Germline mutation in MSH6 associated with multiple malignant neoplasms in a patient With Muir-Torre syndrome. J Clin Oncol 30 (22): e195-8, 2012.
- Murphy HR, Armstrong R, Cairns D, et al.: Muir-Torre Syndrome: expanding the genotype and phenotype--a further family with a MSH6 mutation. Fam Cancer 7 (3): 255-7, 2008.
- Arnold A, Payne S, Fisher S, et al.: An individual with Muir-Torre syndrome found to have a pathogenic MSH6 gene mutation. Fam Cancer 6 (3): 317-21, 2007.
- Mangold E, Rahner N, Friedrichs N, et al.: MSH6 mutation in Muir-Torre syndrome: could this be a rare finding? Br J Dermatol 156 (1): 158-62, 2007.
- Kastrinos F, Stoffel EM, Balmaña J, et al.: Phenotype comparison of MLH1 and MSH2 mutation carriers in a cohort of 1,914 individuals undergoing clinical genetic testing in the United States. Cancer Epidemiol Biomarkers Prev 17 (8): 2044-51, 2008.
- Lamba AR, Moore AY, Moore T, et al.: Defective DNA mismatch repair activity is common in sebaceous neoplasms, and may be an ineffective approach to screen for Lynch syndrome. Fam Cancer 14 (2): 259-64, 2015.
- Syngal S, Brand RE, Church JM, et al.: ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 110 (2): 223-62; quiz 263, 2015.
- Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review (CSR) 1975-2014. National Cancer Institute.
Also available online . Last accessed May 24, 2024. - Jenkins MA, Baglietto L, Dowty JG, et al.: Cancer risks for mismatch repair gene mutation carriers: a population-based early onset case-family study. Clin Gastroenterol Hepatol 4 (4): 489-98, 2006.
- Barrow E, Robinson L, Alduaij W, et al.: Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet 75 (2): 141-9, 2009.
- Engel C, Loeffler M, Steinke V, et al.: Risks of less common cancers in proven mutation carriers with lynch syndrome. J Clin Oncol 30 (35): 4409-15, 2012.
- Watson P, Vasen HF, Mecklin JP, et al.: The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer 123 (2): 444-9, 2008.
- Capelle LG, Van Grieken NC, Lingsma HF, et al.: Risk and epidemiological time trends of gastric cancer in Lynch syndrome carriers in the Netherlands. Gastroenterology 138 (2): 487-92, 2010.
- Aarnio M, Sankila R, Pukkala E, et al.: Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 81 (2): 214-8, 1999.
- van der Post RS, Kiemeney LA, Ligtenberg MJ, et al.: Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet 47 (7): 464-70, 2010.
- Cloyd JM, Chun YS, Ikoma N, et al.: Clinical and Genetic Implications of DNA Mismatch Repair Deficiency in Biliary Tract Cancers Associated with Lynch Syndrome. J Gastrointest Cancer 49 (1): 93-96, 2018.
- Yang KY, Caughey AB, Little SE, et al.: A cost-effectiveness analysis of prophylactic surgery versus gynecologic surveillance for women from hereditary non-polyposis colorectal cancer (HNPCC) Families. Fam Cancer 10 (3): 535-43, 2011.
- Ponti G, Losi L, Pedroni M, et al.: Value of MLH1 and MSH2 mutations in the appearance of Muir-Torre syndrome phenotype in HNPCC patients presenting sebaceous gland tumors or keratoacanthomas. J Invest Dermatol 126 (10): 2302-7, 2006.
- Schwartz RA, Torre DP: The Muir-Torre syndrome: a 25-year retrospect. J Am Acad Dermatol 33 (1): 90-104, 1995.
- Dunlop MG, Farrington SM, Carothers AD, et al.: Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet 6 (1): 105-10, 1997.
- Kastrinos F, Mukherjee B, Tayob N, et al.: Risk of pancreatic cancer in families with Lynch syndrome. JAMA 302 (16): 1790-5, 2009.
- Jensen UB, Sunde L, Timshel S, et al.: Mismatch repair defective breast cancer in the hereditary nonpolyposis colorectal cancer syndrome. Breast Cancer Res Treat 120 (3): 777-82, 2010.
- Shanley S, Fung C, Milliken J, et al.: Breast cancer immunohistochemistry can be useful in triage of some HNPCC families. Fam Cancer 8 (3): 251-5, 2009.
- Walsh MD, Buchanan DD, Cummings MC, et al.: Lynch syndrome-associated breast cancers: clinicopathologic characteristics of a case series from the colon cancer family registry. Clin Cancer Res 16 (7): 2214-24, 2010.
- Buerki N, Gautier L, Kovac M, et al.: Evidence for breast cancer as an integral part of Lynch syndrome. Genes Chromosomes Cancer 51 (1): 83-91, 2012.
- Win AK, Lindor NM, Young JP, et al.: Risks of primary extracolonic cancers following colorectal cancer in lynch syndrome. J Natl Cancer Inst 104 (18): 1363-72, 2012.
- Harkness EF, Barrow E, Newton K, et al.: Lynch syndrome caused by MLH1 mutations is associated with an increased risk of breast cancer: a cohort study. J Med Genet 52 (8): 553-6, 2015.
- Win AK, Lindor NM, Jenkins MA: Risk of breast cancer in Lynch syndrome: a systematic review. Breast Cancer Res 15 (2): R27, 2013.
- Goldberg M, Bell K, Aronson M, et al.: Association between the Lynch syndrome gene MSH2 and breast cancer susceptibility in a Canadian familial cancer registry. J Med Genet 54 (11): 742-746, 2017.
- Lu HM, Li S, Black MH, et al.: Association of Breast and Ovarian Cancers With Predisposition Genes Identified by Large-Scale Sequencing. JAMA Oncol 5 (1): 51-57, 2019.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 1.2021. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2020.
Available online with free registration. Last accessed May 24, 2024. - Ryan S, Jenkins MA, Win AK: Risk of prostate cancer in Lynch syndrome: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev 23 (3): 437-49, 2014.
- Pritchard CC, Mateo J, Walsh MF, et al.: Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med 375 (5): 443-53, 2016.
- De Jong AE, Morreau H, Van Puijenbroek M, et al.: The role of mismatch repair gene defects in the development of adenomas in patients with HNPCC. Gastroenterology 126 (1): 42-8, 2004.
- Johnson PM, Gallinger S, McLeod RS: Surveillance colonoscopy in individuals at risk for hereditary nonpolyposis colorectal cancer: an evidence-based review. Dis Colon Rectum 49 (1): 80-93; discussion 94-5, 2006.
- Lindor NM, Petersen GM, Hadley DW, et al.: Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 296 (12): 1507-17, 2006.
- Reitmair AH, Cai JC, Bjerknes M, et al.: MSH2 deficiency contributes to accelerated APC-mediated intestinal tumorigenesis. Cancer Res 56 (13): 2922-6, 1996.
- Järvinen HJ, Aarnio M, Mustonen H, et al.: Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 118 (5): 829-34, 2000.
- Järvinen HJ, Mecklin JP, Sistonen P: Screening reduces colorectal cancer rate in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 108 (5): 1405-11, 1995.
- Engel C, Rahner N, Schulmann K, et al.: Efficacy of annual colonoscopic surveillance in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol 8 (2): 174-82, 2010.
- Vasen HF, Abdirahman M, Brohet R, et al.: One to 2-year surveillance intervals reduce risk of colorectal cancer in families with Lynch syndrome. Gastroenterology 138 (7): 2300-6, 2010.
- Järvinen HJ, Renkonen-Sinisalo L, Aktán-Collán K, et al.: Ten years after mutation testing for Lynch syndrome: cancer incidence and outcome in mutation-positive and mutation-negative family members. J Clin Oncol 27 (28): 4793-7, 2009.
- Lindberg LJ, Wegen-Haitsma W, Ladelund S, et al.: Risk of multiple colorectal cancer development depends on age and subgroup in individuals with hereditary predisposition. Fam Cancer 18 (2): 183-191, 2019.
- Hurlstone DP, Karajeh M, Cross SS, et al.: The role of high-magnification-chromoscopic colonoscopy in hereditary nonpolyposis colorectal cancer screening: a prospective "back-to-back" endoscopic study. Am J Gastroenterol 100 (10): 2167-73, 2005.
- Lecomte T, Cellier C, Meatchi T, et al.: Chromoendoscopic colonoscopy for detecting preneoplastic lesions in hereditary nonpolyposis colorectal cancer syndrome. Clin Gastroenterol Hepatol 3 (9): 897-902, 2005.
- Müller A, Beckmann C, Westphal G, et al.: Prevalence of the mismatch-repair-deficient phenotype in colonic adenomas arising in HNPCC patients: results of a 5-year follow-up study. Int J Colorectal Dis 21 (7): 632-41, 2006.
- Yurgelun MB, Goel A, Hornick JL, et al.: Microsatellite instability and DNA mismatch repair protein deficiency in Lynch syndrome colorectal polyps. Cancer Prev Res (Phila) 5 (4): 574-82, 2012.
- Monahan KJ, Bradshaw N, Dolwani S, et al.: Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut 69 (3): 411-444, 2020.
- Seppälä TT, Latchford A, Negoi I, et al.: European guidelines from the EHTG and ESCP for Lynch syndrome: an updated third edition of the Mallorca guidelines based on gene and gender. Br J Surg 108 (5): 484-498, 2021.
- Stjepanovic N, Moreira L, Carneiro F, et al.: Hereditary gastrointestinal cancers: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann Oncol 30 (10): 1558-1571, 2019.
- Hendriks YM, de Jong AE, Morreau H, et al.: Diagnostic approach and management of Lynch syndrome (hereditary nonpolyposis colorectal carcinoma): a guide for clinicians. CA Cancer J Clin 56 (4): 213-25, 2006 Jul-Aug.
- Dove-Edwin I, Boks D, Goff S, et al.: The outcome of endometrial carcinoma surveillance by ultrasound scan in women at risk of hereditary nonpolyposis colorectal carcinoma and familial colorectal carcinoma. Cancer 94 (6): 1708-12, 2002.
- Rijcken FE, Mourits MJ, Kleibeuker JH, et al.: Gynecologic screening in hereditary nonpolyposis colorectal cancer. Gynecol Oncol 91 (1): 74-80, 2003.
- Renkonen-Sinisalo L, Bützow R, Leminen A, et al.: Surveillance for endometrial cancer in hereditary nonpolyposis colorectal cancer syndrome. Int J Cancer 120 (4): 821-4, 2007.
- Yang K, Allen B, Conrad P, et al.: Awareness of gynecologic surveillance in women from hereditary non-polyposis colorectal cancer families. Fam Cancer 5 (4): 405-9, 2006.
- Collins VR, Meiser B, Ukoumunne OC, et al.: The impact of predictive genetic testing for hereditary nonpolyposis colorectal cancer: three years after testing. Genet Med 9 (5): 290-7, 2007.
- Schmeler KM, Lynch HT, Chen LM, et al.: Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med 354 (3): 261-9, 2006.
- Kwon JS, Sun CC, Peterson SK, et al.: Cost-effectiveness analysis of prevention strategies for gynecologic cancers in Lynch syndrome. Cancer 113 (2): 326-35, 2008.
- Aarnio M, Salovaara R, Aaltonen LA, et al.: Features of gastric cancer in hereditary non-polyposis colorectal cancer syndrome. Int J Cancer 74 (5): 551-5, 1997.
- Farha N, Hrabe J, Sleiman J, et al.: Clinically actionable findings on surveillance EGD in asymptomatic patients with Lynch syndrome. Gastrointest Endosc 95 (1): 105-114, 2022.
- Canto MI, Harinck F, Hruban RH, et al.: International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 62 (3): 339-47, 2013.
- Burn J, Gerdes AM, Macrae F, et al.: Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 378 (9809): 2081-7, 2011.
- Burn J, Bishop DT, Mecklin JP, et al.: Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N Engl J Med 359 (24): 2567-78, 2008.
- Burn J, Sheth H, Elliott F, et al.: Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: a double-blind, randomised, placebo-controlled trial. Lancet 395 (10240): 1855-1863, 2020.
- Mathers JC, Elliott F, Macrae F, et al.: Cancer Prevention with Resistant Starch in Lynch Syndrome Patients in the CAPP2-Randomized Placebo Controlled Trial: Planned 10-Year Follow-up. Cancer Prev Res (Phila) 15 (9): 623-634, 2022.
- Yurgelun MB, Chan AT: Aspirin for Lynch syndrome: a legacy of prevention. Lancet 395 (10240): 1817-1818, 2020.
- Movahedi M, Bishop DT, Macrae F, et al.: Obesity, Aspirin, and Risk of Colorectal Cancer in Carriers of Hereditary Colorectal Cancer: A Prospective Investigation in the CAPP2 Study. J Clin Oncol 33 (31): 3591-7, 2015.
- Burn J, Mathers JC, Bishop DT: Chemoprevention in Lynch syndrome. Fam Cancer 12 (4): 707-18, 2013.
- Ait Ouakrim D, Dashti SG, Chau R, et al.: Aspirin, Ibuprofen, and the Risk of Colorectal Cancer in Lynch Syndrome. J Natl Cancer Inst 107 (9): , 2015.
- Reyes-Uribe L, Wu W, Gelincik O, et al.: Naproxen chemoprevention promotes immune activation in Lynch syndrome colorectal mucosa. Gut 70 (3): 555-566, 2021.
- de Vos tot Nederveen Cappel WH, Buskens E, van Duijvendijk P, et al.: Decision analysis in the surgical treatment of colorectal cancer due to a mismatch repair gene defect. Gut 52 (12): 1752-5, 2003.
- Natarajan N, Watson P, Silva-Lopez E, et al.: Comparison of extended colectomy and limited resection in patients with Lynch syndrome. Dis Colon Rectum 53 (1): 77-82, 2010.
- Maeda T, Cannom RR, Beart RW, et al.: Decision model of segmental compared with total abdominal colectomy for colon cancer in hereditary nonpolyposis colorectal cancer. J Clin Oncol 28 (7): 1175-80, 2010.
- Hiatt MJ, Casey MJ, Lynch HT, et al.: Efficacy of proximal colectomy for surgical management of right-sided first colorectal cancer in Lynch Syndrome mutation carriers. Am J Surg 216 (1): 99-105, 2018.
- Rodríguez-Bigas MA, Vasen HF, Pekka-Mecklin J, et al.: Rectal cancer risk in hereditary nonpolyposis colorectal cancer after abdominal colectomy. International Collaborative Group on HNPCC. Ann Surg 225 (2): 202-7, 1997.
- de Rosa N, Rodriguez-Bigas MA, Chang GJ, et al.: DNA Mismatch Repair Deficiency in Rectal Cancer: Benchmarking Its Impact on Prognosis, Neoadjuvant Response Prediction, and Clinical Cancer Genetics. J Clin Oncol 34 (25): 3039-46, 2016.
- Lee JS, Petrelli NJ, Rodriguez-Bigas MA: Rectal cancer in hereditary nonpolyposis colorectal cancer. Am J Surg 181 (3): 207-10, 2001.
- Kalady MF, Lipman J, McGannon E, et al.: Risk of colonic neoplasia after proctectomy for rectal cancer in hereditary nonpolyposis colorectal cancer. Ann Surg 255 (6): 1121-5, 2012.
- Olsen KØ, Juul S, Bülow S, et al.: Female fecundity before and after operation for familial adenomatous polyposis. Br J Surg 90 (2): 227-31, 2003.
- Nieuwenhuis MH, Douma KF, Bleiker EM, et al.: Female fertility after colorectal surgery for familial adenomatous polyposis: a nationwide cross-sectional study. Ann Surg 252 (2): 341-4, 2010.
- Guillem JG, Wood WC, Moley JF, et al.: ASCO/SSO review of current role of risk-reducing surgery in common hereditary cancer syndromes. J Clin Oncol 24 (28): 4642-60, 2006.
- Vasen HF, Blanco I, Aktan-Collan K, et al.: Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut 62 (6): 812-23, 2013.
- Rodriguez-Bigas MA, Möeslein G: Surgical treatment of hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome). Fam Cancer 12 (2): 295-300, 2013.
- Samowitz WS, Curtin K, Ma KN, et al.: Microsatellite instability in sporadic colon cancer is associated with an improved prognosis at the population level. Cancer Epidemiol Biomarkers Prev 10 (9): 917-23, 2001.
- Koopman M, Kortman GA, Mekenkamp L, et al.: Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer 100 (2): 266-73, 2009.
- Popat S, Hubner R, Houlston RS: Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 23 (3): 609-18, 2005.
- Hutchins G, Southward K, Handley K, et al.: Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 29 (10): 1261-70, 2011.
- Roth AD, Tejpar S, Delorenzi M, et al.: Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 28 (3): 466-74, 2010.
- Liu GC, Liu RY, Yan JP, et al.: The Heterogeneity Between Lynch-Associated and Sporadic MMR Deficiency in Colorectal Cancers. J Natl Cancer Inst 110 (9): 975-984, 2018.
- Boland CR, Goel A: Microsatellite instability in colorectal cancer. Gastroenterology 138 (6): 2073-2087.e3, 2010.
- Hawn MT, Umar A, Carethers JM, et al.: Evidence for a connection between the mismatch repair system and the G2 cell cycle checkpoint. Cancer Res 55 (17): 3721-5, 1995.
- Carethers JM, Hawn MT, Chauhan DP, et al.: Competency in mismatch repair prohibits clonal expansion of cancer cells treated with N-methyl-N'-nitro-N-nitrosoguanidine. J Clin Invest 98 (1): 199-206, 1996.
- Aebi S, Kurdi-Haidar B, Gordon R, et al.: Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res 56 (13): 3087-90, 1996.
- Carethers JM, Chauhan DP, Fink D, et al.: Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology 117 (1): 123-31, 1999.
- Elsaleh H, Joseph D, Grieu F, et al.: Association of tumour site and sex with survival benefit from adjuvant chemotherapy in colorectal cancer. Lancet 355 (9217): 1745-50, 2000.
- Ribic CM, Sargent DJ, Moore MJ, et al.: Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 349 (3): 247-57, 2003.
- Sinicrope FA, Foster NR, Thibodeau SN, et al.: DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J Natl Cancer Inst 103 (11): 863-75, 2011.
- Fink D, Nebel S, Aebi S, et al.: The role of DNA mismatch repair in platinum drug resistance. Cancer Res 56 (21): 4881-6, 1996.
- Tougeron D, Mouillet G, Trouilloud I, et al.: Efficacy of Adjuvant Chemotherapy in Colon Cancer With Microsatellite Instability: A Large Multicenter AGEO Study. J Natl Cancer Inst 108 (7): , 2016.
- Kim JE, Hong YS, Kim HJ, et al.: Microsatellite Instability was not Associated with Survival in Stage III Colon Cancer Treated with Adjuvant Chemotherapy of Oxaliplatin and Infusional 5-Fluorouracil and Leucovorin (FOLFOX). Ann Surg Oncol 24 (5): 1289-1294, 2017.
- Oh SY, Kim DY, Kim YB, et al.: Oncologic outcomes after adjuvant chemotherapy using FOLFOX in MSI-H sporadic stage III colon cancer. World J Surg 37 (10): 2497-503, 2013.
- Le DT, Uram JN, Wang H, et al.: PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 372 (26): 2509-20, 2015.
- Overman MJ, McDermott R, Leach JL, et al.: Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 18 (9): 1182-1191, 2017.
- André T, Shiu KK, Kim TW, et al.: Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med 383 (23): 2207-2218, 2020.
- Diaz LA, Shiu KK, Kim TW, et al.: Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): final analysis of a randomised, open-label, phase 3 study. Lancet Oncol 23 (5): 659-670, 2022.
- Overman MJ, Lonardi S, Wong KYM, et al.: Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol 36 (8): 773-779, 2018.
- Ludford K, Cohen R, Svrcek M, et al.: Pathological Tumor Response Following Immune Checkpoint Blockade for Deficient Mismatch Repair Advanced Colorectal Cancer. J Natl Cancer Inst 113 (2): 208-211, 2021.
- Zhou XP, Waite KA, Pilarski R, et al.: Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. Am J Hum Genet 73 (2): 404-11, 2003.
- Mester J, Eng C: When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet 163C (2): 114-21, 2013.
- Eng C: PTEN: one gene, many syndromes. Hum Mutat 22 (3): 183-98, 2003.
- Marsh DJ, Kum JB, Lunetta KL, et al.: PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet 8 (8): 1461-72, 1999.
- Pilarski R, Eng C: Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet 41 (5): 323-6, 2004.
- Eng C: PTEN Hamartoma Tumor Syndrome (PHTS). In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp.
Available online . Last accessed November 13, 2024. - Pilarski R, Burt R, Kohlman W, et al.: Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst 105 (21): 1607-16, 2013.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2.2022. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2022.
Available online with free registration. Last accessed May 24, 2024. - Hampel H, Bennett RL, Buchanan A, et al.: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 17 (1): 70-87, 2015.
- Ngeow J, Liu C, Zhou K, et al.: Detecting Germline PTEN Mutations Among At-Risk Patients With Cancer: An Age- and Sex-Specific Cost-Effectiveness Analysis. J Clin Oncol 33 (23): 2537-44, 2015.
- Tan MH, Mester JL, Ngeow J, et al.: Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 18 (2): 400-7, 2012.
- Bubien V, Bonnet F, Brouste V, et al.: High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet 50 (4): 255-63, 2013.
- Heald B, Mester J, Rybicki L, et al.: Frequent gastrointestinal polyps and colorectal adenocarcinomas in a prospective series of PTEN mutation carriers. Gastroenterology 139 (6): 1927-33, 2010.
- Peutz JL: Very remarkable case of familial polyposis of mucous membrane of intestinal tract and nasopharynx accompanied by peculiar pigmentations of skin and mucous membrane. Ned Tijdschr Geneeskd 10: 134-146, 1921.
- Jeghers H, McKusick VA, Katz KH: Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med 241 (26): 1031-6, 1949.
- Spigelman AD, Murday V, Phillips RK: Cancer and the Peutz-Jeghers syndrome. Gut 30 (11): 1588-90, 1989.
- Aretz S, Stienen D, Uhlhaas S, et al.: High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat 26 (6): 513-9, 2005.
- Hemminki A, Markie D, Tomlinson I, et al.: A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391 (6663): 184-7, 1998.
- Jenne DE, Reimann H, Nezu J, et al.: Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 18 (1): 38-43, 1998.
- Boudeau J, Kieloch A, Alessi DR, et al.: Functional analysis of LKB1/STK11 mutants and two aberrant isoforms found in Peutz-Jeghers Syndrome patients. Hum Mutat 21 (2): 172, 2003.
- Lim W, Hearle N, Shah B, et al.: Further observations on LKB1/STK11 status and cancer risk in Peutz-Jeghers syndrome. Br J Cancer 89 (2): 308-13, 2003.
- Giardiello FM, Brensinger JD, Tersmette AC, et al.: Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 119 (6): 1447-53, 2000.
- Lim W, Olschwang S, Keller JJ, et al.: Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology 126 (7): 1788-94, 2004.
- van Lier MG, Wagner A, Mathus-Vliegen EM, et al.: High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol 105 (6): 1258-64; author reply 1265, 2010.
- Srivatsa PJ, Keeney GL, Podratz KC: Disseminated cervical adenoma malignum and bilateral ovarian sex cord tumors with annular tubules associated with Peutz-Jeghers syndrome. Gynecol Oncol 53 (2): 256-64, 1994.
- Scully RE: Sex cord tumor with annular tubules a distinctive ovarian tumor of the Peutz-Jeghers syndrome. Cancer 25 (5): 1107-21, 1970.
- Westerman AM, Entius MM, de Baar E, et al.: Peutz-Jeghers syndrome: 78-year follow-up of the original family. Lancet 353 (9160): 1211-5, 1999.
- Mehenni H, Resta N, Park JG, et al.: Cancer risks in LKB1 germline mutation carriers. Gut 55 (7): 984-90, 2006.
- Gruber SB, Entius MM, Petersen GM, et al.: Pathogenesis of adenocarcinoma in Peutz-Jeghers syndrome. Cancer Res 58 (23): 5267-70, 1998.
- Wang ZJ, Ellis I, Zauber P, et al.: Allelic imbalance at the LKB1 (STK11) locus in tumours from patients with Peutz-Jeghers' syndrome provides evidence for a hamartoma-(adenoma)-carcinoma sequence. J Pathol 188 (1): 9-13, 1999.
- Miyoshi H, Nakau M, Ishikawa TO, et al.: Gastrointestinal hamartomatous polyposis in Lkb1 heterozygous knockout mice. Cancer Res 62 (8): 2261-6, 2002.
- Nakau M, Miyoshi H, Seldin MF, et al.: Hepatocellular carcinoma caused by loss of heterozygosity in Lkb1 gene knockout mice. Cancer Res 62 (16): 4549-53, 2002.
- Takeda H, Miyoshi H, Kojima Y, et al.: Accelerated onsets of gastric hamartomas and hepatic adenomas/carcinomas in Lkb1+/-p53-/- compound mutant mice. Oncogene 25 (12): 1816-20, 2006.
- Amos CI, Keitheri-Cheteri MB, Sabripour M, et al.: Genotype-phenotype correlations in Peutz-Jeghers syndrome. J Med Genet 41 (5): 327-33, 2004.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Colorectal. Version 2.2023 . Plymouth Meeting, PA: National Comprehensive Cancer Network, 2023.
Available with free registration. Last accessed March 13, 2024. - Boland CR, Idos GE, Durno C, et al.: Diagnosis and Management of Cancer Risk in the Gastrointestinal Hamartomatous Polyposis Syndromes: Recommendations From the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 162 (7): 2063-2085, 2022.
- Urquhart P, Grimpen F, Lim GJ, et al.: Capsule endoscopy versus magnetic resonance enterography for the detection of small bowel polyps in Peutz-Jeghers syndrome. Fam Cancer 13 (2): 249-55, 2014.
- Latchford AR, Neale K, Phillips RK, et al.: Juvenile polyposis syndrome: a study of genotype, phenotype, and long-term outcome. Dis Colon Rectum 55 (10): 1038-43, 2012.
- Veale AM, McColl I, Bussey HJ, et al.: Juvenile polyposis coli. J Med Genet 3 (1): 5-16, 1966.
- Chow E, Macrae F: A review of juvenile polyposis syndrome. J Gastroenterol Hepatol 20 (11): 1634-40, 2005.
- Jass JR, Williams CB, Bussey HJ, et al.: Juvenile polyposis--a precancerous condition. Histopathology 13 (6): 619-30, 1988.
- Howe JR, Bair JL, Sayed MG, et al.: Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet 28 (2): 184-7, 2001.
- Zhou XP, Woodford-Richens K, Lehtonen R, et al.: Germline mutations in BMPR1A/ALK3 cause a subset of cases of juvenile polyposis syndrome and of Cowden and Bannayan-Riley-Ruvalcaba syndromes. Am J Hum Genet 69 (4): 704-11, 2001.
- Aytac E, Sulu B, Heald B, et al.: Genotype-defined cancer risk in juvenile polyposis syndrome. Br J Surg 102 (1): 114-8, 2015.
- Brosens LA, van Hattem A, Hylind LM, et al.: Risk of colorectal cancer in juvenile polyposis. Gut 56 (7): 965-7, 2007.
- MacFarland SP, Ebrahimzadeh JE, Zelley K, et al.: Phenotypic Differences in Juvenile Polyposis Syndrome With or Without a Disease-causing SMAD4/BMPR1A Variant. Cancer Prev Res (Phila) 14 (2): 215-222, 2021.
- Gallione CJ, Repetto GM, Legius E, et al.: A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 363 (9412): 852-9, 2004.
- Rosner G, Petel-Galil Y, Laish I, et al.: Adenomatous Polyposis Phenotype in BMPR1A and SMAD4 Variant Carriers. Clin Transl Gastroenterol 13 (10): e00527, 2022.
- Aretz S, Stienen D, Uhlhaas S, et al.: High proportion of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis syndrome. J Med Genet 44 (11): 702-9, 2007.
- O'Malley M, LaGuardia L, Kalady MF, et al.: The prevalence of hereditary hemorrhagic telangiectasia in juvenile polyposis syndrome. Dis Colon Rectum 55 (8): 886-92, 2012.
- Dahdaleh FS, Carr JC, Calva D, et al.: Juvenile polyposis and other intestinal polyposis syndromes with microdeletions of chromosome 10q22-23. Clin Genet 81 (2): 110-6, 2012.
- Stanich PP, Pearlman R, Hinton A, et al.: Prevalence of Germline Mutations in Polyposis and Colorectal Cancer-Associated Genes in Patients With Multiple Colorectal Polyps. Clin Gastroenterol Hepatol 17 (10): 2008-2015.e3, 2019.
- Yurgelun MB, Hornick JL, Curry VK, et al.: Therapy-associated polyposis as a late sequela of cancer treatment. Clin Gastroenterol Hepatol 12 (6): 1046-50, 2014.
- Rigter LS, Kallenberg FG, Bastiaansen B, et al.: A case series of intestinal adenomatous polyposis of unidentified etiology; a late effect of irradiation? BMC Cancer 16 (1): 862, 2016.
- Rigter LS, Spaander MCW, Aleman BMP, et al.: High prevalence of advanced colorectal neoplasia and serrated polyposis syndrome in Hodgkin lymphoma survivors. Cancer 125 (6): 990-999, 2019.
- Palles C, Cazier JB, Howarth KM, et al.: Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 45 (2): 136-44, 2013.
- Briggs S, Tomlinson I: Germline and somatic polymerase ε and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol 230 (2): 148-53, 2013.
- Cancer Genome Atlas Network: Comprehensive molecular characterization of human colon and rectal cancer. Nature 487 (7407): 330-7, 2012.
- Wang F, Zhao Q, Wang YN, et al.: Evaluation of POLE and POLD1 Mutations as Biomarkers for Immunotherapy Outcomes Across Multiple Cancer Types. JAMA Oncol 5 (10): 1504-1506, 2019.
- Mur P, García-Mulero S, Del Valle J, et al.: Role of POLE and POLD1 in familial cancer. Genet Med 22 (12): 2089-2100, 2020.
- Buchanan DD, Stewart JR, Clendenning M, et al.: Risk of colorectal cancer for carriers of a germ-line mutation in POLE or POLD1. Genet Med 20 (8): 890-895, 2018.
- Hamzaoui N, Alarcon F, Leulliot N, et al.: Genetic, structural, and functional characterization of POLE polymerase proofreading variants allows cancer risk prediction. Genet Med 22 (9): 1533-1541, 2020.
- Weren RD, Ligtenberg MJ, Kets CM, et al.: A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet 47 (6): 668-71, 2015.
- Broderick P, Dobbins SE, Chubb D, et al.: Validation of Recently Proposed Colorectal Cancer Susceptibility Gene Variants in an Analysis of Families and Patients-a Systematic Review. Gastroenterology 152 (1): 75-77.e4, 2017.
- Beck SH, Jelsig AM, Yassin HM, et al.: Intestinal and extraintestinal neoplasms in patients with NTHL1 tumor syndrome: a systematic review. Fam Cancer 21 (4): 453-462, 2022.
- Chan JM, Clendenning M, Joseland S, et al.: Rare germline variants in the AXIN2 gene in families with colonic polyposis and colorectal cancer. Fam Cancer 21 (4): 399-413, 2022.
- Leclerc J, Beaumont M, Vibert R, et al.: AXIN2 germline testing in a French cohort validates pathogenic variants as a rare cause of predisposition to colorectal polyposis and cancer. Genes Chromosomes Cancer 62 (4): 210-222, 2023.
- Lammi L, Arte S, Somer M, et al.: Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet 74 (5): 1043-50, 2004.
- Marvin ML, Mazzoni SM, Herron CM, et al.: AXIN2-associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. Am J Med Genet A 155A (4): 898-902, 2011.
- Rivera B, Perea J, Sánchez E, et al.: A novel AXIN2 germline variant associated with attenuated FAP without signs of oligondontia or ectodermal dysplasia. Eur J Hum Genet 22 (3): 423-6, 2014.
- Jaeger E, Leedham S, Lewis A, et al.: Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat Genet 44 (6): 699-703, 2012.
- Meijers-Heijboer H, Wijnen J, Vasen H, et al.: The CHEK2 1100delC mutation identifies families with a hereditary breast and colorectal cancer phenotype. Am J Hum Genet 72 (5): 1308-14, 2003.
- Cybulski C, Górski B, Huzarski T, et al.: CHEK2 is a multiorgan cancer susceptibility gene. Am J Hum Genet 75 (6): 1131-5, 2004.
- de Jong MM, Nolte IM, Te Meerman GJ, et al.: Colorectal cancer and the CHEK2 1100delC mutation. Genes Chromosomes Cancer 43 (4): 377-82, 2005.
- Uson PLS, Riegert-Johnson D, Boardman L, et al.: Germline Cancer Susceptibility Gene Testing in Unselected Patients With Colorectal Adenocarcinoma: A Multicenter Prospective Study. Clin Gastroenterol Hepatol 20 (3): e508-e528, 2022.
- Breen KE, Katona BW, Catchings A, et al.: An updated counseling framework for moderate-penetrance colorectal cancer susceptibility genes. Genet Med 24 (12): 2587-2590, 2022.
- Bychkovsky BL, Agaoglu NB, Horton C, et al.: Differences in Cancer Phenotypes Among Frequent CHEK2 Variants and Implications for Clinical Care-Checking CHEK2. JAMA Oncol 8 (11): 1598-1606, 2022.
- Mundt E, Mabey B, Rainville I, et al.: Breast and colorectal cancer risks among over 6,000 CHEK2 pathogenic variant carriers: A comparison of missense versus truncating variants. Cancer Genet 278-279: 84-90, 2023.
- Hazewinkel Y, López-Cerón M, East JE, et al.: Endoscopic features of sessile serrated adenomas: validation by international experts using high-resolution white-light endoscopy and narrow-band imaging. Gastrointest Endosc 77 (6): 916-24, 2013.
- Guarinos C, Juárez M, Egoavil C, et al.: Prevalence and characteristics of MUTYH-associated polyposis in patients with multiple adenomatous and serrated polyps. Clin Cancer Res 20 (5): 1158-68, 2014.
- Crockett SD, Snover DC, Ahnen DJ, et al.: Sessile serrated adenomas: an evidence-based guide to management. Clin Gastroenterol Hepatol 13 (1): 11-26.e1, 2015.
- Boparai KS, Mathus-Vliegen EM, Koornstra JJ, et al.: Increased colorectal cancer risk during follow-up in patients with hyperplastic polyposis syndrome: a multicentre cohort study. Gut 59 (8): 1094-100, 2010.
- Clendenning M, Young JP, Walsh MD, et al.: Germline Mutations in the Polyposis-Associated Genes BMPR1A, SMAD4, PTEN, MUTYH and GREM1 Are Not Common in Individuals with Serrated Polyposis Syndrome. PLoS One 8 (6): e66705, 2013.
- Yan HHN, Lai JCW, Ho SL, et al.: RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut 66 (9): 1645-1656, 2017.
- Quintana I, Mejías-Luque R, Terradas M, et al.: Evidence suggests that germline RNF43 mutations are a rare cause of serrated polyposis. Gut 67 (12): 2230-2232, 2018.
- Taupin D, Lam W, Rangiah D, et al.: A deleterious RNF43 germline mutation in a severely affected serrated polyposis kindred. Hum Genome Var 2: 15013, 2015.
- Dekker E, Bleijenberg A, Balaguer F, et al.: Update on the World Health Organization Criteria for Diagnosis of Serrated Polyposis Syndrome. Gastroenterology 158 (6): 1520-1523, 2020.
- Leggett BA, Devereaux B, Biden K, et al.: Hyperplastic polyposis: association with colorectal cancer. Am J Surg Pathol 25 (2): 177-84, 2001.
- Rashid A, Houlihan PS, Booker S, et al.: Phenotypic and molecular characteristics of hyperplastic polyposis. Gastroenterology 119 (2): 323-32, 2000.
- Place RJ, Simmang CL: Hyperplastic-adenomatous polyposis syndrome. J Am Coll Surg 188 (5): 503-7, 1999.
- Hyman NH, Anderson P, Blasyk H: Hyperplastic polyposis and the risk of colorectal cancer. Dis Colon Rectum 47 (12): 2101-4, 2004.
- Koide N, Saito Y, Fujii T, et al.: A case of hyperplastic polyposis of the colon with adenocarcinomas in hyperplastic polyps after long-term follow-up. Endoscopy 34 (6): 499-502, 2002.
- Jeevaratnam P, Cottier DS, Browett PJ, et al.: Familial giant hyperplastic polyposis predisposing to colorectal cancer: a new hereditary bowel cancer syndrome. J Pathol 179 (1): 20-5, 1996.
- Bengoechea O, Martínez-Peñuela JM, Larrínaga B, et al.: Hyperplastic polyposis of the colorectum and adenocarcinoma in a 24-year-old man. Am J Surg Pathol 11 (4): 323-7, 1987.
- McCann BG: A case of metaplastic polyposis of the colon associated with focal adenomatous change and metachronous adenocarcinomas. Histopathology 13 (6): 700-2, 1988.
- Muller C, Yamada A, Ikegami S, et al.: Risk of Colorectal Cancer in Serrated Polyposis Syndrome: A Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol 20 (3): 622-630.e7, 2022.
- Kanth P, Yu Z, Keener MB, et al.: Cancer Risk in Patients With and Relatives of Serrated Polyposis Syndrome and Sporadic Sessile Serrated Lesions. Am J Gastroenterol 117 (2): 336-342, 2022.
- Biller LH, Ukaegbu C, Dhingra TG, et al.: A Multi-Institutional Cohort of Therapy-Associated Polyposis in Childhood and Young Adulthood Cancer Survivors. Cancer Prev Res (Phila) 13 (3): 291-298, 2020.
- Anthony E, Reece JC, Milanzi E, et al.: Body Mass Index, sex, non-steroidal anti-inflammatory drug medications, smoking and alcohol are differentially associated with World Health Organisation criteria and colorectal cancer risk in people with Serrated Polyposis Syndrome: an Australian case-control study. BMC Gastroenterol 22 (1): 489, 2022.
- Hurlstone DP, Cross SS, Slater R, et al.: Detecting diminutive colorectal lesions at colonoscopy: a randomised controlled trial of pan-colonic versus targeted chromoscopy. Gut 53 (3): 376-80, 2004.
- Saitoh Y, Waxman I, West AB, et al.: Prevalence and distinctive biologic features of flat colorectal adenomas in a North American population. Gastroenterology 120 (7): 1657-65, 2001.
- Hurlstone DP, Cross SS, Adam I, et al.: Endoscopic morphological anticipation of submucosal invasion in flat and depressed colorectal lesions: clinical implications and subtype analysis of the kudo type V pit pattern using high-magnification-chromoscopic colonoscopy. Colorectal Dis 6 (5): 369-75, 2004.
- Dacosta RS, Wilson BC, Marcon NE: New optical technologies for earlier endoscopic diagnosis of premalignant gastrointestinal lesions. J Gastroenterol Hepatol 17 (Suppl): S85-104, 2002.
- Rembacken BJ, Fujii T, Cairns A, et al.: Flat and depressed colonic neoplasms: a prospective study of 1000 colonoscopies in the UK. Lancet 355 (9211): 1211-4, 2000.
- Tsuda S, Veress B, Tóth E, et al.: Flat and depressed colorectal tumours in a southern Swedish population: a prospective chromoendoscopic and histopathological study. Gut 51 (4): 550-5, 2002.
- Rex DK, Helbig CC: High yields of small and flat adenomas with high-definition colonoscopes using either white light or narrow band imaging. Gastroenterology 133 (1): 42-7, 2007.
- Soetikno RM, Kaltenbach T, Rouse RV, et al.: Prevalence of nonpolypoid (flat and depressed) colorectal neoplasms in asymptomatic and symptomatic adults. JAMA 299 (9): 1027-35, 2008.
- Stoffel EM, Turgeon DK, Stockwell DH, et al.: Chromoendoscopy detects more adenomas than colonoscopy using intensive inspection without dye spraying. Cancer Prev Res (Phila) 1 (7): 507-13, 2008.
- Le Rhun M, Coron E, Parlier D, et al.: High resolution colonoscopy with chromoscopy versus standard colonoscopy for the detection of colonic neoplasia: a randomized study. Clin Gastroenterol Hepatol 4 (3): 349-54, 2006.
- Brooker JC, Saunders BP, Shah SG, et al.: Total colonic dye-spray increases the detection of diminutive adenomas during routine colonoscopy: a randomized controlled trial. Gastrointest Endosc 56 (3): 333-8, 2002.
- Stoffel EM, Turgeon DK, Stockwell DH, et al.: Missed adenomas during colonoscopic surveillance in individuals with Lynch Syndrome (hereditary nonpolyposis colorectal cancer). Cancer Prev Res (Phila) 1 (6): 470-5, 2008.
- Hüneburg R, Lammert F, Rabe C, et al.: Chromocolonoscopy detects more adenomas than white light colonoscopy or narrow band imaging colonoscopy in hereditary nonpolyposis colorectal cancer screening. Endoscopy 41 (4): 316-22, 2009.
- Wallace MH, Frayling IM, Clark SK, et al.: Attenuated adenomatous polyposis coli: the role of ascertainment bias through failure to dye-spray at colonoscopy. Dis Colon Rectum 42 (8): 1078-80, 1999.
- Dekker E, Boparai KS, Poley JW, et al.: High resolution endoscopy and the additional value of chromoendoscopy in the evaluation of duodenal adenomatosis in patients with familial adenomatous polyposis. Endoscopy 41 (8): 666-9, 2009.
- Sakamoto H, Yamamoto H, Hayashi Y, et al.: Nonsurgical management of small-bowel polyps in Peutz-Jeghers syndrome with extensive polypectomy by using double-balloon endoscopy. Gastrointest Endosc 74 (2): 328-33, 2011.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
Page Footer
I want to...
Audiences
Secure Member Sites
The Cigna Group Information
Disclaimer
Individual and family medical and dental insurance plans are insured by Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc., and Cigna HealthCare of Texas, Inc. Group health insurance and health benefit plans are insured or administered by CHLIC, Connecticut General Life Insurance Company (CGLIC), or their affiliates (see
All insurance policies and group benefit plans contain exclusions and limitations. For availability, costs and complete details of coverage, contact a licensed agent or Cigna sales representative. This website is not intended for residents of New Mexico.