Genetics of Endocrine and Neuroendocrine Neoplasias (PDQ®): Genetics - Health Professional Information [NCI]
Multiple Endocrine Neoplasia Type 1
Clinical Description
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant syndrome, with an estimated prevalence of about 1 in 30,000 individuals.[
-
Parathyroid tumors and primary hyperparathyroidism (PHPT) . -
Duodenopancreatic neuroendocrine tumors (NETs) . -
Pituitary tumors .
A clinical diagnosis of MEN1 may be made when an individual has two of the three major endocrine tumors listed above, especially if he/she was diagnosed with these tumors at a young age. Alternatively, familial MEN1 may be defined as having at least one MEN1 case in the family plus at least one first-degree relative (FDR) with one of these three tumors, or two FDRs with a germline pathogenic variant.[
Initial clinical presentation of symptoms typically occurs between the ages of 20 and 30 years. However, in many cases, an MEN1 diagnosis may not be confirmed for many years after initial symptoms occur. The age-related penetrance of MEN1 is 45% to 73% by age 30 years, 82% by age 50 years, and 96% by age 70 years.[
Parathyroid Tumors and PHPT
The most common features and often the first presenting signs of MEN1 are parathyroid tumors, which result in PHPT. These tumors occur in 80% to 100% of patients by age 50 years.[
Individuals with MEN1-associated PHPT will have elevated parathyroid hormone (PTH) and calcium levels in the blood. The clinical manifestations of PHPT are mainly the result of hypercalcemia. Mild hypercalcemia may go undetected and have few or no symptoms. More severe hypercalcemia can result in the following:
- Constipation.
- Nausea and vomiting.
- Dehydration.
- Decreased appetite and abdominal pain.
- Anorexia.
- Diuresis.
- Kidney stones.
- Increased bone resorption with resultant increased risk of bone fracture.
- Lethargy.
- Depression.
- Confusion.
- Hypertension.
- Shortened QT interval.
Since MEN1-associated hypercalcemia is directly related to the presence of parathyroid tumors, surgical removal of these tumors may normalize calcium and PTH levels. This can help relieve an individual's symptoms. However, there have been high recurrence rates of parathyroid tumors after surgery in some series.[
Duodenopancreatic NETs
Duodenopancreatic NETs are the second most common endocrine manifestation in MEN1, occurring in 30% to 80% of patients by age 40 years.[
Duodenopancreatic NETs seen in MEN1 include the following:
- Gastrinomas.
- Nonfunctioning NETs.
- Insulinomas.
- Vasoactive intestinal peptide tumors (VIPomas).
- Glucagonomas.
- Somatostatinomas.
| Tumor type | Estimated Penetrance | Symptoms |
|---|---|---|
| MEN1 = multiple endocrine neoplasia type 1. | ||
| Gastrinoma | ≤70%[ |
Peptic ulcer disease and esophagitis |
| Diarrhea | ||
| Abdominal pain | ||
| Weight loss | ||
| Nonfunctioning | 20%–55%[ |
Local compressive symptoms: abdominal pain, jaundice, anorexia, weight loss |
| Insulinoma | 10%[ |
Whipple's triad: symptomatic hypoglycemia reversed by glucose administration with associated elevation of insulin, C-peptide, and proinsulin levels |
| Vasoactive intestinal peptide | 1%[ |
Watery diarrhea |
| Hypokalemia | ||
| Achlorhydria | ||
| Glucagonoma | 1%[ |
Diabetes mellitus |
| Diarrhea | ||
| Depression | ||
| Necrolytic migratory erythema | ||
| Thromboembolic disease | ||
| Somatostatinoma | <1%[ |
Diabetes mellitus |
| Diarrhea/steatorrhea | ||
| Gallbladder disease | ||
| Hypochlorhydria | ||
| Weight loss | ||
Gastrinomas represent 50% of the gastrointestinal NETs in MEN1 and are the major cause of morbidity and mortality in MEN1 patients.[
Originally, nonfunctioning duodenopancreatic NETs were thought to be uncommon in individuals with MEN1. However, recognition of these tumors has increased with advanced genetic testing and improved imaging techniques. For example, a prospective study showed that MEN1 pathogenic variant carriers had a nonfunctioning duodenopancreatic NET frequency of 55% by age 39 years when they underwent endoscopic ultrasonography of the pancreas.[
Pituitary Tumors
Approximately 15% to 50% of MEN1 patients will develop a pituitary tumor.[
| Tumor type | Estimated Penetrance | Symptoms |
|---|---|---|
| MEN1 = multiple endocrine neoplasia type 1. | ||
| Prolactinoma | 20%[ |
Galactorrhea |
| Amenorrhea/infertility | ||
| Hypogonadism | ||
| Somatotropinoma | 10%[ |
Coarse facial features |
| Soft tissue overgrowth: enlargement of hands/feet | ||
| Hyperhidrosis | ||
| Corticotropinoma | <5%[ |
Weight gain |
| Hypertension | ||
| Flushing | ||
| Easy bruising/bleeding | ||
| Hyperglycemia | ||
Other MEN1-Associated Tumors
Other manifestations of MEN1 include carcinoids of the foregut (5%–10% of MEN1 patients). These are typically bronchial or thymic and are sometimes gastric. Skin lesions are also common and can include facial angiofibromas (up to 80% of MEN1 patients) and collagenomas (~75% of MEN1 patients).[
- Thyroid adenoma.
- Pheochromocytoma.
- Spinal ependymoma.
- Meningioma.
- Leiomyoma (e.g., esophageal, lung, and uterine).
Making the Diagnosis of MEN1
MEN1 is often difficult to diagnose in the absence of a significant family history or a positive genetic test for a pathogenic variant in the MEN1 gene. One study of 560 individuals with MEN1 showed a significant delay between the time of the first presenting symptom and the diagnosis of MEN1.[
Furthermore, identification of an MEN1-associated tumor is not sufficient to make the clinical diagnosis of MEN1 and may not trigger a referral to an endocrinologist. The median time between the first presenting symptom and diagnosis of MEN1 ranges from 7.6 years to 12 years.[
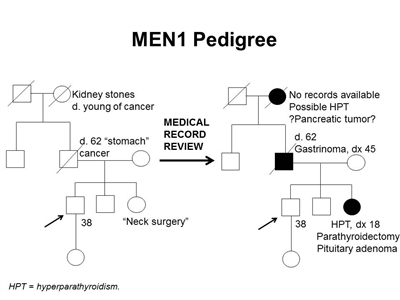
Figure 1. MEN1 pedigree. MEN1 can be very difficult to identify in a pedigree. The pedigree on the left was constructed based on self-report, and the pedigree on the right depicts the same family following a review of available medical records. This pedigree shows some of the features of a family with an MEN1 pathogenic variant across four generations, including affected family members with hyperparathyroidism, a pituitary adenoma, gastrinoma, and a suspected pancreatic tumor. The tumors in MEN1 typically occur at an earlier age than their sporadic counterparts. MEN1 families may exhibit some or all of these features. As an autosomal dominant syndrome, transmission can occur through maternal or paternal lineages, as depicted in the figure.
Since many of the tumors in MEN1 are underdiagnosed or misdiagnosed, identifying an MEN1 gene pathogenic variant in the proband early in the disease process can allow for early detection and treatment of tumors and earlier identification of at-risk family members. Many studies have been performed to determine the prevalence of MEN1 gene pathogenic variants among patients with apparently sporadic MEN1-related tumors.[
- Gastrinoma at any age in the individual or an FDR.
- Multifocal duodenopancreatic NETs at any age.
- PHPT before age 30 or 40 years.
- Multiglandular parathyroid adenomas/hyperplasia or recurrent PHPT.
- Presence of one of the three main MEN1 tumors plus one of the less common tumors/findings.
- Presence of two or more features (e.g., adrenal adenomas and carcinoid tumor).
- Combination of at least two of the following in one individual: parathyroid adenoma; thymic, bronchial, or foregut carcinoid tumor; duodenopancreatic NET; pituitary tumor; adrenal tumor.
- Parathyroid adenoma and a family history of hyperparathyroidism, pituitary adenoma, duodenopancreatic NET, or foregut carcinoid tumor.
- Multiple primary duodenopancreatic NETs in the same person.
Molecular Genetics of MEN1
The MEN1 gene is located on chromosome 11q13 and encodes the protein menin.[
Genetic Testing and Differential Diagnosis for MEN1
Genetic testing for MEN1 pathogenic variants is recommended for individuals meeting
A multigene panel that includes MEN1 and other genes associated with an increased risk of endocrine tumors may also be used. Such genetic testing can be used to distinguish between MEN1 and other forms of hereditary hyperparathyroidism, such as familial isolated hyperparathyroidism (FIHP), hyperparathyroidism–jaw tumor syndrome (HPT-JT), and familial hypocalciuric hypercalcemia (FHH). The hyperparathyroidism in FHH is not primary hyperparathyroidism, which is seen in MEN1, HPT-JT and FIHP. HPT-JT, which is caused by germline pathogenic variants in the CDC73 gene, is associated with PHPT, ossifying lesions of the maxilla and mandible, and renal lesions, usually bilateral renal cysts, hamartomas, and in some cases, Wilms tumor.[
Genetic diagnosis will help guide management for patients with early-onset hyperparathyroidism. This is especially crucial, since many of the above conditions have different management guidelines that correspond with their features. For example, distinguishing between MEN1 and FHH can be critical for a patient's disease management. Removing the parathyroid glands in FHH does not correct the hyperparathyroidism that is seen in patients with MEN1. This could result in an unnecessary surgery that would not relieve the patient's symptoms. In addition, HPT-JT is unique because it increases parathyroid carcinoma risk. Hence, individuals with this syndrome have different management guidelines than individuals with other forms of hereditary hyperparathyroidism.[
| Condition | Gene(s) | Major Clinical Features |
|---|---|---|
| FHH = familial hypocalciuric hypercalcemia; FIHP = familial isolated hyperparathyroidism; HPT-JT = hyperparathyroidism–jaw tumor syndrome; MEN1 = multiple endocrine neoplasia type 1 (gene is italicized); NETs = neuroendocrine tumors; PHPT = primary hyperparathyroidism. | ||
| MEN1 | MEN1 | PHPT, pituitary adenomas, duodenopancreatic NETs[ |
| FIHP | MEN1,CDC73 | PHPT[ |
| HPT-JT | CDC73 | PHPT; osteomas of maxilla and mandible; renal cysts or hamartomas; and rarely, Wilms tumor and parathyroid carcinoma[ |
| FHH | CASR(type 1),GNA11(type 2),AP2S1(type 3) | Hyperparathyroidism (not primary)[ |
Surveillance
Screening and surveillance for MEN1 may include a combination of biochemical tests and imaging techniques.
Traditionally, magnetic resonance imaging (MRI) was used for surveillance and staging. However, ongoing studies have evaluated the role of MRI in functional imaging, including gallium Ga 68-DOTATATE (68Ga-DOTATATE) positron emission tomography (PET)–computed tomography (CT) scanning. A multicenter retrospective study examined 108 MEN1 patients undergoing PET-CT for screening, staging, restaging, or targeted radiotherapy selection. This study demonstrated that PET-CT has the potential to increase diagnostic sensitivity when searching for MEN1-associated NETs.[
A study analyzed thoracic screening techniques in 50 patients with MEN1. It found that when patients with MEN1 underwent functional imaging with fluorine F 18-fludeoxyglucose (18F-FDG) PET-CT screening, they had a similar number of lung nodules as individuals in the general population. However, when lesions in MEN1 patients were FDG-avid, they were more likely to progress during the follow-up period. Therefore, further observation and follow up of FDG-avid lesions may be warranted in patients with MEN1.[
Recommendations for MEN1 surveillance are summarized in
| Biochemical Test or Procedure | Condition Screened For | Age Screening Initiated (y) | Frequency |
|---|---|---|---|
| CT = computed tomography; MEN1 = multiple endocrine neoplasia type 1; MRI = magnetic resonance imaging; NETs = neuroendocrine tumors; PHPT = primary hyperparathyroidism; PTH = parathyroid hormone. | |||
| a Adapted from Brandi et al.[ |
|||
| b The recommendations for abdominal imaging differ between two published guidelines for the diagnosis and management of MEN1.[ |
|||
| c The age to initiate screening and the screening frequency for pituitary tumors may be debatable because the clinical significance of small, nonfunctional tumors is unclear;[ |
|||
| d Adapted from Niederle et al.[ |
|||
| e Adapted from Shirali et al.[ |
|||
| Serum prolactin and/or insulin-like growth factor 1 | Pituitary tumors | 5 | Every 1 y |
| Fasting total serum calcium and/or ionized calcium and PTH | Parathyroid tumors and PHPT | 8 | Every 1 y |
| Fasting serum gastrin | Duodenopancreatic gastrinoma | 20 | Every 1 y |
| Chromogranin A, pancreatic polypeptide, glucagon, and vasointestinal polypeptided | Duodenopancreatic NETs | 10–16 | Up to every 3 years (consider every 3 years if asymptomatic; consider shorter screening intervals depending on the clinical scenario) |
| Fasting glucose and insulin | Insulinoma | 5 | Every 1 y |
| Brain MRIc | Pituitary tumors | 5 | Every 3–5 y based on biochemical results |
| Chest MRIe | Thymic and bronchial NETs | <20 | About every 3 years. Consider more frequent screening for men, smokers, or individuals with a positive family history. Baseline chest MRI is done prior to parathyroidectomy |
| Abdominal CT or MRIb[ |
Duodenopancreatic NETs | 20 | Every 3–5 y based on biochemical results |
| Abdominal CT, MRI, or endoscopic ultrasonographyb[ |
Duodenopancreatic NETs | <10 | Every 1 y |
Interventions
Surgical management of MEN1 is complex and controversial, given the multifocal and multiglandular nature of the disease. Patients with MEN1 have a high risk of tumor recurrence, even after surgery. Additionally, these patients may have an increased risk of developing venous thromboembolisms.[
Treatment for parathyroid tumors
Once evidence of parathyroid disease is established biochemically, surgical removal of the hyperfunctional parathyroid tissue is recommended to achieve eucalcemia and euparathyroidism. However, the timing and the amount of parathyroid and thymus gland tissue that is removed during surgery remains controversial.[
Some groups reserve surgical intervention for symptomatic patients, with continued annual biochemical screening for those without objective signs of disease. Subtotal parathyroidectomy (removal of 3–3.5 glands) is commonly suggested as the initial surgical treatment when a provider decides to proceed with surgery.[
Total parathyroidectomy with autotransplantation of parathyroid tissue to a distant site, such as the forearm, is a less commonly recommended option. Likelihood of cancer recurrence is lowered with total parathyroidectomy. However, this procedure also renders the patient aparathyroid for a period of time while the autotransplanted tissue becomes functional. This can cause a permanent PTH deficiency (no detectable PTH in the body).[
Treatment for duodenopancreatic NETs
The timing and extent of surgery for duodenopancreatic NETs are controversial and depend on many factors, including severity of symptoms, extent of disease, functional component, location and necessity of simple enucleation, subtotal or total pancreatectomy, and pancreaticoduodenectomy (Whipple procedure). Surgical enucleation has been associated with higher recurrence compared with distal pancreatectomy, and a decreased rate of endocrine insufficiency compared with a Whipple procedure.[
Individuals with MEN1 who are diagnosed with NETs often have multiple tumors of various types throughout the pancreas and duodenum, some of which can be identified using magnetic resonance imaging or computed tomography (CT). Combining functional tracer accumulation with anatomic imaging improves tumor localization. 68Ga-DOTATATE PET-CT demonstrates excellent sensitivity in mapping duodenopancreatic NET disease. This modality may guide the initial workup and appears to be superior to standard somatostatin octreotide, especially for lesions smaller than 10 mm.[
In the current era of effective treatment for hyperfunctional hormone excess states, most MEN1-related deaths are due to the malignant nature of duodenopancreatic NETs. A less common but important risk of death is from malignant thymic carcinoid tumors. Indicators of a poor MEN1 prognosis include elevated fasting serum gastrin, the presence of functional hormonal syndromes, liver or distant metastases, aggressive duodenopancreatic NET growth, large duodenopancreatic NET size, or the need for multiple parathyroidectomies. The most common cause of non-MEN1–related death in this patient cohort is from cardiovascular disease.[
Other duodenopancreatic NETs
Glucagonomas, VIPomas, and somatostatinomas are rare but often have higher rates of malignancy than other duodenopancreatic NETs.[
Insulinomas
Medical management of insulinoma using diet and medication is often unsuccessful; the mainstay of treatment for this tumor is surgical resection.[
Gastrinomas
Most MEN1-associated gastrinomas originate in the duodenum. These tumors are typically multifocal and cause hyper-secretion of gastrin, with resultant peptic ulcer disease (Zollinger-Ellison syndrome).[
Several published series have shown a positive correlation between primary tumor size and rate of distant metastasis. One retrospective study showed that 61% of patients with tumors larger than 3 cm had liver metastases.[
The type of surgery for gastrinoma depends on many factors. A Whipple procedure is typically discouraged as an initial surgery, given the high postoperative morbidity and long-term complications, such as diabetes mellitus and malabsorption. Less extensive operations have been described with varying results. At a minimum, duodenectomy with intraoperative palpation and/or ultrasonography to locate and excise duodenal tumors and peri-pancreatic lymph node dissection are performed.[
Nonfunctioning NETs
Approximately 50% of individuals with MEN1 will develop nonfunctioning NETs.[
Pituitary tumors
Medical therapy to suppress hypersecretion is often the first line of therapy for MEN1-associated pituitary tumors. In one series of 136 patients, medical therapy was successful in approximately one-half of patients with secreting tumors (49 of 116, 42%), and successful suppression was correlated with smaller tumor size.[
References:
- Agarwal SK, Ozawa A, Mateo CM, et al.: The MEN1 gene and pituitary tumours. Horm Res 71 (Suppl 2): 131-8, 2009.
- Trump D, Farren B, Wooding C, et al.: Clinical studies of multiple endocrine neoplasia type 1 (MEN1) QJM 89 (9): 653-69, 1996.
- Chandrasekharappa SC, Guru SC, Manickam P, et al.: Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 276 (5311): 404-7, 1997.
- Brandi ML, Gagel RF, Angeli A, et al.: Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 86 (12): 5658-71, 2001.
- Niederle B, Selberherr A, Bartsch DK, et al.: Multiple Endocrine Neoplasia Type 1 and the Pancreas: Diagnosis and Treatment of Functioning and Non-Functioning Pancreatic and Duodenal Neuroendocrine Neoplasia within the MEN1 Syndrome - An International Consensus Statement. Neuroendocrinology 111 (7): 609-630, 2021.
- Carty SE, Helm AK, Amico JA, et al.: The variable penetrance and spectrum of manifestations of multiple endocrine neoplasia type 1. Surgery 124 (6): 1106-13; discussion 1113-4, 1998.
- Goudet P, Dalac A, Le Bras M, et al.: MEN1 disease occurring before 21 years old: a 160-patient cohort study from the Groupe d'étude des Tumeurs Endocrines. J Clin Endocrinol Metab 100 (4): 1568-77, 2015.
- Thakker RV, Newey PJ, Walls GV, et al.: Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 97 (9): 2990-3011, 2012.
- Goudet P, Murat A, Binquet C, et al.: Risk factors and causes of death in MEN1 disease. A GTE (Groupe d'Etude des Tumeurs Endocrines) cohort study among 758 patients. World J Surg 34 (2): 249-55, 2010.
- Chandrasekharappa SC, Teh BT: Clinical and molecular aspects of multiple endocrine neoplasia type 1. Front Horm Res 28: 50-80, 2001.
- Shariq OA, Lines KE, English KA, et al.: Multiple endocrine neoplasia type 1 in children and adolescents: Clinical features and treatment outcomes. Surgery 171 (1): 77-87, 2022.
- del Pozo C, García-Pascual L, Balsells M, et al.: Parathyroid carcinoma in multiple endocrine neoplasia type 1. Case report and review of the literature. Hormones (Athens) 10 (4): 326-31, 2011 Oct-Dec.
- Christakis I, Busaidy NL, Cote GJ, et al.: Parathyroid carcinoma and atypical parathyroid neoplasms in MEN1 patients; A clinico-pathologic challenge. The MD Anderson case series and review of the literature. Int J Surg 31: 10-6, 2016.
- Singh Ospina N, Sebo TJ, Thompson GB, et al.: Prevalence of parathyroid carcinoma in 348 patients with multiple endocrine neoplasia type 1 - case report and review of the literature. Clin Endocrinol (Oxf) 84 (2): 244-249, 2016.
- Norton JA, Venzon DJ, Berna MJ, et al.: Prospective study of surgery for primary hyperparathyroidism (HPT) in multiple endocrine neoplasia-type 1 and Zollinger-Ellison syndrome: long-term outcome of a more virulent form of HPT. Ann Surg 247 (3): 501-10, 2008.
- Hellman P, Skogseid B, Oberg K, et al.: Primary and reoperative parathyroid operations in hyperparathyroidism of multiple endocrine neoplasia type 1. Surgery 124 (6): 993-9, 1998.
- Schreinemakers JM, Pieterman CR, Scholten A, et al.: The optimal surgical treatment for primary hyperparathyroidism in MEN1 patients: a systematic review. World J Surg 35 (9): 1993-2005, 2011.
- Christakis I, Qiu W, Hyde SM, et al.: Genotype-phenotype pancreatic neuroendocrine tumor relationship in multiple endocrine neoplasia type 1 patients: A 23-year experience at a single institution. Surgery 163 (1): 212-217, 2018.
- Donegan D, Singh Ospina N, Rodriguez-Gutierrez R, et al.: Long-term outcomes in patients with multiple endocrine neoplasia type 1 and pancreaticoduodenal neuroendocrine tumours. Clin Endocrinol (Oxf) 86 (2): 199-206, 2017.
- Norton JA, Krampitz G, Jensen RT: Multiple Endocrine Neoplasia: Genetics and Clinical Management. Surg Oncol Clin N Am 24 (4): 795-832, 2015.
- Thomas-Marques L, Murat A, Delemer B, et al.: Prospective endoscopic ultrasonographic evaluation of the frequency of nonfunctioning pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. Am J Gastroenterol 101 (2): 266-73, 2006.
- Lévy-Bohbot N, Merle C, Goudet P, et al.: Prevalence, characteristics and prognosis of MEN 1-associated glucagonomas, VIPomas, and somatostatinomas: study from the GTE (Groupe des Tumeurs Endocrines) registry. Gastroenterol Clin Biol 28 (11): 1075-81, 2004.
- Pipeleers-Marichal M, Somers G, Willems G, et al.: Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med 322 (11): 723-7, 1990.
- Weber HC, Venzon DJ, Lin JT, et al.: Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: a prospective long-term study. Gastroenterology 108 (6): 1637-49, 1995.
- Tonelli F, Giudici F, Fratini G, et al.: Pancreatic endocrine tumors in multiple endocrine neoplasia type 1 syndrome: review of literature. Endocr Pract 17 (Suppl 3): 33-40, 2011 Jul-Aug.
- Triponez F, Dosseh D, Goudet P, et al.: Epidemiology data on 108 MEN 1 patients from the GTE with isolated nonfunctioning tumors of the pancreas. Ann Surg 243 (2): 265-72, 2006.
- Corbetta S, Pizzocaro A, Peracchi M, et al.: Multiple endocrine neoplasia type 1 in patients with recognized pituitary tumours of different types. Clin Endocrinol (Oxf) 47 (5): 507-12, 1997.
- Darling TN, Skarulis MC, Steinberg SM, et al.: Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol 133 (7): 853-7, 1997.
- Ventura M, Melo M, Carrilho F: Outcome and long-term follow-up of adrenal lesions in multiple endocrine neoplasia type 1. Arch Endocrinol Metab 63 (5): 516-523, 2019.
- Machens A, Schaaf L, Karges W, et al.: Age-related penetrance of endocrine tumours in multiple endocrine neoplasia type 1 (MEN1): a multicentre study of 258 gene carriers. Clin Endocrinol (Oxf) 67 (4): 613-22, 2007.
- Pieterman CR, Schreinemakers JM, Koppeschaar HP, et al.: Multiple endocrine neoplasia type 1 (MEN1): its manifestations and effect of genetic screening on clinical outcome. Clin Endocrinol (Oxf) 70 (4): 575-81, 2009.
- Waldmann J, Bartsch DK, Kann PH, et al.: Adrenal involvement in multiple endocrine neoplasia type 1: results of 7 years prospective screening. Langenbecks Arch Surg 392 (4): 437-43, 2007.
- Gibril F, Schumann M, Pace A, et al.: Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore) 83 (1): 43-83, 2004.
- McKeeby JL, Li X, Zhuang Z, et al.: Multiple leiomyomas of the esophagus, lung, and uterus in multiple endocrine neoplasia type 1. Am J Pathol 159 (3): 1121-7, 2001.
- Vortmeyer AO, Lubensky IA, Skarulis M, et al.: Multiple endocrine neoplasia type 1: atypical presentation, clinical course, and genetic analysis of multiple tumors. Mod Pathol 12 (9): 919-24, 1999.
- Yamazaki M, Suzuki S, Kosugi S, et al.: Delay in the diagnosis of multiple endocrine neoplasia type 1: typical symptoms are frequently overlooked. Endocr J 59 (9): 797-807, 2012.
- Lourenço DM, Toledo RA, Coutinho FL, et al.: The impact of clinical and genetic screenings on the management of the multiple endocrine neoplasia type 1. Clinics (Sao Paulo) 62 (4): 465-76, 2007.
- van Leeuwaarde RS, van Nesselrooij BP, Hermus AR, et al.: Impact of Delay in Diagnosis in Outcomes in MEN1: Results From the Dutch MEN1 Study Group. J Clin Endocrinol Metab 101 (3): 1159-65, 2016.
- Strømsvik N, Nordin K, Berglund G, et al.: Living with multiple endocrine neoplasia type 1: decent care-insufficient medical and genetic information: a qualitative study of MEN 1 patients in a Swedish hospital. J Genet Couns 16 (1): 105-17, 2007.
- Marini F, Giusti F, Tonelli F, et al.: Management impact: effects on quality of life and prognosis in MEN1. Endocr Relat Cancer 24 (10): T227-T242, 2017.
- Roy PK, Venzon DJ, Shojamanesh H, et al.: Zollinger-Ellison syndrome. Clinical presentation in 261 patients. Medicine (Baltimore) 79 (6): 379-411, 2000.
- Bardram L, Stage JG: Frequency of endocrine disorders in patients with the Zollinger-Ellison syndrome. Scand J Gastroenterol 20 (2): 233-8, 1985.
- Uchino S, Noguchi S, Sato M, et al.: Screening of the Men1 gene and discovery of germ-line and somatic mutations in apparently sporadic parathyroid tumors. Cancer Res 60 (19): 5553-7, 2000.
- Scheithauer BW, Laws ER, Kovacs K, et al.: Pituitary adenomas of the multiple endocrine neoplasia type I syndrome. Semin Diagn Pathol 4 (3): 205-11, 1987.
- Newey PJ, Thakker RV: Role of multiple endocrine neoplasia type 1 mutational analysis in clinical practice. Endocr Pract 17 (Suppl 3): 8-17, 2011 Jul-Aug.
- Hampel H, Bennett RL, Buchanan A, et al.: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 17 (1): 70-87, 2015.
- Bashford MT, Kohlman W, Everett J, et al.: Addendum: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 21 (12): 2844, 2019.
- Larsson C, Skogseid B, Oberg K, et al.: Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 332 (6159): 85-7, 1988.
- Bassett JH, Forbes SA, Pannett AA, et al.: Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet 62 (2): 232-44, 1998.
- Lemos MC, Thakker RV: Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 29 (1): 22-32, 2008.
- Concolino P, Costella A, Capoluongo E: Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new germline variants reported in the last nine years. Cancer Genet 209 (1-2): 36-41, 2016 Jan-Feb.
- Brandi ML, Agarwal SK, Perrier ND, et al.: Multiple Endocrine Neoplasia Type 1: Latest Insights. Endocr Rev 42 (2): 133-170, 2021.
- Giraud S, Zhang CX, Serova-Sinilnikova O, et al.: Germ-line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am J Hum Genet 63 (2): 455-67, 1998.
- Wautot V, Vercherat C, Lespinasse J, et al.: Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: search for correlation between phenotype and the functional domains of the MEN1 protein. Hum Mutat 20 (1): 35-47, 2002.
- van den Broek MFM, van Nesselrooij BPM, Pieterman CRC, et al.: Clues For Genetic Anticipation In Multiple Endocrine Neoplasia Type 1. J Clin Endocrinol Metab 105 (7): , 2020.
- Thevenon J, Bourredjem A, Faivre L, et al.: Unraveling the intrafamilial correlations and heritability of tumor types in MEN1: a Groupe d'étude des Tumeurs Endocrines study. Eur J Endocrinol 173 (6): 819-26, 2015.
- Agarwal SK, Kester MB, Debelenko LV, et al.: Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet 6 (7): 1169-75, 1997.
- Klein RD, Salih S, Bessoni J, et al.: Clinical testing for multiple endocrine neoplasia type 1 in a DNA diagnostic laboratory. Genet Med 7 (2): 131-8, 2005.
- Teh BT, Farnebo F, Kristoffersson U, et al.: Autosomal dominant primary hyperparathyroidism and jaw tumor syndrome associated with renal hamartomas and cystic kidney disease: linkage to 1q21-q32 and loss of the wild type allele in renal hamartomas. J Clin Endocrinol Metab 81 (12): 4204-11, 1996.
- Carpten JD, Robbins CM, Villablanca A, et al.: HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet 32 (4): 676-80, 2002.
- Marx SJ: Multiple endocrine neoplasia type 1. In: Vogelstein B, Kinzler KW, eds.: The Genetic Basis of Human Cancer. McGraw-Hill, 1998, pp 489-506.
- Warner J, Epstein M, Sweet A, et al.: Genetic testing in familial isolated hyperparathyroidism: unexpected results and their implications. J Med Genet 41 (3): 155-60, 2004.
- Mizusawa N, Uchino S, Iwata T, et al.: Genetic analyses in patients with familial isolated hyperparathyroidism and hyperparathyroidism-jaw tumour syndrome. Clin Endocrinol (Oxf) 65 (1): 9-16, 2006.
- Cetani F, Pardi E, Borsari S, et al.: Molecular pathogenesis of primary hyperparathyroidism. J Endocrinol Invest 34 (7 Suppl): 35-9, 2011.
- Miedlich S, Lohmann T, Schneyer U, et al.: Familial isolated primary hyperparathyroidism--a multiple endocrine neoplasia type 1 variant? Eur J Endocrinol 145 (2): 155-60, 2001.
- Cetani F, Pardi E, Ambrogini E, et al.: Genetic analyses in familial isolated hyperparathyroidism: implication for clinical assessment and surgical management. Clin Endocrinol (Oxf) 64 (2): 146-52, 2006.
- Raue F, Frank-Raue K: Primary hyperparathyroidism--what the nephrologist should know--an update. Nephrol Dial Transplant 22 (3): 696-9, 2007.
- Romero Arenas MA, Morris LF, Rich TA, et al.: Preoperative multiple endocrine neoplasia type 1 diagnosis improves the surgical outcomes of pediatric patients with primary hyperparathyroidism. J Pediatr Surg 49 (4): 546-50, 2014.
- Thakker RV: Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol 386 (1-2): 2-15, 2014.
- Christensen SE, Nissen PH, Vestergaard P, et al.: Familial hypocalciuric hypercalcaemia: a review. Curr Opin Endocrinol Diabetes Obes 18 (6): 359-70, 2011.
- Nesbit MA, Hannan FM, Howles SA, et al.: Mutations affecting G-protein subunit α11 in hypercalcemia and hypocalcemia. N Engl J Med 368 (26): 2476-2486, 2013.
- Nesbit MA, Hannan FM, Howles SA, et al.: Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet 45 (1): 93-7, 2013.
- Mennetrey C, Le Bras M, Bando-Delaunay A, et al.: Value of Somatostatin Receptor PET/CT in Patients With MEN1 at Various Stages of Their Disease. J Clin Endocrinol Metab 107 (5): e2056-e2064, 2022.
- So A, Pointon O, Hodgson R, et al.: An assessment of 18 F-FDG PET/CT for thoracic screening and risk stratification of pulmonary nodules in multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf) 88 (5): 683-691, 2018.
- van den Broek MFM, de Laat JM, van Leeuwaarde RS, et al.: The Management of Neuroendocrine Tumors of the Lung in MEN1: Results From the Dutch MEN1 Study Group. J Clin Endocrinol Metab 106 (2): e1014-e1027, 2021.
- Langer P, Kann PH, Fendrich V, et al.: Prospective evaluation of imaging procedures for the detection of pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. World J Surg 28 (12): 1317-22, 2004.
- de Laat JM, Dekkers OM, Pieterman CR, et al.: Long-Term Natural Course of Pituitary Tumors in Patients With MEN1: Results From the DutchMEN1 Study Group (DMSG). J Clin Endocrinol Metab 100 (9): 3288-96, 2015.
- Shirali AS, Pieterman CRC, Lewis MA, et al.: It's not a mystery, it's in the history: Multidisciplinary management of multiple endocrine neoplasia type 1. CA Cancer J Clin 71 (5): 369-380, 2021.
- Lee ME, Ortega-Sustache YM, Agarwal SK, et al.: Patients With MEN1 Are at an Increased Risk for Venous Thromboembolism. J Clin Endocrinol Metab 106 (2): e460-e468, 2021.
- Pieterman CR, van Hulsteijn LT, den Heijer M, et al.: Primary hyperparathyroidism in MEN1 patients: a cohort study with longterm follow-up on preferred surgical procedure and the relation with genotype. Ann Surg 255 (6): 1171-8, 2012.
- Nilubol N, Weinstein LS, Simonds WF, et al.: Limited Parathyroidectomy in Multiple Endocrine Neoplasia Type 1-Associated Primary Hyperparathyroidism: A Setup for Failure. Ann Surg Oncol 23 (2): 416-23, 2016.
- Lairmore TC, Govednik CM, Quinn CE, et al.: A randomized, prospective trial of operative treatments for hyperparathyroidism in patients with multiple endocrine neoplasia type 1. Surgery 156 (6): 1326-34; discussion 1334-5, 2014.
- Landry JP, Pieterman CRC, Clemente-Gutierrez U, et al.: Evaluation of risk factors, long-term outcomes, and immediate and delayed autotransplantation to minimize postsurgical hypoparathyroidism in multiple endocrine neoplasia type 1 (MEN1): A retrospective cohort study. Surgery 171 (5): 1240-1246, 2022.
- Ratnayake CBB, Loveday BP, Windsor JA, et al.: Patient characteristics and clinical outcomes following initial surgical intervention for MEN1 associated pancreatic neuroendocrine tumours: A systematic review and exploratory meta-analysis of the literature. Pancreatology 19 (3): 462-471, 2019.
- Kishi Y, Shimada K, Nara S, et al.: Basing treatment strategy for non-functional pancreatic neuroendocrine tumors on tumor size. Ann Surg Oncol 21 (9): 2882-8, 2014.
- Nell S, Verkooijen HM, Pieterman CRC, et al.: Management of MEN1 Related Nonfunctioning Pancreatic NETs: A Shifting Paradigm: Results From the DutchMEN1 Study Group. Ann Surg 267 (6): 1155-1160, 2018.
- Qiu W, Christakis I, Silva A, et al.: Utility of chromogranin A, pancreatic polypeptide, glucagon and gastrin in the diagnosis and follow-up of pancreatic neuroendocrine tumours in multiple endocrine neoplasia type 1 patients. Clin Endocrinol (Oxf) 85 (3): 400-7, 2016.
- Ramundo V, Del Prete M, Marotta V, et al.: Impact of long-acting octreotide in patients with early-stage MEN1-related duodeno-pancreatic neuroendocrine tumours. Clin Endocrinol (Oxf) 80 (6): 850-5, 2014.
- Triponez F, Goudet P, Dosseh D, et al.: Is surgery beneficial for MEN1 patients with small (< or = 2 cm), nonfunctioning pancreaticoduodenal endocrine tumor? An analysis of 65 patients from the GTE. World J Surg 30 (5): 654-62; discussion 663-4, 2006.
- Bettini R, Partelli S, Boninsegna L, et al.: Tumor size correlates with malignancy in nonfunctioning pancreatic endocrine tumor. Surgery 150 (1): 75-82, 2011.
- Triponez F, Sadowski SM, Pattou F, et al.: Long-term Follow-up of MEN1 Patients Who Do Not Have Initial Surgery for Small ≤2 cm Nonfunctioning Pancreatic Neuroendocrine Tumors, an AFCE and GTE Study: Association Francophone de Chirurgie Endocrinienne & Groupe d'Etude des Tumeurs Endocrines. Ann Surg 268 (1): 158-164, 2018.
- Kornaczewski Jackson ER, Pointon OP, Bohmer R, et al.: Utility of FDG-PET Imaging for Risk Stratification of Pancreatic Neuroendocrine Tumors in MEN1. J Clin Endocrinol Metab 102 (6): 1926-1933, 2017.
- Brunner SM, Weber F, Werner JM, et al.: Neuroendocrine tumors of the pancreas: a retrospective single-center analysis using the ENETS TNM-classification and immunohistochemical markers for risk stratification. BMC Surg 15: 49, 2015.
- Bartsch DK, Langer P, Wild A, et al.: Pancreaticoduodenal endocrine tumors in multiple endocrine neoplasia type 1: surgery or surveillance? Surgery 128 (6): 958-66, 2000.
- Bartsch DK, Fendrich V, Langer P, et al.: Outcome of duodenopancreatic resections in patients with multiple endocrine neoplasia type 1. Ann Surg 242 (6): 757-64, discussion 764-6, 2005.
- Norton JA, Jensen RT: Role of surgery in Zollinger-Ellison syndrome. J Am Coll Surg 205 (4 Suppl): S34-7, 2007.
- Lopez CL, Waldmann J, Fendrich V, et al.: Long-term results of surgery for pancreatic neuroendocrine neoplasms in patients with MEN1. Langenbecks Arch Surg 396 (8): 1187-96, 2011.
- Drymousis P, Raptis DA, Spalding D, et al.: Laparoscopic versus open pancreas resection for pancreatic neuroendocrine tumours: a systematic review and meta-analysis. HPB (Oxford) 16 (5): 397-406, 2014.
- Morgat C, Vélayoudom-Céphise FL, Schwartz P, et al.: Evaluation of (68)Ga-DOTA-TOC PET/CT for the detection of duodenopancreatic neuroendocrine tumors in patients with MEN1. Eur J Nucl Med Mol Imaging 43 (7): 1258-66, 2016.
- Lastoria S, Marciello F, Faggiano A, et al.: Role of (68)Ga-DOTATATE PET/CT in patients with multiple endocrine neoplasia type 1 (MEN1). Endocrine 52 (3): 488-94, 2016.
- Imamura M, Komoto I, Ota S, et al.: Biochemically curative surgery for gastrinoma in multiple endocrine neoplasia type 1 patients. World J Gastroenterol 17 (10): 1343-53, 2011.
- Tonelli F, Fratini G, Nesi G, et al.: Pancreatectomy in multiple endocrine neoplasia type 1-related gastrinomas and pancreatic endocrine neoplasias. Ann Surg 244 (1): 61-70, 2006.
- Lewis MA, Thompson GB, Young WF: Preoperative assessment of the pancreas in multiple endocrine neoplasia type 1. World J Surg 36 (6): 1375-81, 2012.
- van Asselt SJ, Brouwers AH, van Dullemen HM, et al.: EUS is superior for detection of pancreatic lesions compared with standard imaging in patients with multiple endocrine neoplasia type 1. Gastrointest Endosc 81 (1): 159-167.e2, 2015.
- Ito T, Igarashi H, Uehara H, et al.: Causes of death and prognostic factors in multiple endocrine neoplasia type 1: a prospective study: comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Medicine (Baltimore) 92 (3): 135-81, 2013.
- Akerström G, Stålberg P: Surgical management of MEN-1 and -2: state of the art. Surg Clin North Am 89 (5): 1047-68, 2009.
- O'Riordain DS, O'Brien T, van Heerden JA, et al.: Surgical management of insulinoma associated with multiple endocrine neoplasia type I. World J Surg 18 (4): 488-93; discussion 493-4, 1994 Jul-Aug.
- Crippa S, Zerbi A, Boninsegna L, et al.: Surgical management of insulinomas: short- and long-term outcomes after enucleations and pancreatic resections. Arch Surg 147 (3): 261-6, 2012.
- Sakurai A, Yamazaki M, Suzuki S, et al.: Clinical features of insulinoma in patients with multiple endocrine neoplasia type 1: analysis of the database of the MEN Consortium of Japan. Endocr J 59 (10): 859-66, 2012.
- Vezzosi D, Cardot-Bauters C, Bouscaren N, et al.: Long-term results of the surgical management of insulinoma patients with MEN1: a Groupe d'étude des Tumeurs Endocrines (GTE) retrospective study. Eur J Endocrinol 172 (3): 309-19, 2015.
- Grant CS: Insulinoma. Best Pract Res Clin Gastroenterol 19 (5): 783-98, 2005.
- Giudici F, Nesi G, Brandi ML, et al.: Surgical management of insulinomas in multiple endocrine neoplasia type 1. Pancreas 41 (4): 547-53, 2012.
- Plöckinger U: Diagnosis and Treatment of Gastrinomas in Multiple Endocrine Neoplasia Type 1 (MEN-1). Cancers (Basel) 4 (1): 39-54, 2012.
- Falconi M, Eriksson B, Kaltsas G, et al.: ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 103 (2): 153-71, 2016.
- Mignon M, Cadiot G: Diagnostic and therapeutic criteria in patients with Zollinger-Ellison syndrome and multiple endocrine neoplasia type 1. J Intern Med 243 (6): 489-94, 1998.
- Cadiot G, Vuagnat A, Doukhan I, et al.: Prognostic factors in patients with Zollinger-Ellison syndrome and multiple endocrine neoplasia type 1. Groupe d'Etude des Néoplasies Endocriniennes Multiples (GENEM and groupe de Recherche et d'Etude du Syndrome de Zollinger-Ellison (GRESZE). Gastroenterology 116 (2): 286-93, 1999.
- Dickson PV, Rich TA, Xing Y, et al.: Achieving eugastrinemia in MEN1 patients: both duodenal inspection and formal lymph node dissection are important. Surgery 150 (6): 1143-52, 2011.
- Akerström G, Stålberg P, Hellman P: Surgical management of pancreatico-duodenal tumors in multiple endocrine neoplasia syndrome type 1. Clinics (Sao Paulo) 67 (Suppl 1): 173-8, 2012.
- Zhang IY, Zhao J, Fernandez-Del Castillo C, et al.: Operative Versus Nonoperative Management of Nonfunctioning Pancreatic Neuroendocrine Tumors. J Gastrointest Surg 20 (2): 277-83, 2016.
- Vergès B, Boureille F, Goudet P, et al.: Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab 87 (2): 457-65, 2002.
- Pieterman CR, Vriens MR, Dreijerink KM, et al.: Care for patients with multiple endocrine neoplasia type 1: the current evidence base. Fam Cancer 10 (1): 157-71, 2011.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
Page Footer
I want to...
Audiences
Secure Member Sites
The Cigna Group Information
Disclaimer
Individual and family medical and dental insurance plans are insured by Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc., and Cigna HealthCare of Texas, Inc. Group health insurance and health benefit plans are insured or administered by CHLIC, Connecticut General Life Insurance Company (CGLIC), or their affiliates (see
All insurance policies and group benefit plans contain exclusions and limitations. For availability, costs and complete details of coverage, contact a licensed agent or Cigna sales representative. This website is not intended for residents of New Mexico.