Management
Diagnosis and Testing
Genetic testing for the FHgene is clinically available and performed by Clinical Laboratory Improvement Amendments (CLIA)-certified laboratories. FH currently is the only gene known to be associated with hereditary leiomyomatosis and renal cell cancer (HLRCC). Most patients with HLRCC have a germline pathogenic variant in FH.
Because the genetic analysis of HLRCC is complex, any interpretation of a variant of unknown significance result needs to be performed with consultation by clinical cancer geneticists, ideally in a center that has significant experience with this disease.
Clinical diagnosis
There is no current consensus on the diagnostic criteria for HLRCC.[1] Available guidelines were created during a previous era of histological classification when FH-deficient papillary renal cell carcinoma (RCC) was classified as type 2 papillary RCC. Suggested guidelines may need to be updated based on this RCC classification change, the availability of immunohistochemical stains to identify loss of FH, and the presence of 2SC in tissue manifestations.
Some experts suggest that a clinical dermatologic diagnosis of HLRCC requires one of the following:[2]
- Multiple cutaneous leiomyomas with at least one histologically confirmed leiomyoma.
- A single leiomyoma in the presence of a positive family history of HLRCC.
More recent comprehensive criteria for diagnosis have been suggested and are often used by experts in the field. Suggested criteria include dermatologic manifestations like multiple cutaneous leiomyomas (histologically confirmed). At least two of the following manifestations must also be present:[3,4]
- Surgical treatment for symptomatic uterine leiomyomas before age 40 years.
- A diagnosis of type 2 papillary RCC before age 40 years (now referred to as FH-deficient papillary RCC).
- A first-degree relative who meets one of these criteria.
Since FH-deficient RCC can resemble collecting duct RCC, the presence of FH-deficient RCC before age 40 years has been suggested as an additional criterion.[5] Patients with seemingly sporadic tumors who have a negative family history and a single, histologically confirmed cutaneous leiomyoma may test positive for the presence of a germline FH pathogenic variant. Although the percentage of germline pathogenic variants in these patient populations is unknown, many centers may refer patients with a single cutaneous leiomyoma for genetic counseling and testing, regardless of family history.[6]
Differential diagnosis
Cutaneous lesions
Cutaneous leiomyomas are rare. The detection of multiple lesions is specific to HLRCC. Because leiomyomas are clinically similar to various cutaneous lesions, histological diagnosis is required to objectively prove the nature of the lesion.
Uterine leiomyomas
Uterine leiomyoma is the most common benign pelvic tumor in women in the general population. Most uterine leiomyomas are sporadic and nonsyndromic.[7]
RCCs
Diagnostic clues of HLRCC may rely on the presence of several phenotypic features in different organs (cutaneous, uterine, and renal). One or more of these characteristic features may be present in the patient or in one or more of their affected biologic relatives.
Although familial RCCs are associated with rather specific renal pathology, the rarity of these syndromes results in few pathologists gaining sufficient experience to recognize their histological features.
The differential diagnoses may include other rare familial RCC syndromes with specific renal pathology, including:
- Hereditary papillary renal carcinoma (HPRC). Predisposition to papillary RCC (formerly known as type 1 papillary RCC). Inheritance is autosomal dominant.[8]
- Birt-Hogg-Dubé syndrome (BHD). A spectrum of renal tumors including renal oncocytoma (benign), chromophobe renal cell cancer (malignant), and a combination of both cell types (so-called oncocytic hybrid tumor).[9] Individuals with BHD can present with cutaneous fibrofolliculomas /trichodiscomas and/or with multiple lung cysts and spontaneous pneumothorax. Inheritance is autosomal dominant.[10,11]
Genetic testing
Genetic testing is used clinically for diagnostic confirmation of at-risk individuals. It is recommended that both pretest and posttest genetic counseling be offered to persons contemplating germline pathogenic variant testing.[12] Laboratories offering genetic testing for use in clinical decision making must be certified under CLIA laws.[13]
Testing strategy
Genetic testing for a germline FH pathogenic variant is indicated for all individuals with HLRCC and for all individuals who are suspected of having HLRCC, regardless of family history. This includes individuals with cutaneous leiomyomas, as described in the Clinical diagnosis section of this summary, and individuals who have renal tumors with histological characteristics consistent with HLRCC.[4,14,15] For more information, see the Histopathology section.
Risk to family members
HLRCC is inherited in an autosomal dominant manner.[16] If a parent of a proband is clinically affected or has a disease-causing variant, the siblings of the proband have a 50% chance of inheriting the pathogenic variant. Each child of an individual with HLRCC has a 50% chance of inheriting the pathogenic variant. The degree of clinical severity is not predictable. Prenatal genetic testing may be available in laboratories offering custom prenatal testing for families in which a pathogenic variant has been identified in an affected family member.
Parents of a proband
- Some individuals diagnosed with HLRCC have an affected parent, while others have unaffected parents, suggesting that some individuals have HLRCC as the result of a de novo pathogenic variant or parental mosaicism.
- The proportion of cases caused by de novo pathogenic variants is unknown, as subtle manifestations in parents have not been systematically evaluated; similarly, not all unaffected parents have undergone FH testing.
- Evaluation of parents of a proband with a suspected de novo pathogenic variant may include genetic testing if the FH disease-causing variant in the proband has been identified.
Although some individuals diagnosed with HLRCC have an affected parent, the family history may appear to be negative because of limited family history, failure to recognize the disorder in family members, early death of the affected parent before the onset of syndrome-related symptoms, or late onset of the disease in the affected parent.[17]
Siblings of a proband
- The risk to the siblings of the proband depends upon the genetic status of the proband's parents.
- If a parent of a proband is clinically affected or has a disease-causing variant, each sibling of the proband is at a 50% risk of inheriting the variant.
- If the disease-causing variant cannot be detected in the DNA of either parent, the risk to siblings is low. However, risk will be greater than that of the general population because there is a possibility of germline mosaicism.
Testing of at-risk family members
Use of genetic testing for early identification of at-risk family members improves diagnostic certainty. It reduces the number of unnecessary costly and stressful screening procedures in at-risk members who have not inherited their family's disease-causing variant.[13,18,19]
Early recognition of clinical manifestations may allow timely intervention, which could, in theory, improve outcome. Therefore, clinical surveillance of asymptomatic at-risk relatives for early RCC detection is reasonable, but additional objective data regarding the impact of screening on syndrome-related mortality are needed.
Related genetic counseling issues
Predicting the phenotype in individuals who have inherited a pathogenic variant
It is not possible to predict whether HLRCC-related symptoms will occur or, if they do, what the age at onset, type, severity, or clinical characteristics will be in individuals who have a pathogenic variant. In an in-depth characterization of clinical and genetic features analyzed within 21 new families, the phenotypes displayed a wide range of clinical presentations and no apparent genotype -phenotype correlations were found.[20]
When neither parent of a proband with an autosomal dominant condition has the disease-causing variant or clinical evidence of the disorder, it is likely that the proband has a de novo pathogenic variant. However, nonmedical explanations include the possibility of alternate paternity or undisclosed adoption. Genetic testing of at-risk family members is appropriate in order to identify the need for continued lifelong clinical surveillance. Interpretation of the pathogenic variant test result is most accurate when a disease-causing variant has been identified in an affected family member. Those who have a disease-causing variant are recommended to undergo lifelong periodic surveillance. Meanwhile, family members and offspring who have not inherited the pathogenic variant are thought to have RCC risks similar to those in the general population; no special management of these individuals is recommended.
Early detection of at-risk individuals affects medical management
Screening for early disease manifestations in HLRCC is an important aspect of clinical care of affected individuals. Although there are no prospective studies comparing specific renal cancer screening practices, the aggressive nature of HLRCC-associated RCC [3] justifies efforts directed at the early identification of cancer. When tumors are small and localized, partial nephrectomy may be a feasible option; however, the infiltrative nature of these tumors has led some groups to suggest a wide margin must be taken to achieve complete resection.[21]
Uterine fibroids often cause significant symptoms related to bleeding and the presence of a large mass, but small fibroids may be asymptomatic. As HLRCC fibroids can lead to hysterectomies and loss of the ability to bear children in affected young women, the goal of screening in women interested in preserving fertility is to limit some of these irreversible complications. Although there are no specific management recommendations related to HLRCC-associated fibroids, various management strategies have proven effective in the treatment of sporadic fibroids. These strategies include use of hormonal therapies, pain medications, percutaneous and endovascular procedures, and surgical options. Early referral to a fertility specialist may be useful to assist with family planning.
Surveillance
It has been suggested that individuals with a suspected or confirmed diagnosis of HLRCC, individuals with heterozygous pathogenic variants in FH regardless of clinical manifestations, and at-risk family members who have not undergone genetic testing undertake the following regular surveillance, performed by physicians familiar with the clinical manifestations of HLRCC.
- Skin. There are some published recommendations to perform skin exams on a regular basis, but there is no consensus regarding frequency of skin exams, and recommendations have not been prospectively validated.
- Uterine. For women with an intact uterus, annual gynecologic consultation is recommended, accompanied by periodic imaging. Imaging may include magnetic resonance imaging (MRI) of the pelvis or ultrasound to assess the severity of uterine leiomyomas and to search for changes suggestive of developing leiomyosarcoma.[6,7,16,22]
- Paraganglioma (PGL)/Pheochromocytoma (PHEO). Although PGL/PHEO surveillance is not included in suggested HLRCC guidelines, one study suggested that surveillance similar to that which is recommended for SDHx carriers could be considered for individuals with unique FH variants associated with increased PGL/PHEO risk (i.e., p.Thr234Ala, p.Ala273Thr, and p.Ala194Thr).[23] This surveillance regimen includes annual biochemical screening (urine or plasma metanephrines), careful physical exam with a focus on the neck and abdominal areas, and whole-body cross-sectional imaging.
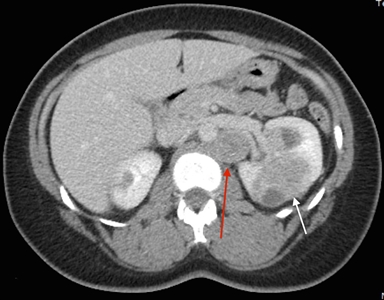
- Renal. In view of the aggressive nature of this disease, annual imaging with either computed tomography (CT) scan with contrast or MRI with gadolinium is warranted even if the initial (baseline) evaluation reveals normal kidneys. Special attention to imaging is warranted in this population because subtle findings, such as a complex cyst, may sometimes represent an aggressive malignancy. The age to initiate renal screening is uncertain, however, because HLRCC has been described in children as young as 10 years. The HLRCC Family Alliance recommends annual imaging beginning at age 8 years in children at risk of HLRCC and those with HLRCC.[1] MRI has the advantage of sparing the patient radiation exposure and for this reason it may be preferred over CT for lifetime surveillance of HLRCC patients.
Any suspicious renal lesion (indeterminate, questionable, or complex cysts) at a previous examination should be closely followed with periodic imaging, preferably using the same modality to allow for comparisons. The use of renal ultrasound examination may be helpful in the characterization of cystic lesions identified on cross-sectional imaging. It should be cautioned that ultrasound examination alone is never sufficient. Renal tumors should be evaluated by a clinician familiar with HLRCC-related renal cancer.[10,24]
Because of the aggressive growth of these tumors, patients warrant regular surveillance with a low threshold for early surgical intervention for solid renal lesions. This strategy differs from that described for several other hereditary RCC syndromes, in which the tumor behavior is more indolent, and for which observation may be a viable option.[10,24,25]
Level of evidence (skin surveillance): 5
Level of evidence (uterine surveillance): 4
Level of evidence (renal surveillance): 4
Treatment of Manifestations
Cutaneous lesions
Cutaneous leiomyomas are most appropriately examined by a dermatologist. Generally, asymptomatic cutaneous leiomyomas require no treatment. Treatment of symptomatic cutaneous leiomyomas may be difficult if a patient has diffuse disease in a wide distribution. Surgical excision may be performed for a solitary painful lesion. Lesions can be treated by cryoablation and/or lasers. Several medications, including calcium channel blockers, alpha blockers, nitroglycerin, antidepressants, and antiepileptic drugs, reportedly reduce leiomyoma-related pain.[26] A small, randomized clinical trial (09-C-0072 [NCT00971620]) showed that intralesional injection of botulinum toxin A (Botox) may improve quality of life.[27]
Level of evidence: 5
Uterine leiomyomas
Uterine leiomyomas are best evaluated by a gynecologist. HLRCC-associated leiomyomas are treated in the same manner as sporadic leiomyomas. However, because of the multiplicity, size, and potential rapid growth observed in HLRCC-related uterine leiomyomas, most women may require medical and/or surgical intervention earlier and more often than would be expected in the general population. Medical therapy (currently including gonadotropin-releasing hormone agonists, anti-hormonal medications, and pain relievers) may be used to initially treat uterine leiomyomas, both to decrease their size in preparation for surgical removal and to provide temporary relief from leiomyoma-related pain. When women desire fertility preservation, myomectomy can be used to remove leiomyomas and simultaneously preserve the uterus. Hysterectomy should be performed only when necessary.[6,7]
Level of evidence: 4
RCCs
Efforts aimed at early detection of HLRCC-related RCC are prudent, given its biological aggressiveness. However, studies do not currently show that early detection is clearly associated with improved survival. Surgical excision of these malignancies at the first sign of disease is recommended, unlike management of other hereditary cancer syndromes. The propensity for lymph node involvement, even with small renal tumors, may necessitate a lymph node dissection for more appropriate staging.[21] Radical nephrectomy or partial nephrectomy with wide margins should be considered in individuals with detectable renal masses, including small, subcentimetric tumors.[10,24,25]
Level of evidence: 4
Therapies under investigation
It has been suggested that hypoxia-inducible factor (HIF1)-alpha overexpression is involved in HLRCC tumorigenesis.[28,29] Therefore, potential targeted therapies for HLRCC-associated tumors may include HIF1-alpha targeting agents, when such agents become clinically available.
Loss of oxidative phosphorylation resulting from biallelic inactivation of FH renders HLRCC tumors almost entirely reliant on aerobic glycolysis for meeting cellular adenosine triphosphate and other bioenergetics requirements. Consequently, targeting aerobic glycolysis is being explored as a therapeutic strategy.[30,31] A phase II study (10-C-0114 [NCT01130519]) examining the combination of bevacizumab and erlotinib for the treatment of advanced HLRCC is ongoing and is based partly on the premise that this combination might inhibit effective glucose delivery to tumor cells.[32]
Other investigations [33] evaluating the known consequences of FH inactivation in HLRCC kidney cancer have confirmed that there are very high levels of NAD(P)H dehydrogenase quinine 1 (NQO1) expression in HLRCC kidney tumors. Expression of this compound is lower in other types of hereditary RCC, including the clear cell RCC associated with von Hippel-Lindau disease and the papillary RCC associated with HPRC. The activation of an oxidative stress response pathway mediated by NRF2, a transcription factor that regulates the transcription of NQO1, could explain NQO1 overexpression in these tumors. Vandetanib, an oral VEGFR2 and EGFR inhibitor with additional activity against Abl-1 kinase, has potent activity against FH-deficient cells in vitro and induces regression of HLRCC-derived xenografts in mice. The activity of vandetanib in this model is mediated, at least in part, by its ability to disrupt the NRF2-mediated cytoprotective oxidative stress response pathway in an Abl-dependent fashion. Furthermore, metformin, an activator of 5'–AMP activated protein kinase (AMPK), was synergistic with vandetanib both in vitro and in mouse xenografts derived from FH-deficient human renal cancer.[34] These data provided the basis for a clinical trial (NCT02495103) to evaluate the efficacy of this combination in HLRCC patients with advanced kidney cancer.
General information about clinical trials is also available from the NCI website.
References:
- HLRCC Family Alliance: The HLRCC Handbook. Version 2.0. Boston, MA: HLRCC Family Alliance, 2013. Available online. Last accessed November 6, 2024.
- Schmidt LS, Linehan WM: Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis 7: 253-60, 2014.
- Smit DL, Mensenkamp AR, Badeloe S, et al.: Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet 79 (1): 49-59, 2011.
- Menko FH, Maher ER, Schmidt LS, et al.: Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer 13 (4): 637-44, 2014.
- Lehtonen HJ: Hereditary leiomyomatosis and renal cell cancer: update on clinical and molecular characteristics. Fam Cancer 10 (2): 397-411, 2011.
- Toro JR, Nickerson ML, Wei MH, et al.: Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 73 (1): 95-106, 2003.
- Stewart L, Glenn GM, Stratton P, et al.: Association of germline mutations in the fumarate hydratase gene and uterine fibroids in women with hereditary leiomyomatosis and renal cell cancer. Arch Dermatol 144 (12): 1584-92, 2008.
- Zbar B, Tory K, Merino M, et al.: Hereditary papillary renal cell carcinoma. J Urol 151 (3): 561-6, 1994.
- Pavlovich CP, Walther MM, Eyler RA, et al.: Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol 26 (12): 1542-52, 2002.
- Linehan WM, Bratslavsky G, Pinto PA, et al.: Molecular diagnosis and therapy of kidney cancer. Annu Rev Med 61: 329-43, 2010.
- Schmidt LS, Linehan WM: Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol 12 (10): 558-69, 2015.
- Riley BD, Culver JO, Skrzynia C, et al.: Essential elements of genetic cancer risk assessment, counseling, and testing: updated recommendations of the National Society of Genetic Counselors. J Genet Couns 21 (2): 151-61, 2012.
- Update: information about obtaining a CLIA certificate. Jt Comm Perspect 26 (12): 6-7, 2006.
- Kopp RP, Stratton KL, Glogowski E, et al.: Utility of prospective pathologic evaluation to inform clinical genetic testing for hereditary leiomyomatosis and renal cell carcinoma. Cancer 123 (13): 2452-2458, 2017.
- Merino MJ, Torres-Cabala C, Pinto P, et al.: The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol 31 (10): 1578-85, 2007.
- Launonen V, Vierimaa O, Kiuru M, et al.: Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A 98 (6): 3387-92, 2001.
- Refae MA, Wong N, Patenaude F, et al.: Hereditary leiomyomatosis and renal cell cancer: an unusual and aggressive form of hereditary renal carcinoma. Nat Clin Pract Oncol 4 (4): 256-61, 2007.
- American Society of Clinical Oncology: American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility. J Clin Oncol 21 (12): 2397-406, 2003.
- Robson ME, Storm CD, Weitzel J, et al.: American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol 28 (5): 893-901, 2010.
- Wei MH, Toure O, Glenn GM, et al.: Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 43 (1): 18-27, 2006.
- Shuch B, Singer EA, Bratslavsky G: The surgical approach to multifocal renal cancers: hereditary syndromes, ipsilateral multifocality, and bilateral tumors. Urol Clin North Am 39 (2): 133-48, v, 2012.
- Patel VM, Handler MZ, Schwartz RA, et al.: Hereditary leiomyomatosis and renal cell cancer syndrome: An update and review. J Am Acad Dermatol 77 (1): 149-158, 2017.
- Zavoshi S, Lu E, Boutros PC, et al.: Fumarate Hydratase Variants and Their Association With Paraganglioma/Pheochromocytoma. Urology 176: 106-114, 2023.
- Grubb RL, Franks ME, Toro J, et al.: Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol 177 (6): 2074-9; discussion 2079-80, 2007.
- Pfaffenroth EC, Linehan WM: Genetic basis for kidney cancer: opportunity for disease-specific approaches to therapy. Expert Opin Biol Ther 8 (6): 779-90, 2008.
- Ritzmann S, Hanneken S, Neumann NJ, et al.: Type 2 segmental manifestation of cutaneous leiomyomatosis in four unrelated women with additional uterine leiomyomas (Reed's Syndrome). Dermatology 212 (1): 84-7, 2006.
- Naik HB, Steinberg SM, Middelton LA, et al.: Efficacy of Intralesional Botulinum Toxin A for Treatment of Painful Cutaneous Leiomyomas: A Randomized Clinical Trial. JAMA Dermatol 151 (10): 1096-102, 2015.
- Isaacs JS, Jung YJ, Mole DR, et al.: HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8 (2): 143-53, 2005.
- Pollard PJ, Brière JJ, Alam NA, et al.: Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14 (15): 2231-9, 2005.
- Xie H, Valera VA, Merino MJ, et al.: LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther 8 (3): 626-35, 2009.
- Yamasaki T, Tran TA, Oz OK, et al.: Exploring a glycolytic inhibitor for the treatment of an FH-deficient type-2 papillary RCC. Nat Rev Urol 8 (3): 165-71, 2011.
- Linehan WM, Srinivasan R, Garcia JA: Non-clear cell renal cancer: disease-based management and opportunities for targeted therapeutic approaches. Semin Oncol 40 (4): 511-20, 2013.
- Linehan WM, Pinto PA, Srinivasan R, et al.: Identification of the genes for kidney cancer: opportunity for disease-specific targeted therapeutics. Clin Cancer Res 13 (2 Pt 2): 671s-679s, 2007.
- Sourbier C, Ricketts CJ, Matsumoto S, et al.: Targeting ABL1-mediated oxidative stress adaptation in fumarate hydratase-deficient cancer. Cancer Cell 26 (6): 840-50, 2014.