Juvenile Myelomonocytic Leukemia Treatment (PDQ®): Treatment - Health Professional Information [NCI]
Pathogenesis and Risk Factors
The pathogenesis of juvenile myelomonocytic leukemia (JMML) has been closely linked to activation of the RAS oncogene pathway, along with related syndromes (see
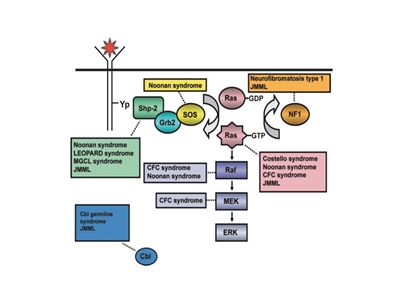
Figure 1. Schematic diagram showing ligand-stimulated Ras activation, the Ras-Erk pathway, and the gene mutations found to date contributing to the neuro-cardio-facio-cutaneous congenital disorders and JMML. NL/MGCL: Noonan-like/multiple giant cell lesion; CFC: cardia-facio-cutaneous; JMML: juvenile myelomonocytic leukemia. Reprinted from Leukemia Research, 33 (3), Rebecca J. Chan, Todd Cooper, Christian P. Kratz, Brian Weiss, Mignon L. Loh, Juvenile myelomonocytic leukemia: A report from the 2nd International JMML Symposium, Pages 355-62, Copyright 2009, with permission from Elsevier.
Syndromes and genetic features associated with an increased risk of developing JMML include the following:[
- Neurofibromatosis type 1 (NF1). Up to 14% of cases of JMML occur in children with NF1.[
7 ] - Noonan syndrome. Noonan syndrome is usually inherited as an autosomal dominant condition but can also arise spontaneously. It is characterized by facial dysmorphism, short stature, webbed neck, and neurocognitive and cardiac abnormalities. Germline variants in PTPN11 are observed in children with Noonan syndrome and in children with JMML.[
8 ,9 ,10 ]Importantly, some children with Noonan syndrome have hematologic features indistinguishable from JMML that self-resolve during infancy, similar to what happens in children with Down syndrome and transient myeloproliferative disorder.[
2 ,10 ]In a large prospective cohort of 641 patients with Noonan syndrome and a germline PTPN11 variant, 36 patients (approximately 6%) showed myeloproliferative features, with 20 patients (approximately 3%) meeting the consensus diagnostic criteria for JMML.[
10 ]- Of the 20 patients meeting the criteria for JMML, 12 patients had severe neonatal manifestations (e.g., life-threatening complications related to congenital heart defects, pleural effusion, leukemia infiltrates, and/or thrombocytopenia), and 10 of 20 patients died during the first month of life.
- Among the remaining eight patients, none required intensive therapy at diagnosis or during follow-up.
- All 16 patients with myeloproliferative features that did not meet JMML criteria were alive, with a median follow-up of 3 years, and no patient received chemotherapy.
- Variants in the CBL gene. CBL is an E3 ubiquitin-protein ligase that is involved in targeting proteins, particularly tyrosine kinases, for proteasomal degradation. Variants in the CBL gene occur in 10% to 15% of JMML cases,[
11 ,12 ] with many of these cases occurring in children with germline CBL variants.[13 ,14 ,15 ]CBL germline variants result in an autosomal dominant developmental disorder that is often characterized by impaired growth, developmental delay, cryptorchidism, and a predisposition to JMML.[
13 ,15 ] Some individuals with CBL germline variants experience spontaneous regression of their JMML but develop vasculitis later in life,[13 ] whereas patients with only somatic CBL variants require therapy.[15 ] JMML arising from germline variants is clinically indistinguishable from JMML arising from somatic variants, which necessitates studies of both normal and leukemic tissue.[15 ]CBL variants are nearly always mutually exclusive of RAS and PTPN11 variants.[11 ]
References:
- Chan RJ, Cooper T, Kratz CP, et al.: Juvenile myelomonocytic leukemia: a report from the 2nd International JMML Symposium. Leuk Res 33 (3): 355-62, 2009.
- Loh ML: Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol 152 (6): 677-87, 2011.
- Bresolin S, Zecca M, Flotho C, et al.: Gene expression-based classification as an independent predictor of clinical outcome in juvenile myelomonocytic leukemia. J Clin Oncol 28 (11): 1919-27, 2010.
- Olk-Batz C, Poetsch AR, Nöllke P, et al.: Aberrant DNA methylation characterizes juvenile myelomonocytic leukemia with poor outcome. Blood 117 (18): 4871-80, 2011.
- Stiller CA, Chessells JM, Fitchett M: Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer 70 (5): 969-72, 1994.
- Choong K, Freedman MH, Chitayat D, et al.: Juvenile myelomonocytic leukemia and Noonan syndrome. J Pediatr Hematol Oncol 21 (6): 523-7, 1999 Nov-Dec.
- Niemeyer CM, Arico M, Basso G, et al.: Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89 (10): 3534-43, 1997.
- Tartaglia M, Niemeyer CM, Fragale A, et al.: Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34 (2): 148-50, 2003.
- Kratz CP, Niemeyer CM, Castleberry RP, et al.: The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 106 (6): 2183-5, 2005.
- Strullu M, Caye A, Lachenaud J, et al.: Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet 51 (10): 689-97, 2014.
- Loh ML, Sakai DS, Flotho C, et al.: Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 114 (9): 1859-63, 2009.
- Muramatsu H, Makishima H, Jankowska AM, et al.: Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood 115 (10): 1969-75, 2010.
- Niemeyer CM, Kang MW, Shin DH, et al.: Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet 42 (9): 794-800, 2010.
- Pérez B, Mechinaud F, Galambrun C, et al.: Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet 47 (10): 686-91, 2010.
- Hecht A, Meyer JA, Behnert A, et al.: Molecular and phenotypic diversity of CBL-mutated juvenile myelomonocytic leukemia. Haematologica 107 (1): 178-186, 2022.
Genomics of Juvenile Myelomonocytic Leukemia (JMML)
Molecular Features of JMML
The genomic landscape of JMML is characterized by variants in one of five genes of the RAS pathway: NF1, NRAS, KRAS, PTPN11, and CBL.[
The variant rate in JMML leukemia cells is very low, but additional variants beyond those of the five RAS pathway genes described above are observed.[
A report describing the genomic landscape of JMML found that 16 of 150 patients (11%) lacked canonical RAS pathway variants. Among these 16 patients, 3 were observed to have in-frame fusions involving receptor tyrosine kinases (DCTN1::ALK, RANBP2::ALK, and TBL1XR1::ROS1 gene fusions). These patients all had monosomy 7 and were aged 56 months or older. One patient with an ALK gene fusion was treated with crizotinib plus conventional chemotherapy and achieved a complete molecular remission and proceeded to allogeneic bone marrow transplant.[
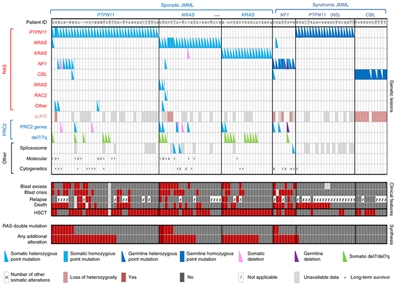
Figure 2. Alteration profiles in individual JMML cases. Germline and somatically acquired alterations with recurring hits in the RAS pathway and PRC2 network are shown for 118 patients with JMML who underwent detailed genetic analysis. Blast excess was defined as a blast count ≥10% but <20% of nucleated cells in the bone marrow at diagnosis. Blast crisis was defined as a blast count ≥20% of nucleated cells in the bone marrow. NS, Noonan syndrome. Reprinted by permission from Macmillan Publishers Ltd: Nature Genetics (Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 [11]: 1334-40, 2015), copyright (2015).
Genomic and Molecular Prognostic Factors
Several genomic factors affect the prognosis of patients with JMML, including the following:
- Number of non–RAS pathway variants. A predictor of prognosis for children with JMML is the number of variants beyond the disease-defining RAS pathway variants.[
1 ,2 ]- One study observed that zero or one somatic alteration (pathogenic variant or monosomy 7) was identified in 64 patients (65.3%) at diagnosis, whereas two or more alterations were identified in 34 patients (34.7%).[
2 ] In multivariate analysis, variant number (2 or more vs. 0 or 1) maintained significance as a predictor of inferior event-free survival (EFS) and overall survival (OS). A higher proportion of patients diagnosed with two or more alterations were older and male, and these patients also demonstrated a higher rate of monosomy 7 or somatic NF1 variants.[2 ] - Another study observed that approximately 60% of patients had one or more additional variants beyond their disease-defining RAS pathway variant. These patients had an inferior OS compared with patients who had no additional variants (3-year OS rate, 61% vs. 85%, respectively).[
1 ] - A third study observed a trend for an inferior OS for patients with two or more variants compared with patients with zero or one variant.[
3 ]
- One study observed that zero or one somatic alteration (pathogenic variant or monosomy 7) was identified in 64 patients (65.3%) at diagnosis, whereas two or more alterations were identified in 34 patients (34.7%).[
- RAS pathway double variants. Although variants in the five canonical RAS pathway genes associated with JMML (NF1, NRAS, KRAS, PTPN11, and CBL) are generally mutually exclusive, 4% to 17% of cases have variants in two of these RAS pathway genes.[
1 ,2 ] This finding has been associated with a poorer prognosis.[1 ,2 ]- Two RAS pathway variants were identified in 11% of JMML patients in one report, and these patients had a significantly inferior EFS rate (14%) compared with patients who had a single RAS pathway variant (62%). Patients with Noonan syndrome were excluded from the analyses.[
2 ] - Similar findings for RAS pathway variants were reported in a second study. This study observed that patients with RAS pathway double variants (15 of 96 patients) had lower survival rates than did patients with either no additional variants or with additional variants beyond the RAS pathway variant.[
1 ]
- Two RAS pathway variants were identified in 11% of JMML patients in one report, and these patients had a significantly inferior EFS rate (14%) compared with patients who had a single RAS pathway variant (62%). Patients with Noonan syndrome were excluded from the analyses.[
- DNA methylation profile.
- One study applied DNA methylation profiling to a discovery cohort of 39 patients with JMML and to a validation cohort of 40 patients. Distinctive subsets of JMML with either high, intermediate, or low methylation levels were observed in both cohorts. Patients with the lowest methylation levels had the highest survival rates, and all but 1 of 15 patients experienced spontaneous resolution in the low methylation cohort. High methylation status was associated with lower EFS rates.[
5 ] - Another study applied DNA methylation profiling to a cohort of 106 patients with JMML. The study observed one subgroup of patients with a hypermethylation profile and one subgroup of patients with a hypomethylation profile. Patients in the hypermethylation group had a significantly lower OS rate than did patients in the hypomethylation group (5-year OS rate, 46% vs. 73%, respectively). Patients in the hypermethylation group also had a significantly poorer 5-year transplant-free survival rate than did patients in the hypomethylation group (2.2%; 95% CI, 0.2%–10.1% vs. 41.2%; 95% CI, 27.1%–54.8%). Hypermethylation status was associated with two or more variants, higher fetal hemoglobin levels, older age, and lower platelet count at diagnosis. All patients with Noonan syndrome were in the hypomethylation group.[
3 ] - A study examined 33 patients with JMML who had CBL variants. The study identified 31 patients with low methylation and 2 patients with intermediate methylation. Both of the children with intermediate methylation relapsed after undergoing HSCT. Because treatment, which included observation only, varied among the 31 patients with low methylation, the impact of the methylation profile on therapeutic decisions and outcomes could not be fully assessed. However, the methylation status was not prognostic of spontaneous resolution.[
6 ]
- One study applied DNA methylation profiling to a discovery cohort of 39 patients with JMML and to a validation cohort of 40 patients. Distinctive subsets of JMML with either high, intermediate, or low methylation levels were observed in both cohorts. Patients with the lowest methylation levels had the highest survival rates, and all but 1 of 15 patients experienced spontaneous resolution in the low methylation cohort. High methylation status was associated with lower EFS rates.[
- LIN28B overexpression. LIN28B overexpression, which is present in approximately one-half of children with JMML, identifies a biologically distinctive subset of JMML. LIN28B is an RNA-binding protein that regulates stem cell renewal.[
7 ]- LIN28B overexpression was positively correlated with high blood fetal hemoglobin level and age (both of which are associated with poor prognosis), and it was negatively correlated with presence of monosomy 7 (also associated with inferior prognosis). Although LIN28B overexpression identifies a subset of patients with increased risk of treatment failure, it was not found to be an independent prognostic factor when other factors such as age and monosomy 7 status are considered.[
7 ] - Another study also observed a subset of JMML patients with elevated LIN28B expression. The study identified LIN28B as the gene for which expression was most strongly associated with hypermethylation status.[
3 ]
- LIN28B overexpression was positively correlated with high blood fetal hemoglobin level and age (both of which are associated with poor prognosis), and it was negatively correlated with presence of monosomy 7 (also associated with inferior prognosis). Although LIN28B overexpression identifies a subset of patients with increased risk of treatment failure, it was not found to be an independent prognostic factor when other factors such as age and monosomy 7 status are considered.[
References:
- Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 (11): 1334-40, 2015.
- Stieglitz E, Taylor-Weiner AN, Chang TY, et al.: The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47 (11): 1326-33, 2015.
- Murakami N, Okuno Y, Yoshida K, et al.: Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 131 (14): 1576-1586, 2018.
- Sakaguchi H, Okuno Y, Muramatsu H, et al.: Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet 45 (8): 937-41, 2013.
- Stieglitz E, Mazor T, Olshen AB, et al.: Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat Commun 8 (1): 2127, 2017.
- Hecht A, Meyer JA, Behnert A, et al.: Molecular and phenotypic diversity of CBL-mutated juvenile myelomonocytic leukemia. Haematologica 107 (1): 178-186, 2022.
- Helsmoortel HH, Bresolin S, Lammens T, et al.: LIN28B overexpression defines a novel fetal-like subgroup of juvenile myelomonocytic leukemia. Blood 127 (9): 1163-72, 2016.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
Page Footer
I want to...
Audiences
Secure Member Sites
The Cigna Group Information
Disclaimer
Individual and family medical and dental insurance plans are insured by Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc., and Cigna HealthCare of Texas, Inc. Group health insurance and health benefit plans are insured or administered by CHLIC, Connecticut General Life Insurance Company (CGLIC), or their affiliates (see
All insurance policies and group benefit plans contain exclusions and limitations. For availability, costs and complete details of coverage, contact a licensed agent or Cigna sales representative. This website is not intended for residents of New Mexico.