Retinoblastoma Treatment (PDQ®): Treatment - Health Professional Information [NCI]
General Information About Retinoblastoma
Retinoblastoma is a pediatric cancer that requires careful integration of multidisciplinary care. Treatment of retinoblastoma aims to save the patient's life and preserve useful vision. For patients presenting with extraocular retinoblastoma, treatment with systemic chemotherapy and radiation therapy is likely to be curative. However, extraorbital disease requires intensive chemotherapy and may include consolidation with high-dose chemotherapy and autologous hematopoietic stem cell rescue with or without radiation therapy. While a large proportion of patients with systemic extra–central nervous system (CNS) metastases can be cured, the prognosis for patients with intracranial disease is dismal.
Incidence
Retinoblastoma is a relatively uncommon tumor of childhood that arises in the retina. It accounts for about 3% of the cancers occurring in children younger than 15 years.
Retinoblastoma is a cancer of the very young child. Two-thirds of all cases of retinoblastoma are diagnosed before age 2 years.[
Anatomy
Retinoblastoma arises in the retina, and it may grow under the retina and/or toward the vitreous cavity. Involvement of the ocular coats and optic nerve occurs as a sequence of events as the tumor progresses.
Focal invasion of the choroid is common, although massive invasion occurs in cases of advanced disease. After invading the choroid, the tumor gains access to systemic circulation and creates the potential for metastases. Further progression through the ocular coats leads to invasion of the sclera and the orbit. Tumors that invade the anterior chamber may gain access to systemic circulation through the canal of Schlemm. Progression through the optic nerve and past the lamina cribrosa increases the risk of systemic and CNS dissemination (see
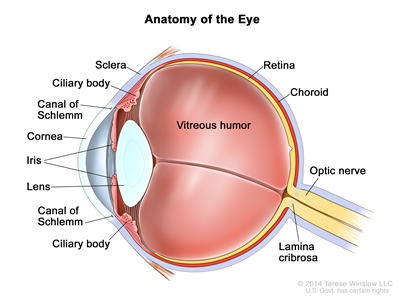
Figure 1. Anatomy of the eye showing the sclera, ciliary body, canal of Schlemm, cornea, iris, lens, vitreous humor, retina, choroid, optic nerve, and lamina cribrosa. The vitreous humor is a gel that fills the center of the eye.
Screening
Consensus reports from the American Association of Ophthalmic Oncologists and Pathologists and the American Association for Cancer Research Childhood Cancer Predisposition Workshop describe screening guidelines for children at risk of developing retinoblastoma.[
In children with a positive family history of retinoblastoma, early-in-life screening by fundus examination is performed under general anesthesia at regular intervals. Examinations are performed according to a schedule based on the absolute estimated risk, as determined by identification of the RB1 variant in the family and the presence of the RB1 variant in the child.[
Infants born to affected parents have a dilated eye examination under anesthesia as soon as possible in the first month of life, and a genetic evaluation is performed. Infants with a positive genetic test are examined under anesthesia on a monthly basis. In infants who do not develop disease, monthly examinations continue throughout the first year. The frequency of those examinations may be decreased progressively during the second and subsequent years. Screening children with a positive family history of retinoblastoma can improve their prognosis, in terms of globe sparing and use of less intensive, ocular-salvage treatments (see
| Relative of Proband | Pretest Risk for Mutant Allele (%) | |
|---|---|---|
| | Bilateral Proband (100) | Unilateral Proband (15) |
| a Reprinted from |
||
| b Pretest risk forRB1mutation in family members of an affected child with |
||
| c Third- and fourth-degree relatives of unilateral probands have calculated risks of 0.003% and 0.001%, respectively, which are less than the normal population risk of 0.007% (1 in 15,000 live births); therefore, the risk is stated at 0.007%. | ||
| Offspring (infant) | 50 | 7.5 |
| Parent | 5 | 0.8 |
| Sibling | 2.5 | 0.4 |
| Niece/nephew | 1.3 | 0.2 |
| Aunt/uncle | 0.1 | 0.007c |
| First cousin | 0.05 | 0.007c |
| General population | 0.007 | |
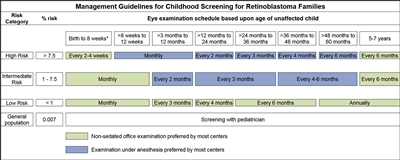
Figure 2. Management guidelines for childhood screening for retinoblastoma. The presented schedules are general guidelines and reflect a schedule for examinations in which no lesions of concern are noted. It may be appropriate to examine some children more frequently. Decisions regarding examination method, examination under anesthesia (EUA) versus nonsedated examination in the office, are complex and best decided by the clinician in discussion with the patient's family. The preference of the majority of the clinical centers involved in the creation of this consensus statement is reflected, but individual centers may make policy decisions based on available resources and expert clinician preference. Examination under anesthesia will be strongly considered for any child who is unable to participate in an office examination sufficiently to allow thorough examination of the retina. *A minority of clinical centers also prefer EUA for high- and intermediate-risk children (calculated risk >1%) from birth to 8 weeks of age. Reprinted from Ophthalmology, Volume 125, Issue 3, Alison H. Skalet, Dan S. Gombos, Brenda L. Gallie, Jonathan W. Kim, Carol L. Shields, Brian P. Marr, Sharon E. Plon, Patricia Chévez-Barrios, Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists, Pages 453–458, Copyright (2018), with permission from Elsevier.
It is common practice to use ophthalmic examinations to screen the parents and siblings of patients with retinoblastoma to exclude an unknown familial disease. However, in the absence of genetic testing, the screening plan for a child with a biological parent who had unilateral retinoblastoma is not well defined.[
Clinical Presentation
Age at presentation correlates with laterality. Patients with bilateral disease present at a younger age, usually in the first 12 months of life.
Most patients present with leukocoria, which is occasionally first noticed after a flash photograph is taken. Strabismus is the second most common presenting sign and usually correlates with macular involvement. Very advanced intraocular tumors present with pain, orbital cellulitis, glaucoma, or buphthalmos.
As the tumor progresses, patients may present with orbital or metastatic disease. Metastases occur in the preauricular and laterocervical lymph nodes, in the CNS, or systemically (commonly in the bones, bone marrow, and liver).
In the United States, Hispanic children and children living in lower socioeconomic conditions have presented with more advanced disease.[
Diagnostic and Staging Evaluation
Diagnostic evaluation of retinoblastoma includes the following:
- Eye examination. Intraocular retinoblastoma is usually diagnosed without pathological confirmation. An examination under anesthesia with a maximally dilated pupil and scleral indentation is required to inspect the entire retina. The number, location, and size of tumors; the presence of retinal detachment and subretinal fluid; and the presence of subretinal and vitreous seeds must be documented in detail.
- Ocular ultrasonography and magnetic resonance imaging (MRI). Bidimensional ocular ultrasonography and MRI can be useful to differentiate retinoblastoma from other causes of leukocoria and in the evaluation of extrascleral and extraocular extension in children with advanced intraocular retinoblastoma. Optic nerve enhancement by MRI does not necessarily indicate involvement. Cautious interpretation of those findings is needed.[
7 ]
Patients with suspected extraocular extension by imaging or high-risk pathology in the enucleated eye (i.e., massive choroidal invasion or involvement of the sclera or the optic nerve beyond the lamina cribrosa) may need to be evaluated for the presence of metastatic disease. Patients presenting with these pathological features in the enucleated eye are at high risk of developing metastases. In these cases, the following procedures may be performed:[
- Bone scintigraphy.
- Bone marrow aspiration and biopsy.
- Lumbar puncture.
Genetics and Genomics of Retinoblastoma
Retinoblastoma is a tumor that occurs in heritable (25%–30%) and nonheritable (70%–75%) forms.
Heritable Retinoblastoma
Heritable retinoblastoma is defined by the presence of a germline pathogenic variant of the RB1 gene. This germline pathogenic variant may have been inherited from an affected progenitor (25% of cases) or may have occurred in a germ cell before conception or in utero during early embryogenesis in patients with sporadic disease (75% of cases). The presence of positive family history or bilateral or multifocal disease is suggestive of heritable disease.
Heritable retinoblastoma may manifest as unilateral or bilateral disease. The penetrance of the RB1 variant (laterality, age at diagnosis, and number of tumors) is probably dependent on concurrent genetic modifiers such as MDM2 and MDM4 polymorphisms.[
Children with heritable retinoblastoma tend to be diagnosed at a younger age than children with the nonheritable form of the disease.[
Nonheritable Retinoblastoma
The genomic landscape of retinoblastoma is driven by alterations in RB1 that lead to biallelic inactivation.[
Recurrent changes in genes other than RB1 are uncommon in retinoblastoma but do occur. Variants or deletions of BCOR and amplification of MYCN are the most frequently reported events.[
Genetic Counseling
Genetic counseling is an integral part of the management of patients with retinoblastoma and their families, regardless of clinical presentation. Counseling includes a discussion of the main forms of retinoblastoma, which helps parents understand the genetic consequences of each form of retinoblastoma and estimate the risk of disease in family members.[
Genetic counseling, however, is not always straightforward. Approximately 10% of children with retinoblastoma have somatic genetic mosaicism, which contributes to the difficulty of genetic counseling.[
Genetic Testing
Blood and tumor samples can be tested to determine whether a patient with retinoblastoma has a germline or somatic variant in the RB1 gene. Once the patient's genetic variant has been identified, other family members can be screened directly for the variant with targeted sequencing.
A multistep assay that includes the following may be performed for a complete genetic evaluation of the RB1 gene:[
- DNA sequencing to identify variants within coding exons and immediate flanking intronic regions plus the promoter regions.
- Duplication/deletion analysis.
- Methylation analysis of the RB1 promoter region on DNA isolated from the tumor.
In cases of somatic mosaicism or cytogenetic abnormalities, the variants may not be easily detected. More exhaustive techniques such as karyotyping, fluorescence in situ hybridization, and methylation analysis of the RB1 promoter may be needed. Deep (2500x) sequencing of an RB1 genomic amplicon from lymphocyte DNA can reveal low-level mosaicism.[
The absence of detectable somatic RB1 variants in approximately 3% of unilateral, nonheritable retinoblastoma cases suggests that alternative genetic mechanisms may underlie the development of retinoblastoma.[
Postdiagnosis Surveillance
Children with a germline RB1 pathogenic variant may continue to develop new tumors for a few years after diagnosis and treatment. For this reason, these patients need to be examined frequently. It is common practice for examinations to occur every 2 to 4 months for at least 28 months.[
A proportion of children who present with unilateral retinoblastoma will eventually develop disease in the opposite eye. Periodic examinations of the unaffected eye are performed until the germline status of the RB1 gene is determined.
Because of the poor prognosis for patients with trilateral retinoblastoma, screening with neuroimaging until age 5 years is a common practice in the monitoring of children with the heritable form of the disease. For more information, see the
Causes of Retinoblastoma-Related Mortality
While retinoblastoma is a highly curable disease, the challenge is to preserve life and to prevent the loss of an eye, blindness, and other serious effects of treatment that reduce the patient's life span or quality of life. With improvements in the diagnosis and management of retinoblastoma over the past several decades, metastatic retinoblastoma is observed less frequently in the United States and other developed nations. As a result, other causes, such as trilateral retinoblastoma and subsequent neoplasms (SNs), have become significant contributors to retinoblastoma-related mortality in the first and subsequent decades of life. In the United States, before the advent of chemoreduction as a means of treating heritable or bilateral disease and the implementation of neuroimaging screening, trilateral retinoblastoma contributed to more than 50% of retinoblastoma-related mortality for patients in the first decade after their diagnosis.[
Trilateral retinoblastoma
Trilateral retinoblastoma is a well-recognized syndrome that occurs in 5% to 15% of patients with heritable retinoblastoma. It is defined by the development of an asynchronous intracranial midline neuroblastic tumor, which typically develops between the ages of 20 and 36 months.[
Because of the poor prognosis and the apparent improved survival with early detection and aggressive treatment of trilateral retinoblastoma, screening with routine neuroimaging could potentially detect most cases within 2 years of the first retinoblastoma diagnosis.[
Although it is not clear whether early diagnosis can impact survival, screening with MRI has been recommended as often as every 6 months for 5 years for patients suspected of having heritable disease or those with unilateral disease and a positive family history.[
A cystic pineal gland, which is commonly detected by surveillance MRI, needs to be distinguished from a cystic variant of pineoblastoma. In children without retinoblastoma, the incidence of pineal cysts has been reported to be 55.8%.[
References:
- Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649.
Also available online . Last accessed December 22, 2023. - National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute.
Available online . Last accessed February 25, 2025. - Skalet AH, Gombos DS, Gallie BL, et al.: Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology 125 (3): 453-458, 2018.
- Kamihara J, Bourdeaut F, Foulkes WD, et al.: Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clin Cancer Res 23 (13): e98-e106, 2017.
- Abramson DH: Re: Skalet et al.: Screening children at risk for retinoblastoma: consensus report from the American Association of Ophthalmic Oncologists and Pathologists (Ophthalmology. 2018;125:453-458). Ophthalmology 125 (9): e63-e64, 2018.
- Truong B, Green AL, Friedrich P, et al.: Ethnic, Racial, and Socioeconomic Disparities in Retinoblastoma. JAMA Pediatr 169 (12): 1096-104, 2015.
- Khurana A, Eisenhut CA, Wan W, et al.: Comparison of the diagnostic value of MR imaging and ophthalmoscopy for the staging of retinoblastoma. Eur Radiol 23 (5): 1271-80, 2013.
- Kaliki S, Shields CL, Rojanaporn D, et al.: High-risk retinoblastoma based on international classification of retinoblastoma: analysis of 519 enucleated eyes. Ophthalmology 120 (5): 997-1003, 2013.
- Castéra L, Sabbagh A, Dehainault C, et al.: MDM2 as a modifier gene in retinoblastoma. J Natl Cancer Inst 102 (23): 1805-8, 2010.
- de Oliveira Reis AH, de Carvalho IN, de Sousa Damasceno PB, et al.: Influence of MDM2 and MDM4 on development and survival in hereditary retinoblastoma. Pediatr Blood Cancer 59 (1): 39-43, 2012.
- Shields CL, Dockery P, Ruben M, et al.: Likelihood of Germline Mutation With Solitary Unilateral Retinoblastoma Based on Patient Age at Presentation: Analysis of 482 Consecutive Patients. J Pediatr Ophthalmol Strabismus 58 (6): 355-364, 2021 Nov-Dec.
- Andreoli MT, Chau FY, Shapiro MJ, et al.: Epidemiological trends in 1452 cases of retinoblastoma from the Surveillance, Epidemiology, and End Results (SEER) registry. Can J Ophthalmol 52 (6): 592-598, 2017.
- Zhang J, Benavente CA, McEvoy J, et al.: A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481 (7381): 329-34, 2012.
- Rushlow DE, Mol BM, Kennett JY, et al.: Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol 14 (4): 327-34, 2013.
- McEvoy J, Nagahawatte P, Finkelstein D, et al.: RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget 5 (2): 438-50, 2014.
- Afshar AR, Pekmezci M, Bloomer MM, et al.: Next-Generation Sequencing of Retinoblastoma Identifies Pathogenic Alterations beyond RB1 Inactivation That Correlate with Aggressive Histopathologic Features. Ophthalmology 127 (6): 804-813, 2020.
- Kooi IE, Mol BM, Massink MP, et al.: Somatic genomic alterations in retinoblastoma beyond RB1 are rare and limited to copy number changes. Sci Rep 6: 25264, 2016.
- Francis JH, Richards AL, Mandelker DL, et al.: Molecular Changes in Retinoblastoma beyond RB1: Findings from Next-Generation Sequencing. Cancers (Basel) 13 (1): , 2021.
- Ewens KG, Bhatti TR, Moran KA, et al.: Phosphorylation of pRb: mechanism for RB pathway inactivation in MYCN-amplified retinoblastoma. Cancer Med 6 (3): 619-630, 2017.
- Richter S, Vandezande K, Chen N, et al.: Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet 72 (2): 253-69, 2003.
- Dommering CJ, Mol BM, Moll AC, et al.: RB1 mutation spectrum in a comprehensive nationwide cohort of retinoblastoma patients. J Med Genet 51 (6): 366-74, 2014.
- Reddy MA, Butt M, Hinds AM, et al.: Prognostic Information for Known Genetic Carriers of RB1 Pathogenic Variants (Germline and Mosaic). Ophthalmol Retina 5 (4): 381-387, 2021.
- Eloy P, Dehainault C, Sefta M, et al.: A Parent-of-Origin Effect Impacts the Phenotype in Low Penetrance Retinoblastoma Families Segregating the c.1981C>T/p.Arg661Trp Mutation of RB1. PLoS Genet 12 (2): e1005888, 2016.
- Clark R: Retinoblastoma: genetic testing and counseling. In: Singh A, Damato B: Clinical Ophthalmic Oncology. Saunders Elsevier, 2007, pp 441-6.
- Amitrano S, Marozza A, Somma S, et al.: Next generation sequencing in sporadic retinoblastoma patients reveals somatic mosaicism. Eur J Hum Genet 23 (11): 1523-30, 2015.
- Sagi M, Frenkel A, Eilat A, et al.: Genetic screening in patients with Retinoblastoma in Israel. Fam Cancer 14 (3): 471-80, 2015.
- Chen Z, Moran K, Richards-Yutz J, et al.: Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum Mutat 35 (3): 384-91, 2014.
- Nichols KE, Houseknecht MD, Godmilow L, et al.: Sensitive multistep clinical molecular screening of 180 unrelated individuals with retinoblastoma detects 36 novel mutations in the RB1 gene. Hum Mutat 25 (6): 566-74, 2005.
- Abramson DH, Mendelsohn ME, Servodidio CA, et al.: Familial retinoblastoma: where and when? Acta Ophthalmol Scand 76 (3): 334-8, 1998.
- Broaddus E, Topham A, Singh AD: Survival with retinoblastoma in the USA: 1975-2004. Br J Ophthalmol 93 (1): 24-7, 2009.
- de Jong MC, Kors WA, de Graaf P, et al.: Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol 15 (10): 1157-67, 2014.
- Rodjan F, de Graaf P, Brisse HJ, et al.: Trilateral retinoblastoma: neuroimaging characteristics and value of routine brain screening on admission. J Neurooncol 109 (3): 535-44, 2012.
- Kivelä T: Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol 17 (6): 1829-37, 1999.
- Sirin S, de Jong MC, Galluzzi P, et al.: MRI-based assessment of the pineal gland in a large population of children aged 0-5 years and comparison with pineoblastoma: part II, the cystic gland. Neuroradiology 58 (7): 713-21, 2016.
- Pham TT, Siebert E, Asbach P, et al.: Magnetic resonance imaging based morphologic evaluation of the pineal gland for suspected pineoblastoma in retinoblastoma patients and age-matched controls. J Neurol Sci 359 (1-2): 185-92, 2015.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
Page Footer
I want to...
Audiences
Secure Member Sites
The Cigna Group Information
Disclaimer
Individual and family medical and dental insurance plans are insured by Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc., and Cigna HealthCare of Texas, Inc. Group health insurance and health benefit plans are insured or administered by CHLIC, Connecticut General Life Insurance Company (CGLIC), or their affiliates (see
All insurance policies and group benefit plans contain exclusions and limitations. For availability, costs and complete details of coverage, contact a licensed agent or Cigna sales representative. This website is not intended for residents of New Mexico.