Wilms Tumor and Other Childhood Kidney Tumors Treatment (PDQ®): Treatment - Health Professional Information [NCI]
Wilms Tumor
Incidence of Wilms Tumor
Wilms tumor is the most frequent tumor of the kidney in infants and children. The incidence of Wilms tumor is 9.7 cases for every 1 million children younger than 15 years and 13.5 cases per 1 million infants.[
The male-to-female ratio in unilateral cases of Wilms tumor is 0.92 to 1.00, but in bilateral cases, there is a female excess (0.60). The mean age at diagnosis is 44 months in unilateral cases and 31 months in bilateral cases of Wilms tumor.[
Syndromes and Other Conditions Associated With Wilms Tumor
Wilms tumor typically develops in otherwise healthy children without any predisposition to developing cancer. However, approximately 10% of children with Wilms tumor have been reported to have a congenital anomaly.[
Children with Wilms tumor may have associated hemihypertrophy and urinary tract anomalies, including cryptorchidism and hypospadias. Children may have recognizable phenotypic syndromes such as overgrowth, aniridia, genetic malformations, and others. These syndromes have provided clues to the genetic basis of the disease. The phenotypic syndromes and other conditions have been grouped into overgrowth and non-overgrowth categories (see
It is important to recognize that the absolute risk of developing Wilms tumor varies with the underlying condition or anomaly. For example, most patients with hemihypertrophy will not develop Wilms tumor.
| Syndrome/Condition | Gene | Overgrowth Phenotype | Non-Overgrowth Phenotype |
|---|---|---|---|
| CLOVES = congenital lipomatous overgrowth, vascular malformations, epidermal nevi, and skeletal/spinal abnormalities; MULIBREY = distinctive abnormalities of the (MU)scles, (LI)ver, (BR)ain, and (EY)es; WAGR = Wilms tumor, aniridia, genitourinary abnormalities, and range of developmental delays. | |||
| a Adapted from Treger et al.[ |
|||
| High Risk of Wilms Tumor (>20%) | |||
| WAGR syndrome (WAGR spectrum) | WT1deletion | X | |
| Denys-Drash syndrome | WT1missense variant | X | |
| Perlman syndrome | DIS3L2variant | X | |
| Fanconi anemia with biallelic variants inBRCA2(FANCD1) orPALB2(FANCN) | BRCA2,PALB2 | X | |
| Premature chromatid separation/mosaic variegated aneuploidy | BiallelicBUB1BorTRIP13variant | X | |
| Moderate Risk of Wilms Tumor (5%–20%) | |||
| Frasier syndrome | WT1intron 9 splice variant | X | |
| Beckwith-Wiedemann syndrome | Uniparental disomy or H19 epivariant | X | |
| Simpson-Golabi-Behmel syndrome | GPC3variant | X | |
| Low Risk of Wilms Tumor (<5%) | |||
| Bloom syndrome | BiallelicBLMvariant | X | |
| DICER1 syndrome | DICER1variant | X | |
| Li-Fraumeni syndrome | TP53,CHEK2 | X | |
| Isolated hemihypertrophy | X | ||
| Hyperparathyroidism-jaw tumor syndrome | CDC73(also known asHRPT2) variant | X | |
| MULIBREY nanism syndrome | TRIM37variant | X | |
| PIK3CA-related segmental overgrowth including CLOVES syndrome | PIK3CAvariant | X | |
| 9q22.3 microdeletion syndrome | 9q22.3 | X | |
| Sotos syndrome | NSD1 | X | |
| Familial Wilms tumor | FWT1 | X | |
| FWT2 | |||
| Genitourinary anomalies | WT1 | X | |
| Sporadic aniridia | WT1 | X | |
| Trisomy 18 | X | ||
For information about the genes associated with Wilms tumor, including WT1 and WT2, see the
Syndromic causes of Wilms tumor
WT1-related syndromes
WT1-related syndromes include the following:
- WAGR syndrome (WAGR spectrum).[
13 ] WAGR syndrome is characterized by the following:- W ilms tumor.
- A niridia.
- G enitourinary abnormalities.
- R ange of developmental delays.
The constellation of WAGR syndrome occurs in association with an interstitial deletion on chromosome 11 (del(11p13)). The prevalence of this deletion is about 0.4% of children with Wilms tumor.[
14 ,15 ] The risk of Wilms tumor development in children with WAGR syndrome is approximately 50%. These children will present earlier (median age, 22 months) and have a higher incidence of bilateral Wilms tumors (37%) than children with nonsyndromic Wilms tumors.[16 ,17 ] A study that used the International WAGR Syndrome Association survey found that 64 of 145 children (44%) developed Wilms tumor. Most children had unilateral stage I or stage II Wilms tumors and favorable histology (59 of 64, 92%). One child developed bilateral Wilms tumors. Two children presented with stage IV disease, both with favorable histology.[18 ] In the International Society of Paediatric Oncology (SIOP) Renal Tumor Study Group experience, 3 of 43 patients developed contralateral tumors, one of which occurred 7 years after initial diagnosis.[17 ] For more information, see theGenomics of Wilms Tumor section. - Denys-Drash syndrome and Frasier syndrome. Genitourinary anomalies such as hypospadias, undescended testis, and others are associated with WT1 variants (prevalence is about 8%–10% of children with Wilms tumor). Children with XY genome with pseudo-hermaphroditism and/or renal disease (glomerulonephritis or nephrotic syndrome) who develop Wilms tumor may have Denys-Drash or Frasier syndrome (characterized by male hermaphroditism, primary amenorrhea, chronic renal failure, and other abnormalities),[
19 ] both of which are associated with variants in the WT1 gene.[20 ] Specifically, germline missense pathogenic variants in the WT1 gene are responsible for most cases of Wilms tumor that occur as part of Denys-Drash syndrome.[21 ,22 ] The risk of Wilms tumor is about 90% for children with Denys-Drash syndrome, and bilateral disease develops in 20% of patients.[22 ,23 ] In Frasier syndrome, splice-site WT1 variants result in an imbalance of WT1 isoforms and a much lower incidence of Wilms tumor.[24 ]
WT2-related syndromes
WT2-related syndromes include the following:
- Beckwith-Wiedemann syndrome. Beckwith-Wiedemann syndrome is an overgrowth syndrome characterized by asymmetric growth of one or more parts of the body, large tongue, omphalocele or umbilical hernia at birth, creases or pits in the skin near the ears, kidney abnormalities, and hypoglycemia (in neonates). In a population-based registry linkage study of all live births in Texas from 1999 to 2017, children with Beckwith-Wiedemann syndrome were 42 times more likely to develop pediatric cancer. Hepatoblastoma was the most common cancer, followed by Wilms tumor. The percentage of children with Beckwith-Wiedemann syndrome diagnosed with cancer was 1.24% by age 5 years, 5.58% by age 10 years, and 10.81% by age 15 years. The presence of any isolated overgrowth feature was associated with the risk of developing cancer (hazard ratio [HR], 4.70). Hepatosplenomegaly (HR, 23.04) and macroglossia (HR, 11.18) had the strongest associations with cancer risk.[
25 ] Approximately 15% of children with Beckwith-Wiedemann syndrome will have bilateral tumors.[26 ]Beckwith-Wiedemann syndrome is caused by altered expression of two gene clusters involved in growth control and cell-cycle progression regulated by two independent imprinting control regions (ICR1 [telomeric ICR] and ICR2 [centromeric ICR]) at chromosome 11p15.5. The two ICRs are characterized by differential methylation of maternal and paternal alleles. A variety of molecular mechanisms are implicated in Beckwith-Wiedemann syndrome pathogenesis, leading to unbalanced expression of imprinted genes within these two domains. Tumor predisposition results primarily from dysregulation at the telomeric domain of 11p15 (ICR1 gain of methylation [ICR1-GoM] and paternal uniparental disomy [UPD]) rather than at the centromeric domain of 11p15 (ICR2 loss of methylation [ICR2-LoM] and CDKN1C variant).[
27 ] Approximately 15% of cases with clear-cut phenotypes have no molecular defects established so far.[28 ,29 ]The molecular subtypes of the syndrome predispose patients to the development of different tumor histotypes.[
30 ,31 ,32 ]The prevalence of Beckwith-Wiedemann syndrome has previously been reported as 1% of children with Wilms tumor.[
26 ,33 ,34 ,35 ] However, a 5-year national Dutch cohort study demonstrated that 16% of patients with Wilms tumor (20 of 126) have Beckwith-Wiedemann syndrome. This study included both patients with clinical diagnoses and patients in which the Beckwith-Wiedemann syndrome phenotype was not apparent, such as 11p15 ICR1 gain of methylation in normal renal parenchymal and peripheral blood. Mosaicism likely accounts for the phenotypically occult cases.[36 ] In aggregate, approximately 10% of patients with Beckwith-Wiedemann syndrome will develop Wilms tumor. However, this incidence varies based on epigenotype. Children with ICR1-GoM have the highest risk of developing Wilms tumor (22%–29%). Children with paternal UPD have a lower risk (7%–17%), and patients with ICR2-LoM and CDKN1C variants have minimal risk.[27 ,31 ,32 ] Beckwith-Wiedemann syndrome patients with hemihypertrophy have a fourfold increased tumor risk over Beckwith-Wiedemann syndrome patients without hemihypertrophy.[37 ] For more information, see theGenomics of Wilms Tumor section.
Other syndromic causes of Wilms tumor
Other syndromic causes of Wilms tumor include the following:
- Perlman syndrome. This is a rare, autosomal recessively inherited, congenital overgrowth syndrome. It is characterized by fetal gigantism, renal dysplasia and nephroblastomatosis, islet cell hypertrophy, multiple congenital anomalies, and intellectual disability. Survivors have a high risk of developing Wilms tumor (75%).[
38 ]Germline inactivating pathogenic variants in DIS3L2 on chromosome 2q37 are associated with Perlman syndrome. Preliminary data suggest that DIS3L2 plays a role in normal kidney development and in a subset of sporadic Wilms tumor cases.[
39 ]Heterozygous DIS3L2 constitutional variants appear to have an association with Wilms tumor predisposition. In a 5-year national Dutch cohort study, 4% of patients with Wilms tumors (5 of 126) had DIS3L2 variants. However, penetrance is likely much lower than in homozygous cases (Perlman syndrome).[
36 ] - Simpson-Golabi-Behmel syndrome. This syndrome is characterized by macroglossia, macrosomia, renal and skeletal abnormalities, and increased risk of embryonal cancers.
The syndrome is caused by variants or deletions in the GPC3 and GPC4 genes, and these genetic aberrations are believed to enhance the risk of Wilms tumor (8%).[
40 ] - CLOVES syndrome. This syndrome is characterized by the following:
- C ongenital L ipomatous O vergrowth.
- V ascular malformations.
- E pidermal nevi.
- S keletal/spinal abnormalities.
This syndrome results from postzygotic, somatic variants in PIK3CA, which may involve large or small regions of the child.[
41 ] - Sotos syndrome. This syndrome is characterized by cerebral gigantism and learning disability, ranging from mild to severe. Sotos syndrome is associated with behavioral problems, congenital cardiac anomalies, neonatal jaundice, and renal anomalies such as Wilms tumor, scoliosis, and seizures.
Variants in the NSD1 gene are the only known cause of Sotos syndrome.[
42 ] - 9q22.3 microdeletion syndrome. This syndrome is characterized by craniofacial abnormalities, metopic craniosynostosis, hydrocephalus, macrosomia, and learning disabilities.
Of 44 described patients with 9q22.3 deletions, 7 developed Wilms tumor, and there was an association with overgrowth in 4 of those 7 patients. Although the size of the deletions was variable, all of them encompassed the PTCH1 gene.[
43 ]; [44 ][Level of evidence C1] According to the authors of this study, surveillance for Wilms tumor should be considered in any patient with 9q22.3 microdeletion syndrome, especially in the presence of overgrowth.[44 ][Level of evidence C1] - Bloom syndrome. This syndrome is characterized by short stature and being thinner than other family members, sun-sensitive skin changes, and an increased risk of Wilms tumor.
Variants in the BLM gene are the only known cause of Bloom syndrome.[
45 ] - Li-Fraumeni syndrome. This syndrome is a rare disorder that greatly increases the risk of developing several types of cancer, particularly in children and young adults. The cancers most often associated with Li-Fraumeni syndrome include breast cancer, osteosarcoma, soft tissue sarcoma, brain tumor, leukemia, adrenocortical carcinoma, and Wilms tumor.
The TP53 gene variant is present in most families with Li-Fraumeni syndrome. The CHEK2 gene variant is also known to cause Li-Fraumeni syndrome.[
46 ] - Alagille syndrome. This syndrome includes congenital cardiopathy; facial dysmorphology; and vertebral, ocular, and renal abnormalities. It has been reported along with Wilms tumor in two patients who had identified variants.[
47 ] - Bohring-Opitz syndrome. This syndrome is a rare genetic condition characterized by distinctive facial features, variable microcephaly, hypertrichosis, nevus flammeus, severe myopia, unusual posture, severe intellectual disability, and feeding issues.
The syndrome is associated with ASXL1 variants and an estimated incidence of Wilms tumor of 7%.[
48 ]
Nonsyndromic causes of Wilms tumor
Nonsyndromic causes of Wilms tumor include the following:
- Familial Wilms tumor. Despite the number of genes that appear to be involved in the development of Wilms tumor, familial Wilms tumor is uncommon, with approximately 2% of patients having a positive family history of Wilms tumor. Siblings of children with Wilms tumor have a less-than-1% chance of developing Wilms tumor.[
49 ,50 ,51 ] The risk of Wilms tumor among offspring of persons who have had unilateral (sporadic) tumors is less than 2%.[52 ]Two distribution loci at 17q12-q21 (FWT1) and 19q13.4 (FWT2) have been identified by genetic linkage studies of families affected by Wilms tumor. Although the genes have yet to be characterized, in siblings with Wilms tumor, loss of function of the transcriptional corepressor TRIM28 was detected, which is located at FWT2.[
53 ,54 ,55 ] Occasionally, Wilms tumor families have germline pathogenic variants in WT1. In these families, most, but not all, of the family members have genitourinary tract malformations.[56 ,57 ]Inactivating variants in CTR9 have been identified in 3 of 35 Wilms tumor families. CTR9 is located at 11p15.3 and is a key component of the polymerase-associated factor 1 (PAF1) complex, which has multiple roles in RNA polymerase II regulation and transcriptional elongation and is implicated in embryonic organogenesis.[
58 ] A few families with familial Wilms tumor have germline microdeletion or microinsertion pathogenic variants in the H19 region of 11p15.3 that result in hypermethylation of the site.[59 ] - Constitutional 11p15 abnormalities. Constitutional 11p15 abnormalities have been identified in lymphocyte DNA of 13 of 437 individuals (3%) with sporadic Wilms tumor without features of growth disorders, including 12% of bilateral cases. All were de novo abnormalities and appeared to be postzygotic, except for one novel microdeletion in a child whose mother had the variant and was not affected; however, a younger brother with the microdeletion had Beckwith-Wiedemann syndrome. This suggests that constitutional 11p15 analysis should be considered in all individuals with Wilms tumor.[
59 ] - Sporadic aniridia. Sporadic aniridia may result from small germline pathogenic deletions of one copy of the PAX6 gene that includes part or all of the adjacent WT1 gene but does not result in genitourinary abnormalities or intellectual disability (i.e., not obviously WAGR syndrome). Therefore, many patients with sporadic aniridia develop Wilms tumor and are candidates for genetic testing. The relative risk of Wilms tumor in sporadic aniridia is 67-fold.[
60 ] About one-half of individuals with sporadic aniridia and PAX6 and WT1 deletions develop Wilms tumor.[61 ] - Isolated hemihypertrophy (also known as lateralized overgrowth or hemihyperplasia). Hemihypertrophy is an asymmetric overgrowth of one or more body parts in the absence of a recognized pattern of malformations, dysplasia, or morphological variants and has been associated with Wilms tumor.[
62 ] It can also be associated with other predisposition syndromes such as Beckwith-Wiedemann syndrome. Clinical signs may not be very evident, and hemihypertrophy may be noted after tumor diagnosis.In patients with isolated hemihypertrophy and paternal uniparental isodisomy of 11p15.5, the risk of Wilms tumor is estimated to be about 8%.[
63 ] - Trisomy 18.[
64 ] - Fanconi anemia with biallelic variants in BRCA2 (FANCD1) or PALB2 (FANCN). BRCA2 and PALB2 play central roles in homologous recombination DNA repair. Biallelic variants in either BRCA2 or PALB2 lead to Fanconi anemia and to increased risks of selected childhood cancers, including Wilms tumor.[
65 ,66 ,67 ] - Maternal pesticide exposure. In a French population study, the maternal use of any household pesticide during pregnancy was associated with a risk of Wilms tumor in children (odds ratio [OR], 1.6). Insecticides were the most commonly reported type of pesticide, and the association with Wilms tumor was stronger when insecticides were used more than once per month.[
68 ][Level of evidence C1]
Genomics of Wilms Tumor
Molecular Features of Wilms Tumor
A Wilms tumor may arise during embryogenesis on the background of an otherwise genomically normal kidney, or it may arise from nongermline somatic genetic precursor lesions residing in histologically and functionally normal kidney tissue. Hypermethylation of H19, a known component of a subset of Wilms tumors, is a very common genetic abnormality found in these normal-appearing areas of precursor lesions.[
One study performed genome-wide sequencing, mRNA and miRNA expression, DNA copy number, and methylation analysis on 117 Wilms tumors, followed by targeted sequencing of 651 Wilms tumors.[
- Wilms tumors commonly arise through more than one genetic event.
- Wilms tumors show differences in gene expression and methylation patterns with different genetic aberrations.
- Wilms tumors have a large number of candidate driver genes, most of which are altered in less than 5% of Wilms tumors.
- Wilms tumors have recurrent variants in genes with common functions, with most involved in either early renal development or epigenetic regulation (e.g., chromatin modifications, transcription elongation, and miRNA).
Approximately one-third of Wilms tumor cases involve variants in WT1, CTNNB1, or AMER1 (WTX).[
Elevated rates of Wilms tumor are observed in patients with a number of genetic disorders, including WAGR (Wilms tumor, aniridia, genitourinary abnormalities, and range of developmental delays) syndrome (WAGR spectrum), Beckwith-Wiedemann syndrome, hemihypertrophy, Denys-Drash syndrome, and Perlman syndrome.[
The genomic and genetic characteristics of Wilms tumor are summarized below.
WT1gene
The WT1 gene is located on the short arm of chromosome 11 (11p13). WT1 is a transcription factor that is required for normal genitourinary development and is important for differentiation of the renal blastema.[
Wilms tumor with a WT1 variant is characterized by the following:
- Evidence of WNT pathway activation by activating variants in the CTNNB1 gene is common.[
80 ,81 ,82 ] - Loss of heterozygosity (LOH) at 11p15 is commonly observed, as paternal uniparental disomy for chromosome 11 represents a common mechanism for losing the remaining normal WT1 allele.[
80 ,83 ] - Nephrogenic rests are benign foci of embryonal kidney cells that abnormally persist into postnatal life. Intralobar nephrogenic rests occur in approximately 20% of Wilms tumor cases. They are observed at high rates in cases with genetic syndromes that have WT1 variants such as WAGR and Denys-Drash syndromes.[
84 ] Intralobar nephrogenic rests are also observed in cases with sporadic WT1 and MLLT1 variants.[85 ,86 ] - WT1 germline pathogenic variants are uncommon (2%–4%) in nonsyndromic Wilms tumor.[
57 ,87 ] - WT1 variants and 11p15 LOH were associated with relapse in patients with very low-risk Wilms tumor in one study of 56 patients who did not receive chemotherapy.[
88 ] These findings need validation but may provide biomarkers for stratifying patients in the future.
Germline WT1 pathogenic variants are more common in children with Wilms tumor and one of the following:
- WAGR syndrome, Denys-Drash syndrome,[
22 ] or Frasier syndrome.[19 ] - Genitourinary anomalies, including hypospadias and cryptorchidism.
- Bilateral Wilms tumor.
- Unilateral Wilms tumor with nephrogenic rests in the contralateral kidney.
- Stromal and rhabdomyomatous differentiation.
Germline WT1 pathogenic single nucleotide variants produce genetic syndromes that are characterized by nephropathy, 46XY disorder of sex development, and varying risks of Wilms tumor.[
- WAGR syndrome. Children with WAGR syndrome are at high risk (approximately 50%) of developing Wilms tumor.[
6 ] WAGR syndrome results from deletions at chromosome 11p13 that involve a set of contiguous genes that include the WT1 and PAX6 genes.Inactivating variants or deletions in the PAX6 gene lead to aniridia, while deletion of WT1 confers the increased risk of Wilms tumor. Loss of the LMO2 gene has been associated with a more frequent development of Wilms tumor in patients with congenital aniridia and WAGR-region deletions.[
91 ][Level of evidence C1] Sporadic aniridia in which WT1 is not deleted is not associated with increased risk of Wilms tumor. Accordingly, children with familial aniridia, generally occurring for many generations, and without renal abnormalities, have a normal WT1 gene and are not at an increased risk of Wilms tumor.[33 ,92 ]Wilms tumor in children with WAGR syndrome is characterized by an excess of bilateral disease, intralobar nephrogenic rests, early age at diagnosis, and stromal-predominant histology in FH tumors.[
16 ] The intellectual disability in WAGR syndrome may be secondary to deletion of other genes, including SLC1A2 or BDNF.[59 ]
- Denys-Drash syndrome. This syndrome is characterized by nephrotic syndrome caused by diffuse mesangial sclerosis, XY pseudohermaphroditism, and increased risk of Wilms tumor (>90%).
WT1 variants in Denys-Drash syndrome are most often missense variants in exons 8 and 9, which code for the DNA binding region of WT1.[
22 ] - Frasier syndrome. This syndrome is characterized by progressive nephropathy caused by focal segmental glomerulosclerosis, gonadoblastoma, and XY pseudohermaphroditism.
WT1 variants in Frasier syndrome typically occur in intron 9 at the KT site, and create an alternative splicing variant, thereby preventing production of the usually more abundant WT1 +KTS isoform.[
24 ]
Studies evaluating genotype/phenotype correlations of WT1 variants have shown that the risk of Wilms tumor is highest for truncating variants (14 of 17 cases, 82%) and lower for missense variants (27 of 67 cases, 42%). The risk is lowest for KTS splice site variants (1 of 27 cases, 4%).[
CTNNB1gene
CTNNB1 is one of the most commonly altered genes in Wilms tumor, reported to occur in 15% of patients with Wilms tumor.[
AMER1(WTX) gene on the X chromosome
AMER1 is located on the X chromosome at Xq11.1. It is altered in 15% to 20% of Wilms tumor cases.[
AMER1 alterations are equally distributed between males and females, and AMER1 inactivation has no apparent effect on clinical presentation or prognosis.[
Imprinting cluster regions (ICRs) on chromosome 11p15 (WT2) and Beckwith-Wiedemann syndrome
A second Wilms tumor locus, WT2, maps to an imprinted region of chromosome 11p15.5. When it is a germline pathogenic variant, it causes Beckwith-Wiedemann syndrome. About 3% of children with Wilms tumor have germline epigenetic or genetic changes at the 11p15.5 growth regulatory locus without any clinical manifestations of overgrowth. Like children with Beckwith-Wiedemann syndrome, these children have an increased incidence of bilateral Wilms tumor or familial Wilms tumor.[
Approximately one-fifth of patients with Beckwith-Wiedemann syndrome who develop Wilms tumor present with bilateral disease, and metachronous bilateral disease is also observed.[
Approximately 80% of patients with Beckwith-Wiedemann syndrome have a molecular defect of the 11p15 domain.[
Several candidate genes at the WT2 locus comprise the two independent imprinted domains: IGF2 and H19; and CDKN1C and KCNQ1OT1.[
A relationship between epigenotype and phenotype has been shown in Beckwith-Wiedemann syndrome, with a different rate of cancer in Beckwith-Wiedemann syndrome according to the type of alteration of the 11p15 region.[
The following four main molecular subtypes of Beckwith-Wiedemann syndrome are characterized by specific genotype-phenotype correlations:
- ICR1 gain of methylation (ICR1-GoM). Five percent to 10% of cases are caused by telomeric ICR1-GoM, which causes both biallelic expression of the IGF2 gene (normally expressed by the paternal allele only) and reduced expression of the oncosuppressor H19 gene. The incidence of Wilms tumor is 22.8%.[
103 ] - ICR2 loss of methylation (ICR2-LoM). Fifty percent of cases with Beckwith-Wiedemann syndrome are caused by ICR2-LoM, resulting in reduced expression of the CDKN1C gene, normally expressed by the maternal chromosome only. Tumor incidence is very low (2.5%).[
103 ] - Uniparental disomy (UPD). Altered expression at both imprinted gene clusters is observed in mosaic UPD of chromosome 11p15.5, accounting for 20% to 25% of the cases. The incidence of Wilms tumor is 6.2%, followed by hepatoblastoma (4.7%) and adrenal carcinoma (1.5%).[
103 ] Fewer than 1% of cases with Beckwith-Wiedemann syndrome are caused by chromosomal rearrangements involving the 11p15 region. - CDKN1C variants. Maternally inheritable CDKN1C loss-of-function variants account for approximately 5% of the cases. This type is associated with a 4.3% incidence of neuroblastoma.[
103 ]
Other tumors such as neuroblastoma or hepatoblastoma were reported in patients with paternal 11p15 isodisomy.[
Loss of imprinting or gene methylation is rarely found at other loci, supporting the specificity of loss of imprinting at 11p15.5.[
Other genes and chromosomal alterations
Additional genes and chromosomal alterations that have been implicated in the pathogenesis and biology of Wilms tumor include the following:
- 1q. Gain of chromosome 1q is associated with an inferior outcome and is the single most powerful predictor of outcome.[
107 ,108 ] Gain of chromosome 1q is one of the most common cytogenetic abnormalities in Wilms tumor and is observed in approximately 30% of tumors.In an analysis of FH Wilms tumor from 1,114 patients from NWTS-5 (COG-Q9401/NCT00002611), 28% of the tumors displayed 1q gain.[
107 ]- The 8-year event-free survival (EFS) rate was 77% for patients with 1q gain and 90% for those lacking 1q gain (P < .001). Within each disease stage, 1q gain was associated with inferior EFS.
- The 8-year overall survival (OS) rate was 88% for those with 1q gain and 96% for those lacking 1q gain (P < .001). OS was significantly inferior in cases with stage I disease (P < .0015) and stage IV disease (P = .011).
- Similar results were reported in the International Society of Paediatric Oncology (SIOP) WT 2001 study of 586 children with Wilms tumor.[
108 ]
One study included a cohort of FH Wilms tumor that was enriched for patients who relapsed. The study found that the prevalence of 1q gain was higher in the relapsed Wilms tumor specimens (75%) than in the matched primary samples (47%).[
109 ] The increased prevalence of 1q gain at relapse supports its association with poor prognosis and disease progression. - 16q and 1p. Additional tumor-suppressor or tumor-progression genes may lie on chromosomes 16q and 1p, as evidenced by LOH for these regions in 17% and 11% of Wilms tumor cases, respectively.[
110 ]- In large NWTS studies, patients with tumor-specific loss of these loci had significantly worse relapse-free survival and OS rates. Combined loss of 1p and 16q are criteria used to select FH Wilms tumor patients for more aggressive therapy in the current Children's Oncology Group (COG) study. However, a U.K. study of more than 400 patients found no significant association between 1p deletion and poor prognosis, but a poor prognosis was associated with 16q LOH.[
111 ] - An Italian study of 125 patients, using treatment quite similar to that in the COG study, found significantly worse prognosis in those with 1p deletions but not 16q deletions.[
112 ]
These conflicting results may arise from the greater prognostic significance of 1q gain described above. LOH of 16q and 1p loses significance as independent prognostic markers in the presence of 1q gain. However, in the absence of 1q gain, LOH of 16q and 1p retains their adverse prognostic impact.[
107 ] The LOH of 16q and 1p appears to arise from complex chromosomal events that result in 1q LOH or 1q gain. The change in 1q appears to be the critical tumorigenic genetic event.[113 ] - In large NWTS studies, patients with tumor-specific loss of these loci had significantly worse relapse-free survival and OS rates. Combined loss of 1p and 16q are criteria used to select FH Wilms tumor patients for more aggressive therapy in the current Children's Oncology Group (COG) study. However, a U.K. study of more than 400 patients found no significant association between 1p deletion and poor prognosis, but a poor prognosis was associated with 16q LOH.[
- miRNAPG. Variants in selected miRNAPG are observed in approximately 20% of Wilms tumor cases and appear to perpetuate the progenitor state.[
70 ,73 ,74 ,75 ,76 ] The products of these genes direct the maturation of miRNAs from the initial pre-miRNA transcripts to functional cytoplasmic miRNAs (see Figure 1).[114 ] The most commonly altered miRNAPG is DROSHA, with a recurrent variant (E1147K) affecting a metal-binding residue of the RNase IIIb domain, representing about 80% of DROSHA-altered tumors. Other miRNAPG that are altered in Wilms tumor include DGCR8, DICER1, TARBP2, DIS3L2, and XPO5. These variants are generally mutually exclusive, and they appear to be deleterious and result in impaired expression of tumor-suppressing miRNAs. A striking sex bias was noted for patients with variants in DGCR8 (located on chromosome 22q11), with 38 of 43 cases (88%) arising in girls.[73 ,74 ]Germline pathogenic variants in miRNAPG are observed for DICER1 and DIS3L2, with variants in the former causing DICER1 syndrome and variants in the latter causing Perlman syndrome.
- DICER1 syndrome is typically caused by inherited truncating variants in DICER1, with tumor formation following acquisition of a missense variant in a domain of the remaining allele of DICER1 (the RNase IIIb domain) responsible for processing miRNAs derived from the 5p arms of pre-miRNAs.[
115 ] Tumors associated with DICER1 syndrome include pleuropulmonary blastoma, cystic nephroma, ovarian sex cord–stromal tumors, multinodular goiter, and embryonal rhabdomyosarcoma.[115 ] Wilms tumor is an uncommon presentation of the DICER1 syndrome. In one study, three families with DICER1 syndrome included children with Wilms tumor, with two of the Wilms tumor cases showing the typical second DICER1 variant in the RNase IIIb domain.[116 ] Another study identified DICER1 variants in 2 of 48 familial Wilms tumor families.[117 ] Large sequencing studies of Wilms tumor cohorts have also observed occasional cases with DICER1 variants.[74 ,75 ] - Perlman syndrome is a rare autosomal recessive overgrowth disorder caused by variants in DIS3L2, which encodes a ribonuclease that is responsible for degrading pre-let-7 miRNA.[
39 ,118 ] Heterozygous germline DIS3L2 pathogenic inactivations are also associated with Wilms tumor development.[36 ] Patients with Perlman syndrome have a poor prognosis, with a high neonatal mortality rate. In a survey of published cases of Perlman syndrome (N = 28), in infants who survived beyond the neonatal period, approximately two-thirds developed Wilms tumor, and all patients showed developmental delay. Fetal macrosomia, ascites, and polyhydramnios are frequent manifestations.[119 ]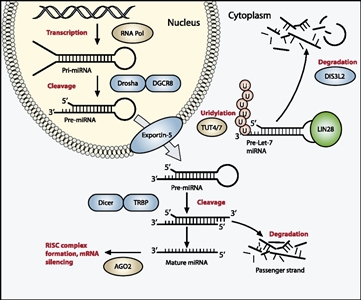
Figure 1. The miRNA processing pathway is commonly mutated in Wilms tumor. Expression of mature miRNA is initiated by RNA polymerase–mediated transcription of DNA-encoded sequences into pri-miRNA, which form a long double-stranded hairpin. This structure is then cleaved by a complex of Drosha and DGCR8 into a smaller pre-miRNA hairpin, which is exported from the nucleus and then cleaved by Dicer (an RNase) and TRBP (with specificity for dsRNA) to remove the hairpin loop and leave two single-stranded miRNAs. The functional strand binds to Argonaute (Ago2) proteins into the RNA-induced silencing complex (RISC), where it guides the complex to its target mRNA, while the nonfunctional strand is degraded. Targeting of mRNAs by this method results in mRNA silencing by mRNA cleavage, translational repression, or deadenylation. Let-7 miRNAs are a family of miRNAs highly expressed in ESCs with tumor suppressor properties. In cases in which LIN28 is overexpressed, LIN28 binds to pre-Let-7 miRNA, preventing DICER from binding and resulting in LIN28-activated polyuridylation by TUT4 or TUT7, causing reciprocal DIS3L2-mediated degradation of Let-7 pre-miRNAs. Genes involved in miRNA processing that have been associated with Wilms tumor are highlighted in blue (inactivating) and green (activating) and include DROSHA, DGCR8, XPO5 (encoding exportin-5), DICER1, TARBP2, DIS3L2, and LIN28. Copyright © 2015 Hohenstein et al.; Published by Cold Spring Harbor Laboratory Press. Genes Dev. 2015 Mar 1; 29(5): 467–482. doi: 10.1101/gad.256396.114. This article is distributed exclusively by Cold Spring Harbor Laboratory Press under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at https://creativecommons.org/licenses/by-nc/4.0/.
- DICER1 syndrome is typically caused by inherited truncating variants in DICER1, with tumor formation following acquisition of a missense variant in a domain of the remaining allele of DICER1 (the RNase IIIb domain) responsible for processing miRNAs derived from the 5p arms of pre-miRNAs.[
- SIX1 and SIX2. SIX1 and SIX2 are highly homologous transcription factors that play key roles in early renal development and are expressed in the metanephric mesenchyme, where they maintain the mesenchymal progenitor population. In patients with Wilms tumors, the frequency of SIX1 variants is 3% to 4%, and the frequency of SIX2 variants is 1% to 3%.[
73 ,74 ]- Virtually all SIX1 and SIX2 variants are in exon 1 and result in a glutamine-to-arginine variant at position 177 (Q177R).
- Variants in WT1, AMER1, and CTNNB1 are infrequent in cases with SIX1, SIX2, or miRNAPG variants. Conversely, SIX1 or SIX2 variants and miRNAPG variants tend to occur together.
- In Wilms tumor, SIX1 and SIX2 variants are associated with the high-risk blastemal subtype and the presence of undifferentiated blastema in chemotherapy-naïve samples.
- In a study of 82 cases of FH Wilms tumor, SIX1 Q177R hotspot variants were identified at a higher rate in tumor specimens at relapse (11 cases; 13.4%) than in those at diagnosis (4%). For 45 cases that had both diagnostic and relapse specimens, there were 6 cases with SIX1 Q177R at relapse, 3 of which did not have SIX1 Q177R at diagnosis. This finding suggests that this variant is not required for tumor development in some individuals with Wilms tumor.[
109 ]
86 ] The altered MLLT1 protein shows altered binding to acetylated histone tails. Patients with MLLT1-altered tumors present at a younger age and have a high prevalence of precursor intralobar nephrogenic rests, supporting a model whereby activating MLLT1 variants early in renal development result in the development of Wilms tumor. - TP53 (tumor suppressor gene). Most anaplastic Wilms tumor cases show variants in the TP53 tumor suppressor gene.[
120 ,121 ,122 ]TP53 may be useful as an unfavorable prognostic marker.[120 ,121 ]In a study of 118 prospectively identified patients with diffuse anaplastic Wilms tumor registered on the NWTS-5 trial, 57 patients (48%) demonstrated TP53 variants, 13 patients (11%) demonstrated TP53 segmental copy number loss without variants, and 48 patients (41%) lacked both (wild-type TP53 [wtTP53]). All TP53 variants were detected by sequencing alone. Patients with stage III or stage IV disease with wtTP53 had a significantly lower relapse rate and mortality rate than did patients with TP53 abnormalities (P = .00006 and P = .00007, respectively). The TP53 status had no effect on patients with stage I or stage II tumors.[
123 ]- In-depth analysis of a subset of 39 patients with diffuse anaplastic Wilms tumor showed that 7 patients (18%) were wtTP53. These wtTP53 tumors demonstrated gene expression evidence of p53 pathway activation. Retrospective pathology review of wtTP53 tumors revealed no or very low volume of anaplasia in six of seven tumors. These data support the key role of TP53 loss in the development of anaplasia in Wilms tumor and support its significant clinical influence in patients who have residual anaplastic disease after surgery.[
123 ]
- In-depth analysis of a subset of 39 patients with diffuse anaplastic Wilms tumor showed that 7 patients (18%) were wtTP53. These wtTP53 tumors demonstrated gene expression evidence of p53 pathway activation. Retrospective pathology review of wtTP53 tumors revealed no or very low volume of anaplasia in six of seven tumors. These data support the key role of TP53 loss in the development of anaplasia in Wilms tumor and support its significant clinical influence in patients who have residual anaplastic disease after surgery.[
- FBXW7. FBXW7, a ubiquitin ligase component, is an established tumor suppressor gene that has been identified as recurrently altered at low rates in Wilms tumor and other malignancies. Variants of this gene have been associated with epithelial-type tumor histology.[
124 ]; [125 ][Level of evidence C1] - TRIM28. TRIM28 encodes a multidomain protein involved in the regulation of many cellular processes and is an autosomal dominant Wilms tumor predisposition gene. TRIM28 accounts for about 8% of familial Wilms tumor and 2% of unselected Wilms tumor.[
55 ,126 ,127 ,128 ]; [125 ][Level of evidence C1]- A strong association between TRIM28 variants and epithelial Wilms tumor has been observed, and most individuals with a TRIM28 variant have a Wilms tumor of predominantly epithelial histology.[
55 ,126 ,127 ]; [125 ][Level of evidence C1] - In a cohort of 91 affected individuals from 49 families with Wilms tumor pedigrees, 33 individuals were identified as having constitutional cancer-predisposing variants, 21 of whom had a variant in TRIM28. There was a strong parent-of-origin effect, with all ten evaluable cases having inherited variants that were maternally transmitted.[
125 ][Level of evidence C1] - Most TRIM28-altered cases have either frameshift, nonsense, or splice-site variants in one allele combined with LOH in the second allele, leading to loss of TRIM28 protein expression in the tumor. Immunohistochemistry staining for loss of TRIM28 protein expression can be used to identify most patients whose tumors have TRIM28 variants.[
128 ]
- A strong association between TRIM28 variants and epithelial Wilms tumor has been observed, and most individuals with a TRIM28 variant have a Wilms tumor of predominantly epithelial histology.[
- 9q22.3 microdeletion syndrome. Patients with 9q22.3 microdeletion syndrome have an increased risk of Wilms tumor.[
43 ] The chromosomal region with germline pathogenic deletion includes PTCH1, the gene that is altered in Gorlin syndrome (nevoid basal cell carcinoma syndrome associated with osteosarcoma). 9q22.3 microdeletion syndrome is characterized by the clinical findings of Gorlin syndrome, as well as developmental delay and/or intellectual disability, metopic craniosynostosis, obstructive hydrocephalus, prenatal and postnatal macrosomia, and seizures. Five patients who presented with Wilms tumor in the context of a constitutional 9q22.3 microdeletion have been reported.[43 ,129 ,130 ] - MYCN. Genomic alterations involving the MYCN network (e.g., MYCN, MAX, MGA, NONO) have been reported to occur in 25% to 30% of Wilms tumor cases.[
109 ] Specific genomic alterations associated with the MYCN network include the following:- MYCN copy number gain was observed in approximately 13% of Wilms tumor cases. MYCN gain was more common in anaplastic cases (7 of 23 cases, 30%) than in nonanaplastic cases (11.2%), and it was associated with poorer relapse-free survival (RFS) and overall survival, independent of histology.[
131 ]MYCN tandem duplication was reported in 11 of 82 (13%) FH Wilms tumor specimens from relapse.[109 ] - Germline pathogenic copy number gain at MYCN has been reported in a bilateral Wilms tumor case,[
131 ] and germline MYCN pathogenic duplication was also reported for a child with prenatal bilateral nephroblastomatosis and a family history of nephroblastoma.[132 ] - Variants at codon 44 (p.P44L) of MYCN are observed in approximately 3% to 4% of Wilms tumor cases at diagnosis [
131 ,133 ] and in 8.5% of cases at relapse.[109 ] In a study of 810 Wilms tumor cases, 24 (3%) had MYCN P44L hotspot variants. RFS was significantly lower (68.6%) in patients with P44L variants than in patients with wild-type MYCN status (87.1%).[133 ] - The MYCN interacting protein MAX was altered at codon 60 (R60Q) in 7 of 782 Wilms tumor cases (0.9%).[
133 ] RFS was significantly lower in patients with the MAX R60Q hotspot variant than in patients with wild-type MAX status.
- MYCN copy number gain was observed in approximately 13% of Wilms tumor cases. MYCN gain was more common in anaplastic cases (7 of 23 cases, 30%) than in nonanaplastic cases (11.2%), and it was associated with poorer relapse-free survival (RFS) and overall survival, independent of histology.[
- CTR9. Inactivating CTR9 germline pathogenic variants were identified in 4 of 36 familial Wilms tumor pedigrees.[
58 ,134 ]CTR9, which is located at chromosome 11p15.3, is a key component of the polymerase-associated factor 1 complex (PAF1c), which has multiple roles in RNA polymerase II regulation and is implicated in embryonic organogenesis and maintenance of embryonic stem cell pluripotency. - REST. Inactivating germline pathogenic variants in REST (encoding RE1-silencing transcription factor) were identified in four familial Wilms tumor pedigrees.[
78 ] REST is a transcriptional repressor that functions in cellular differentiation and embryonic development. Most REST variants clustered within the portion of REST encoding the DNA-binding domain, and functional analyses showed that these variants compromise REST transcriptional repression. When screened for REST variants, 9 of 519 individuals with Wilms tumor who had no history of relatives with the disease tested positive for the variant; some had parents who also tested positive.[78 ] These observations indicate that REST is a Wilms tumor predisposition gene associated with approximately 2% of Wilms tumor.
Figure 2 summarizes the genomic landscape of a selected cohort of Wilms tumor patients selected because they experienced relapse despite showing FH.[

Figure 2. Unsupervised analysis of gene expression data. Non-negative Matrix Factorization (NMF) analysis of 75 FH Wilms tumor resulted in six clusters. Five of six MLLT1 mutant tumors with available gene expression data occurred in NMF cluster 3, and two were accompanied by CTNNB1 mutations. This cluster also contained a substantial number of tumors with retention of imprinting of 11p15 (including all MLLT1-mutant tumors), in contrast to other clusters, where most cases showed 11p15 loss of heterozygosity or retention of imprinting. Almost all miRNAPG-mutated cases were in NMF cluster 2, and most WT1, WTX, and CTNNB1 mutations were in NMF clusters 3 and 4. Copyright © 2015 Perlman, E., Gadd, S., Arold, S. et al. MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology Wilms tumours. Nat Commun 6, 10013 (2015). https://doi.org/10.1038/ncomms10013 This article is distributed by Nature Publishing Group, a division of Macmillan Publishers Limited under a Creative Commons Attribution 4.0 International License, as described at https://creativecommons.org/licenses/by/4.0/.
Genomic alterations in Wilms tumor at relapse
Wilms tumor at relapse appears to maintain most of the genomic alterations present at diagnosis, although there may be changes in the prevalence of alterations in specific genes between diagnosis and relapse.[
- The prevalence of 1q gain in relapsed Wilms tumor specimens (75%) was higher than that observed for tumors at diagnosis (47%).[
109 ] The increased prevalence of 1q gain at relapse is consistent with its association with poor prognosis and disease progression. - SIX1 Q177R hotspot variants were identified at a higher rate in tumor specimens at relapse (11 of 82 cases; 13.4%) than in those at diagnosis (4%).[
109 ] For 45 cases with both diagnostic and relapse specimens, there were 6 cases with SIX1 Q177R at relapse, 3 of which did not have SIX1 Q177R at diagnosis. This is consistent with SIX1 Q177R not being an early tumorigenesis event in some cases.[109 ] - Genomic alterations in genes associated with the MYCN network were present in approximately 30% of Wilms tumor cases at relapse.[
109 ] The most common MYCN network alterations were MYCN tandem duplication (13%) and MYCN P44L hotspot variants (11%).
Recurrent and refractory Wilms tumors from 56 pediatric patients underwent tumor sequencing in the National Cancer Institute–Children's Oncology Group (NCI-COG) Pediatric MATCH trial. This process revealed genomic alterations that were considered actionable for treatment in MATCH study arms in 6 of 56 tumors (10.7%). BRCA2 variants were found in 2 of 56 tumors (3.6%).[
Genomic alterations in adults with Wilms tumor
Wilms tumor in patients older than 16 years is rare, with an incidence rate of less than 0.2 cases per 1 million people per year.[
A study of 14 patients with a Wilms tumor diagnosis who were older than 16 years (range, 17–46 years; median age, 31 years) evaluated exonic variants for 1,425 cancer-related genes.[
- Five patients (36%) harbored BRAF V600E variants.[
136 ] While BRAF V600E variants are extremely uncommon in pediatric Wilms tumor, they are present in 90% of metanephric adenomas of the kidney, a typically benign condition arising almost exclusively in adults.[137 ] - All five adult cases of Wilms tumor with BRAF V600E had better-differentiated areas identical to metanephric adenoma adjacent to areas consistent in appearance with epithelial Wilms tumor.
- Two of the five cases with BRAF V600E variants had TERT promoter variants in addition to BRAF variants.
- ASXL1 variants were observed in 4 of 14 cases, including 1 of 5 cases with BRAF V600E variants and 3 of 9 cases without BRAF V600E variants. ASXL1 variants are not common in pediatric Wilms tumor (approximately 2% of cases).[
70 ] - For the nine tumors that did not have BRAF variants, some had genomic alterations associated with Wilms tumor in children (e.g., 1q gain and variants in WT1 [n = 2]).
Another report described renal tumors that had histological overlap between metanephric adenoma and epithelial Wilms tumor.[
Bilateral Wilms Tumor
Approximately 5% to 10% of individuals with Wilms tumor have bilateral or multicentric tumors. The prevalence of bilateral involvement is higher in individuals with genetic predisposition syndromes than in those without predisposition syndromes. For example, in 545 cases of bilateral Wilms tumors, bona fide germline pathogenic variants were found in 22% of patients.[
Bilateral disease can be synchronous (both kidneys affected at the same time) or metachronous (one affected after the other) and occurs in 6.3% and 0.85% of patients with Wilms tumor, respectively.[
Bilateral Wilms tumors with WT1 variants are associated with early presentation in pediatric patients (age 10 months vs. age 39 months for those without a variant) and a high frequency of WT1 nonsense variant in exon 8. Three percent of patients with bilateral Wilms tumor have affected family members.[
Genomic analysis of kidney tissue in bilateral Wilms tumor indicates that a clonal expansion early in the nephrogenesis of normal-appearing but genetically aberrant precursor lesions occurred before the divergence of left and right kidney primordia.[
Screening Children Predisposed to Wilms Tumor
The primary purpose of screening is to enable earlier detection of a small and localized tumor (stage I or II), improve prognosis, and use less intensive treatment (such as to facilitate nephron-sparing surgery).[
Tumor screening programs for each overgrowth syndrome have been suggested. These programs were based on published age, incidence of tumor type, and recommendations from the 2016 American Association for Cancer Research (AACR) Childhood Cancer Predisposition Workshop. Although data about different cancer risks based on genetic or epigenetic subgroups for certain syndromes are emerging, and subgroup-specific recommendations have been developed in Europe, these practices have not been adopted in the United States. The AACR workshop committee proposed a uniform screening approach for all syndromes associated with a greater-than-1% risk of Wilms tumor. Additional screening for hepatoblastoma by serum alpha-fetoprotein (AFP) measurement and ultrasonography is also recommended for patients with Beckwith-Wiedemann syndrome, trisomy 18, and Simpson-Golabi-Behmel syndrome.[
Based on a literature search of patients with Beckwith-Wiedemann spectrum and Wilms tumor where the age at diagnosis was compared against data collected through the Surveillance, Epidemiology, and End Results (SEER) Program, screening patients with Beckwith-Wiedemann spectrum seems to significantly decrease the age and stage at the time of diagnosis in this population. Screening until age 7 years is effective in detecting close to 95% of all Wilms tumors in the Beckwith-Wiedemann spectrum. Screening until age 30 months may also prove useful for patients with ICR2-LoM, consistent with the recommendations for hepatoblastoma screening in this population.[
- Beckwith-Wiedemann syndrome. Approximately 8% of patients with Beckwith-Wiedemann syndrome will develop a malignancy, with the most common being either Wilms tumor or hepatoblastoma, although adrenal tumors can also occur.[
103 ]Screening for hepatoblastoma or adrenal tumors with abdominal ultrasonography and serum AFP usually begins at birth or when the syndrome is diagnosed and continues until age 4 years. After age 4 years, most hepatoblastomas will have occurred, and imaging may be limited to renal ultrasonography, which is quicker and does not require fasting before the exam.[
145 ]Screening for Wilms tumor usually continues until age 8 years. Physical examination by a specialist (geneticist or pediatric oncologist) is recommended twice per year, and ongoing education regarding tumor manifestations, reinforcing the rationale for screening and compliance with the screening regimen, is discussed.[
143 ]Proposed screening guidelines for Wilms tumor are available for patients with Beckwith-Wiedemann syndrome who have undergone molecular subtyping.[
103 ] The four main molecular subtypes of Beckwith-Wiedemann syndrome (ICR1-GoM, ICR2-LoM, UPD, and CDKN1C variant) are characterized by specific genotype-phenotype correlations, including tumor risk. For more information about the molecular subtypes, see theGenomics of Wilms Tumor section.Proposed screening for specific molecular subtypes of Beckwith-Wiedemann syndrome is as follows:
- Patients with a defect of the ICR1 region (ICR1-GoM) and UPD should undergo abdominal ultrasonography every 3 months until age 8 to 10 years. A clinical examination of the abdomen and muscle mass occurs monthly for the first year and then at 3-month intervals, between ultrasonography scans, until age 6 years.
- For patients with loss of imprinting at ICR2 (ICR2-LoM), an abdominal ultrasonography is performed at the time of clinical or molecular diagnosis. Only patients with organomegaly or severe hemihypertrophy require surveillance by ultrasonography scans. Monthly clinical examinations are performed for the first 2 years, followed by clinical examinations every 3 to 6 months until age 6 years.
- Patients with a CDKN1C variant are not at high risk of developing Wilms tumor. There are no data to support routine screening.
- WAGR Syndrome (or WAGR Spectrum). Patients with WAGR spectrum tend to experience an earlier age of initial Wilms tumor development compared with patients with nonsyndromic Wilms tumor. Using data from the WAGR Syndrome Patient Registry (n = 91), the reported median age at initial development of Wilms tumor or isolated nephrogenic rests was 19 months (range, 11–28 months). All patients with reported Wilms tumor developed their initial Wilms tumor by age 8 years, and 95% of patients developed their tumor by age 5 years.[
13 ] Other investigators have reported that approximately 20% of patients developed their Wilms tumor after age 4 years.[15 ,16 ]Multiple patients have been diagnosed with the development of Wilms tumor past the age of 7 to 8 years and/or relapse occurring years after initial diagnosis. Some cases labeled as relapses have been de novo disease in the contralateral kidney.[
16 ] In the WAGR Syndrome Patient Registry, late presentation of relapse occurred in one participant at age 19 years, 7 months, which was more than 17 years from their first Wilms tumor diagnosis and represented the third occurrence.[13 ]Investigators from SIOP have reported about the benefit of surveillance in a cohort of 43 patients with WAGR Syndrome and Wilms tumor/nephroblastomatosis enrolled in SIOP treatment studies. Of 39 patients, 27 (69%) were asymptomatic and the tumors were detected by surveillance, whereas 12 patients (31%) presented with a palpable/visible abdominal mass and/or other symptoms. Of these 12 patients, 2 had not been diagnosed with WAGR syndrome. Tumors detected by surveillance had a significantly decreased volume compared with tumors that were symptomatic (18 mL vs. 375 mL; P = .001), which enabled a high rate of nephron-sparing surgery (85%).[
146 ] The authors recommend the use of preoperative chemotherapy as treatment for patients with WAGR syndrome in order to facilitate nephron-sparing surgery. This surgery can improve outcomes for patients with chronic kidney disease associated with WAGR syndrome.[147 ] Preoperative chemotherapy has been reported to decrease tumor size in 50% of WAGR patients.[146 ]Surveillance options for the WAGR population at age 8 years and older should be discussed with the patient's family and multidisciplinary health care team to determine the appropriate follow-up schedule for Wilms tumor monitoring. Factors such as the patient's previous medical history and presence of nephrogenic rests and nephroblastomatosis should be considered.[
13 ] - Hemihypertrophy (also known as lateralized overgrowth or hemihyperplasia). Children with isolated hemihypertrophy are also at risk of developing liver tumors, adrenal tumors, and Wilms tumor (risk, 3%–4%). Screening with abdominal ultrasonography and serum AFP is suggested until age 4 years. After age 4 years, most hepatoblastomas will have occurred, and imaging may be limited to renal ultrasonography, which is quicker and does not require fasting before the exam.[
143 ]Hemihypertrophy can occur as part of a syndrome (most commonly Beckwith-Wiedemann syndrome) or an isolated phenomenon. The Beckwith-Wiedemann Syndrome International Consensus Group suggested that individuals with florid Beckwith-Wiedemann syndrome phenotype and those with isolated hemihypertrophy who have similar molecular findings as those with Beckwith-Wiedemann syndrome should be considered part of the Beckwith-Wiedemann Syndrome spectrum and managed according to the subtype of Beckwith-Wiedemann syndrome. Molecular testing may be considered for patients with isolated hemihypertrophy based on the clinical scoring system proposed by the Beckwith-Wiedemann Syndrome International Consensus Group.[
148 ]Children with isolated hemihypertrophy and negative molecular tests may not need surveillance because the risk may be very low. However, more studies of large cohorts of molecularly tested children with isolated hemihypertrophy are needed to determine the risk.[
92 ,148 ] - Sporadic aniridia. Newborns born with sporadic aniridia should undergo molecular testing for deletion analysis of PAX6 and WT1, which are consistent with WAGR syndrome. Approximately 30% of patients with sporadic aniridia have WAGR.[
149 ] If a deletion of WT1 is observed, the child should be screened with ultrasonography every 3 months until age 8 years, and the parents should be educated about the need for early identification and treatment of Wilms tumor.[92 ,150 ,151 ] - Children of survivors of bilateral Wilms tumor. Although the risk of Wilms tumor in the children of survivors of bilateral Wilms tumor is unknown and likely varies with the gene in which the variant occurred, some experts recommend screening such children with serial ultrasonography examinations every 3 months until age 8 years.[
77 ] - Bohring-Opitz syndrome. Bohring-Opitz syndrome is a rare genetic condition associated with ASXL1 variants. Overall, about 7% of individuals with Bohring-Opitz syndrome develop Wilms tumors.[
152 ] Screening with abdominal ultrasonography every 3 to 4 months in the first 8 years of life has been suggested.[48 ] - Simpson-Golabi-Behmel syndrome. Affected males with Simpson-Golabi-Behmel syndrome with GPC3 variants or deletions have an approximate 10% risk of Wilms tumor. Regular age-dependent screening for tumors, including abdominal ultrasonography, urinalysis, and biochemical markers, has been recommended for males with Simpson-Golabi-Behmel syndrome, although the true benefit has not been determined. It was previously thought that carrier females were not at increased risk of Wilms tumor and did not require surveillance. However, there are reports of rare cases where Simpson-Golabi-Behmel syndrome may have a significant clinical expression in females and Wilms tumors have occurred.[
92 ] In clearly affected females, screening should be considered for embryonic tumors, including Wilms tumors.[153 ] - Klippel-Trénaunay syndrome. The risk of Wilms tumor in children with Klippel-Trénaunay syndrome (a unilateral limb overgrowth syndrome) was no different than the risk in the general population when assessed using the National Wilms Tumor Study (NWTS) database. Routine ultrasonography surveillance is not recommended.[
154 ] - Perlman syndrome. Perlman syndrome is a rare congenital overgrowth syndrome that has an autosomal recessive inheritance pattern. A molecular diagnosis can be made by the presence of inactivating variants in DIS3L2 on chromosome 2q37.1. Fifty-three percent of children will die in the neonatal period. The kidneys show nephroblastomatosis in about 75% of cases. The incidence of Wilms tumor is 64% in infants who survive beyond the neonatal period.[
39 ] It is recommended that these patients be offered regular surveillance similar to that offered to patients with Beckwith-Wiedemann syndrome.[143 ] - DICER1 syndrome. Cystic nephroma is seen in 10% of families presenting with pleuropulmonary blastoma, typically occurring before age 4 years. Rare progression to anaplastic sarcoma of the kidney may occur. DICER1 syndrome includes an elevated risk of Wilms tumor, which is not a consequence of a prior cystic nephroma. Surveillance consists of abdominal ultrasonography, which starts with the first chest computed tomography (CT) screening for pleuropulmonary blastoma, and it is done every 6 to 12 months to age 8 years. Surveillance may be continued annually until age 12 years, depending on the individual patient.[
155 ] Thirteen years is the oldest reported age of a Wilms tumor diagnosis in a DICER1 variant carrier.[117 ,156 ] Surveillance aims to identify cystic nephromas when they are small and nephron-sparing surgery is still possible, since tumors that progress to anaplastic sarcoma of the kidney have higher morbidity rates. Because anaplastic sarcoma of the kidney can be diagnosed at wider age ranges than cystic nephroma, extending abdominal ultrasonography screening to age 12 years should be considered. This time frame is the highest risk period for anaplastic sarcoma of the kidney (90% of germline cases presumably detected).[157 ] - Germline pathogenic or likely pathogenic variant in Wilms tumor predisposition genes (CTR9, REST, TP53, TRIM28) in the absence of syndrome features or a family history suggestive of any specific cancer predisposition syndrome. Surveillance should be considered throughout the period of increased Wilms tumor risk (typically up to age 8 years), but may vary depending on the condition.[
158 ]
Genetic counseling
Wilms tumor develops in association with an underlying germline predisposition in 10% to 15% of cases. A genetics referral is recommended for all children with Wilms tumor who have a positive family history of cancer, bilateral kidney involvement, or syndrome-specific features.[
The McGill Interactive Pediatric OncoGenetic Guidelines (MIPOGG) study aims to develop an eHealth tool to assist physicians in identifying children at increased risk of having a cancer predisposition syndrome. Based on a thorough literature review, a decisional algorithm specific to Wilms tumor was developed. This algorithm consists of five tumor-specific criteria (age <2 years, bilaterality/multifocality, stromal-predominant histology, nephrogenic rests, and overgrowth features) and universal criteria, including features of family history suspicious for a cancer predisposition syndrome and congenital anomalies. This tool was applied retrospectively to 180 consecutive pediatric patients with Wilms tumor, diagnosed and/or treated at The Hospital for Sick Children (1997–2016) who underwent targeted molecular diagnostic testing.[
- Application of the algorithm generated a binary recommendation for or against genetic referral for cancer predisposition syndrome evaluations. The algorithm identified 100% of children with Wilms tumor and a confirmed cancer predisposition syndrome (n = 27).
- Age younger than 2 years, bilaterality/multifocality, and congenital anomalies were strongly associated with pathogenic variants in WT1.
- Presence of more than one overgrowth feature was strongly associated with Beckwith-Wiedemann syndrome.
- Stromal-predominant histology did not contribute to cancer predisposition syndrome recognition (and has been removed from the essential criteria). In combination with other suspicious features, stromal histology may increase the likelihood of identifying a WT1-related disorder.
- Plans are under way to test this tool prospectively in patients diagnosed with Wilms tumor who undergo comprehensive genomic sequencing of their germline DNA.
If a child is found to harbor a pathogenic or likely pathogenic variant in a Wilms tumor predisposition gene, then their parents and close relatives can also be offered testing. Affected individuals should be counseled about the risk of additional neoplasms and oncologic manifestations, as appropriate, as well as the risk to future offspring.[
Clinical Features of Wilms Tumor
Most Wilms tumor patients present asymptomatically with an abdominal mass noticed by a parent or pediatrician on a well-child visit. In children with known predisposing clinical syndromes, renal tumors can be found during routine screening. Clinical findings may include the following:
- A lump, swelling, or pain in the abdomen. Most children present with a nontender, large flank mass that is noted when they are bathed or dressed. If noted on physical examination, the mass does not move with respiration in contrast to splenomegaly. Abdominal pain is present in 40% of children.
- Blood in the urine. Gross hematuria occurs in about 18% of children with Wilms tumor at presentation, and microscopic hematuria is seen in 24% of patients.[
160 ] - Hypertension. About 25% of children have hypertension at presentation, which is attributed to activation of the renin-angiotensin system.
- Hypercalcemia. Symptomatic hypercalcemia can sometimes be seen at presentation of rhabdoid tumors.
- Constitutional symptoms such as fever, anorexia, and weight loss occur in 10% of cases.
Children with Wilms tumor or other renal malignancies may also seek medical attention as a result of the following:
- Vascular obstruction or metastasis, including pulmonary symptoms such as dyspnea caused by lung metastasis.
- Abdominal pain caused by liver metastasis, prominent abdominal wall vessels, or varicocele due to inferior vena cava obstruction.
- Pulmonary embolus (rare).
- Rapid abdominal enlargement, anemia, and severe pain may be seen in the few children who develop subcapsular hemorrhage.
Diagnostic and Staging Evaluation for Wilms Tumor
The Children's Oncology Group Diagnostic Imaging Committee and the Society for Pediatric Radiology Oncology Committee have published a white paper with recommendations for the imaging of pediatric renal tumors.[
- Physical examination and history. Children with a renal mass are carefully assessed for signs of associated syndromes such as aniridia, developmental delay, hypospadias, cryptorchidism, pseudohermaphrodism, overgrowth, and hemihypertrophy.
- Complete blood count.
- Liver function test.
- Renal function test.
- Urinalysis.
- Abdominal imaging.
- Abdominal x-ray.
- Ultrasonography exam of the abdomen. Ultrasonography exam of the abdomen is often performed before a more definitive CT scan with contrast or magnetic resonance imaging (MRI) with contrast of the abdomen. This procedure is unnecessary after the definitive diagnostic study has been performed.[
161 ] - CT scan with contrast (oral contrast is not necessary) or MRI of abdomen and pelvis (with and without intravenous contrast).[
161 ]- CT scan of the abdomen will confirm the renal origin of the mass and determine whether there are bilateral tumors.[
162 ] About 5% of renal masses thought to be Wilms tumor based on clinical and radiological findings are diagnosed as another condition.[163 ] - A review of children with bilateral Wilms tumor demonstrated that only 0.25% of bilateral tumors were missed with modern helical CT scans, all of which were small tumors.[
164 ] - Preoperative assessment by imaging of intravascular extension of Wilms tumor is essential to guide management. Four percent of Wilms tumor patients present with inferior vena cava or atrial involvement and 11% with renal vein involvement, which may lead to differences in management. Embolization of a caval thrombus to the pulmonary artery is rare but can be lethal, and the presence of a thrombus must be identified preoperatively to prevent this occurrence and guide treatment. A report from the COG shows that CT can accurately identify cavoatrial thrombus, obviating the need for ultrasonography if CT has already been performed.[
165 ] - Ascites beyond the cul-de-sac is most predictive of preoperative Wilms tumor rupture, regardless of attenuation. In the presence of ascites, fat stranding around the tumor and the presence of retroperitoneal fluid are highly predictive of rupture.[
162 ] - The concern about CT is radiation exposure, but the procedure is quick, allows continuous imaging of the chest and abdomen, has moderate specificity for detection of preoperative spill, may help distinguish nephrogenic rests from Wilms tumor, and provides excellent pulmonary detail.[
162 ,166 ] - The main drawback with abdominal MRI is that moderate to deep sedation is often required in young children. However, it provides excellent organ detail in patients with bilateral involvement or liver metastases. Abdominal MRI is preferred for better assessment of potential nephrogenic rests and their distinction from true Wilms tumor.[
167 ] If the decision is to perform an abdominal MRI, then a CT scan of the lungs should be done first to avoid obscuration of the lung bases by atelectasis.[168 ] MRI is the preferred imaging modality in children with known bilateral Wilms tumors or known bilateral tumor predisposition.[168 ]
- CT scan of the abdomen will confirm the renal origin of the mass and determine whether there are bilateral tumors.[
- CT scan of chest. Approximately 15% to 20% of patients will present with metastases. The common sites of metastases for Wilms tumor are the lungs (85%), liver (10%), and bone and spine (rarely). CT scanning provides the most sensitive method of detecting metastatic lung nodules. The use of iodinated intravenous contrast is preferred for baseline chest CTs because it allows simultaneous evaluation of the lung parenchyma, regional vasculature, and other mediastinal structures.[
161 ] Approximately 7.5% of patients may present with a pleural effusion.[169 ][Level of evidence C1] - Chest x-ray is unnecessary if chest CT is performed initially.
- Fluorine F 18-fludeoxyglucose (18F-FDG) positron emission tomography (PET)-CT. PET scanning is not routinely used in Wilms tumor, although Wilms tumor is 18F-FDG avid. 18F-FDG PET-CT imaging adds clinically applicable information to conventional CT scan imaging. PET-CT may be particularly helpful in patients with bilateral disease or those receiving preoperative chemotherapy. 18F-FDG PET-CT highlights FDG-avid areas in the tumor and metastases, which corresponds to histologically confirmed active disease.[
170 ] - Bone scan or cross-sectional imaging of other sites is reserved for patients with signs or symptoms of distant extrapulmonary metastases.
- von Willebrand disease work-up. About 4% of patients presenting with Wilms tumor have an acquired form of von Willebrand disease, although many are asymptomatic. von Willebrand multimers bind to Wilms tumor, reducing the plasma concentration to low levels.[
171 ] Some clinicians recommend evaluation for von Willebrand disease before surgery, because, although uncommon, it may be associated with substantial bleeding risks and can be managed preemptively. Acquired von Willebrand disease in the context of Wilms tumor will usually resolve once chemotherapy is started or the tumor is resected.[172 ] - Biopsy or resection. In children with a renal mass that clinically appears to be resectable or stage I or stage II Wilms tumor, a biopsy is not performed so that tumor cells are not spread during the biopsy. A biopsy would upstage such a patient to stage III. Nephrectomy (in North America) or chemotherapy (in Europe) is performed instead. Therefore, the diagnostic pathology is first seen when the nephrectomy specimen is examined.
Biopsy of a renal mass may be indicated if the mass is atypical by radiographic appearance for Wilms tumor, and the patient is not going to undergo immediate nephrectomy. If a primary nephrectomy cannot be performed, a biopsy, either open or with multiple cores, is required. The contraindications to primary nephrectomy are the following:
- Extension of tumor thrombus to the level of the hepatic veins. These patients should be considered for tumor resection after neoadjuvant chemotherapy when there is evidence of regression of the vena caval thrombus regardless of the degree of response of the primary tumor.
- The tumor involves contiguous structures whereby the only means of removing the kidney tumor requires removal of the other structure (e.g., spleen, pancreas, colon but excluding the adrenal gland and diaphragm). While Wilms tumors are frequently adherent to adjacent organs, in most cases, there is not frank invasion by the tumor and it can be dissected freely from the organs. Radical en bloc resection (e.g., partial hepatectomy) is not generally warranted. If removal of a small section of diaphragm, psoas muscle, or tip of the pancreas allows the tumor to be removed intact, this is considered safe and appropriate.
- The surgeon's judgment that nephrectomy would result in significant or unnecessary morbidity/mortality, significant tumor spill, or residual tumor.[
173 ] - If there is pulmonary compromise because of extensive pulmonary metastases or, in rare cases, hepatic disease.
If a child undergoes a biopsy as the first procedure, they are considered stage III because they have gross residual tumors.
Biopsy tissue from inoperable Wilms tumor obtained before chemotherapy may be used for histological review and initial treatment decisions. However, the use of biopsy to determine histology in an inoperable tumor remains controversial because biopsy may cause local tumor spread and the histological classification of the Wilms tumor cannot be determined by biopsy.[
173 ]Anaplastic histology can be difficult to detect in any biopsy sample because of tumor heterogeneity. Data from NWTS-4 and NWTS-5 (COG-Q9401/NCT00002611) demonstrated that, because of the histological heterogeneity of Wilms tumor, a significant number of patients have anaplastic histology that is missed during an up-front biopsy whether it be a core needle biopsy or an incisional biopsy [
174 ] but revealed at the time of definitive surgery after chemotherapy.Detection of a contralateral renal lesion in a child with Wilms tumor can change the stage and initial management of the patient, indicating a role for a renal-sparing approach without up-front surgery. The detection of contralateral renal lesions is important at baseline imaging because routine intraoperative exploration of the contralateral kidney is no longer recommended based on the results of the NWTS-4 study.[
164 ,175 ] Treatment as a bilateral Wilms tumor should be considered if the initial imaging studies suggests a bilateral process. If the origin of the other lesion is indeterminate, a pathological assessment of that lesion should be considered before proceeding with a nephrectomy.[164 ,175 ]Children who have bilateral Wilms tumor are often treated without a biopsy.[
176 ] Biopsy can be avoided if the child is of typical age and the tumor has the usual radiographic appearance. This was assessed on the COG AREN0534 (NCT00945009) study where 187 of 189 patients with Wilms tumor were treated initially without a biopsy. If after 6 weeks of therapy, response was less than 30% by RECIST1.1 criteria, bilateral biopsies were performed to assess for anaplasia, stromal differentiation, and rhabdomyomatous changes. If anaplasia was detected, the chemotherapy treatment was changed. If stromal differentiation or rhabdomyomatous changes were detected, further chemotherapy was unlikely to result in tumor shrinkage and definitive surgery was the suggested approach.[176 ]
For patients with suspected Wilms tumor, additional preoperative staging studies are performed to assess lymph node status, intravascular extension, and rupture of Wilms tumor.[
- Lymph node sampling is required to locally stage all Wilms tumor patients. Lymph nodes have been shown to be of major prognostic value for both short-term and long-term survival. Gross inspection is notoriously inaccurate, with a false-negative rate of 31.3% and a false-positive rate of 18.1%.[
177 ] - Intravascular extension of the Wilms tumor. Preoperative assessment of intravascular extension of Wilms tumor is essential to guide management. The presence of intravenous tumor thrombus in the lumen of the renal vein, inferior vena cava, and right atrium has been reported in up to 11.3% of Wilms tumor patients and may lead to differences in management.
In North America, local staging of Wilms tumor is performed with CT or MRI of the abdomen and pelvis. Contrast-enhanced CT for Wilms tumor patients has high sensitivity and specificity for detection of cavoatrial tumor thrombus that may impact surgical approach. Routine Doppler evaluation may be done after CT has been performed but is not necessarily required.[
165 ] If the tumor is at or above the hepatic veins, a biopsy with preoperative chemotherapy is suggested because of the lower rate of serious intraoperative complications. Before surgical approach to the renal mass is performed, large tumor thrombi need to be controlled, especially when they extend above the hepatic vein, to avoid embolization of the tumor. In some cases, cardiopulmonary bypass is required.[178 ] - Wilms tumors can rupture before surgery. The term rupture is used to imply a break in the tumor capsule before surgery, whereas the term spill refers to a break in the tumor during surgery. Based on their similar diagnostic performances, either CT or MRI can be used to detect rupture. Although imaging findings of rupture have high specificity (88%), the diagnosis of rupture has to be confirmed at surgery. Imaging alone cannot be used for initial staging because of the low sensitivity and specificity for preoperative rupture and lymph node status.[
168 ]
Prognosis and Prognostic Factors for Wilms Tumor
Wilms tumor is a curable disease in most affected children. Since the 1980s, the 5-year survival rate for Wilms tumor with favorable histology (FH) has been consistently greater than 90%.[
The prognosis for patients with Wilms tumor depends on the following:[
- Histopathological features of the tumor (
FH vs.anaplastic histology ). For more information, see theHistological Findings in Wilms Tumor section. - Stage of disease at diagnosis.
- Molecular features of the tumor such as 1q gain and loss of heterozygosity of 1p and 16q. 1q gain, affecting 28% of Wilms tumors, is the most powerful predictor of outcome and is associated with an adverse outcome.[
107 ,108 ,110 ] Loss of heterozygosity of 11p15 and loss of imprinting of 11p15 is associated with relapse in very low-risk patients who do not receive chemotherapy.[110 ,185 ] For more information, see theGenomics of Wilms Tumor section. - Age. Age at presentation is commonly between 2 and 5 years, and the incidence of Wilms tumor in children older than age 10 years is rare. Older age is associated with an adverse prognosis.[
186 ,187 ]
Older Adolescents and Adults With Wilms Tumor
Wilms tumor in patients older than 16 years is rare, with an incidence rate of around 0.2 cases per 1 million per year in patients aged 15 to 39 years.[
Wilms tumor occurring in adults differs from that occurring in children in several ways. Adults rarely present with bilateral disease (<1%). More adult patients had additional primary malignancies (both before and after the diagnosis of Wilms tumor) compared with their pediatric counterparts.[
A situation that is specific to adults is the diagnosis of Wilms tumor in pregnant women. This diagnosis is made incidentally during the ultrasonography monitoring of the pregnancy or because of clinical symptoms such as abdominal pain and fever.[
The outcomes for adolescent and young adult (AYA) patients (aged 15 to 39 years) and adult patients are inferior to the outcomes for children.
- In an analysis of patients with Wilms tumor in the Surveillance, Epidemiology, and End Results (SEER) database, AYA patients (n = 104) had a statistically worse 5-year OS rate (69% vs. 94%; P < .001) than did pediatric patients (n = 2,574).[
193 ][Level of evidence C1]
The inferior outcome of the adult patients may be multifactorial, including differences in tumor biology between children and adults, incorrect diagnosis, inadequate staging (e.g., more likely to be staged as localized disease or to not receive lymph node sampling), undertreatment/poor compliance (e.g., not receiving radiation therapy), unfamiliarity of medical oncologists and pathologists with Wilms tumors in adults (possibly leading to diagnostic error and delay), delays in initiating the appropriate risk-adapted therapy, and lack of specific treatment protocols for adults.[
Treatment of adults with Wilms tumor
As Wilms tumor rarely occurs in adults, there is no standard treatment protocol. Better results have been reported for adults when they are treated in pediatric trials.
A Wilms tumor in an adult represents a therapeutic emergency because of the tumor's rapid growth and because urologists and oncologists are more familiar with the indolent growth of renal cell carcinoma.
The NWTS Group reported the outcomes for adult patients with Wilms tumor from the NWTS-1, -2, and -3 trials.[
- The 3-year OS rate for adults on the NWTS-1 trial was 24% (compared with 74% in children) and improved to a 5-year OS rate of 82.6% on the NWTS-3 trial, although the number of adult patients treated on each trial was 31 or fewer.
- These data suggest that many adults with Wilms tumor, if treated appropriately, can expect to be cured, especially if the tumor has not spread and/or is completely resected.
For adults with refractory or recurrent disease, screening for potential therapeutic targets in the tumor should be considered.[
The following recommendations from the renal tumor committees of the International Society of Paediatric Oncology (SIOP) and COG encourage a uniform approach to improve outcome for adults with Wilms tumor.[
- Consult with a pediatric oncologist who has experience with the treatment of Wilms tumor as soon as a histological diagnosis is suspected.
- Avoid delaying the start of chemotherapy. Ideally, chemotherapy, and radiation therapy if necessary, should be started by day 14 postnephrectomy, although delaying the start until day 30 is acceptable.
- Be alert for toxicity of vincristine (neurotoxicity) and dactinomycin (hepatic toxicity) in adults.
- Register patients in pediatric renal tumor trials if studies are available and the patients are eligible.
In a series of 14 adult Wilms tumors that were evaluated by expanded targeted sequencing, 5 (36%) demonstrated BRAF V600E variants. These tumors contained areas that were morphologically identical to BRAF V600E–altered metanephric adenoma. All of the BRAF V600E–altered Wilms tumors in this cohort occurred in patients older than 30 years. Identifying a BRAF V600E variant has therapeutic significance because these patients may respond to therapy with BRAF/MEK inhibitors.[
Histological Findings in Wilms Tumor
Although most patients with a histological diagnosis of Wilms tumor do well with current treatment, approximately 10% of patients have histopathological features that are associated with a worse prognosis, and in some types, with a high incidence of relapse and death. Wilms tumor can be separated into the following two prognostic groups based on tumor and kidney histopathology:
-
Favorable histology (FH) . -
Anaplastic histology .
Favorable histology (FH)
Histologically, Wilms tumor mimics the triphasic development of a normal kidney consisting of blastemal, epithelial (tubules), and stromal cell types. Not all tumors are triphasic, and monophasic patterns may present diagnostic difficulties.
While associations between histological features and prognosis or responsiveness to therapy have been suggested, with the exception of anaplasia, none of these features have reached statistical significance in North American treatment algorithms. Therefore, histological features do not direct the initial therapy.[
In the AREN03B2 (NCT00898365) study, patients with stage I disease were analyzed based on epithelial histology (n = 177) and treatment. When analyzed by epithelial histology, the 4-year EFS rate was 96%, and the OS rate was 100%. When these patients were analyzed according to treatment, patients treated with vincristine and dactinomycin (regimen EE-4A) (n = 117) had a 4-year EFS rate of 96%, compared with a 4-year EFS rate of 98% for patients who underwent nephrectomy only (n = 57) (P = .549).[
In a series of 14 adults (aged 17–46 years) with Wilms tumors, targeted tumor sequencing revealed BRAF V600E variants in 5 of the tumors. All of these tumors had better-differentiated areas that were identical to metanephric adenoma, in combination with epithelial Wilms tumor. Adults who have tumors with this histological manifestation may benefit from sequencing of their tumors.[
Anaplastic histology
Anaplastic histology accounts for about 10% of Wilms tumor cases. Anaplastic histology is the single most important histological predictor of response and survival in patients with Wilms tumor. Tumors occurring in older patients (aged 10–16 years) have a higher incidence of anaplastic histology.[
The following two histological criteria must be present to confirm the diagnosis of anaplasia:
- Presence of multipolar polyploid mitotic figures with marked nuclear enlargement.
- Hyperchromasia.
Changes on 17p consistent with variants in the TP53 gene have been associated with foci of anaplastic histology.[
Anaplasia correlates best with responsiveness to therapy rather than to tumor aggressiveness. It is most consistently associated with poor prognosis when it is diffusely distributed and when identified at advanced stages. These tumors are more resistant to the chemotherapy traditionally used in children with FH Wilms tumor.[
GermlineTRIM28testing
To identify patients with germline pathogenic variants in TRIM28, routine assessment of Wilms tumors by immunohistochemistry with the anti-KAP1 antibody should be performed to look for TRIM28 loss. Even though most TRIM28-altered tumors are epithelial (predominant) Wilms tumors, testing should be considered for all subtypes, because other histological subtypes have been reported to have the TRIM28 variant. Subsequently, genetic analysis of TRIM28 in blood-derived DNA can be performed in all patients who display loss of TRIM28 in the tumor.[
Nephrogenic rests
Nephrogenic rests are abnormally retained (past 36 weeks) embryonic kidney precursor cells arranged in clusters. Nephrogenic rests are found in about 1% of unselected pediatric autopsies, 35% of kidneys with unilateral Wilms tumor, and nearly 100% of kidneys with bilateral Wilms tumor.[
The term nephroblastomatosis is defined as the presence of diffuse or multifocal nephrogenic rests. In unilateral Wilms tumors, nephrogenic rests are usually only detectable by histology, whereas in bilateral Wilms tumor, the proliferating nephrogenic rests may be large enough to be seen on imaging.[
Distinguishing between nephrogenic rests and Wilms tumors by imaging is challenging because there is an overlap in their appearance. A retrospective study evaluated 52 young children (aged <5 years) with nephrogenic rests and small Wilms tumors (all lesions <5 cm) that had been surgically sampled and pathologically evaluated before any medical intervention. The investigators found that a Wilms tumor diagnosis should be favored over a nephrogenic rest diagnosis when a renal mass is spherical, exophytic, and larger than 1.75 cm in maximal diameter. Homogeneity by imaging favors the diagnosis of perilobar nephrogenic rests, whereas intralobar rests and Wilms tumors are more likely to be inhomogeneous.[
Diffuse hyperplastic perilobar nephroblastomatosis represents one unique category of nephroblastomatosis that forms a thick rind around one or both kidneys and is considered a preneoplastic condition. Distinguishing between Wilms tumor and diffuse hyperplastic perilobar nephrogenic rests may be a challenge, and it is critical to examine the juncture between the lesion and the surrounding renal parenchyma. Incisional biopsies are of no diagnostic value unless they include the margin between the lesion and the normal renal parenchyma.[
The type and percentage of nephrogenic rests vary in patients with unilateral or bilateral disease. Patients with bilateral Wilms tumor have a higher proportion of perilobar rests (52%) than of intralobar or combined rests (32%) and higher relative proportions of rests, compared with patients with unilateral tumors (18% perilobar and 20% intralobar or both).[
Patients with any type of nephrogenic rest in a kidney removed for nephroblastoma are considered at increased risk of tumor formation in the remaining kidney. This risk decreases with patient age.[
For information about the treatment of bilateral diffuse hyperplastic perilobar nephroblastomatosis, see the
Extrarenal nephrogenic rests are rare and may develop into extrarenal Wilms tumor.[
Stage Information for Wilms Tumor
Both the results of the imaging studies and the surgical and pathological findings at nephrectomy are used to determine the stage of disease. The stage is the same for tumors with FH or anaplastic histology. Thus, the stage information is characterized by a statement of both criteria (for example, stage II, FH or stage II, anaplastic histology).[
The staging system was originally developed by the NWTS Group and is still used by the COG. The staging system used in North America and incidence by stage are outlined below.[
Stage I
In stage I Wilms tumor (43% of patients), all of the following criteria must be met:
- Tumor is limited to the kidney and is completely resected.
- The renal capsule is intact.
- The tumor is not ruptured or biopsied before being removed.
- No involvement of renal sinus vessels.
- No evidence of the tumor at or beyond the margins of resection.
- All lymph nodes sampled are negative.
For a tumor to qualify for certain therapeutic protocols such as very low-risk stage I, regional lymph nodes must be examined microscopically.
Stage II
In stage II Wilms tumor (20% of patients), the tumor is completely resected, and there is no evidence of tumor at or beyond the margins of resection. The tumor extends beyond the kidney as evidenced by any one of the following criteria:
- There is regional extension of the tumor (i.e., penetration of the renal capsule, or extensive invasion of the soft tissue of the renal sinus, as discussed below).
- Blood vessels in the nephrectomy specimen outside the renal parenchyma, including those of the renal sinus, contain tumor cells. Margins are clear.
- Vascular extension of tumor is considered stage II only if it is completely removed en bloc in the nephrectomy specimen.
All lymph nodes sampled are negative.
Rupture or spillage confined to the flank, including biopsy of the tumor, is now included in stage III by the COG Renal Tumor Committee (COG RTC); however, data to support this approach are controversial.[
Stage III
In stage III Wilms tumor (21% of patients), there is postsurgical residual nonhematogenous tumor that is confined to the abdomen. Any one of the following may occur:
- Lymph nodes in the abdomen or pelvis are involved by tumor. (Lymph node involvement in the thorax or other extra-abdominal sites is a criterion for stage IV.)
- The tumor has penetrated through the peritoneal surface.
- Tumor implants are found on the peritoneal surface.
- Gross or microscopic tumor remains postoperatively (e.g., tumor cells are found at the margin of surgical resection on microscopic examination).
- The tumor is not completely resectable because of local infiltration into vital structures.
- Tumor rupture before surgery or any spill during surgery is considered stage III.
- Biopsy is performed, regardless of type—Tru-cut biopsy, open biopsy, or fine-needle aspiration—before the tumor is removed.
- The tumor is removed in more than one piece (e.g., tumor cells are found in a separately excised adrenal gland; a tumor thrombus in the renal vein is removed separately from the nephrectomy specimen). Extension of the primary tumor in the vena cava into the thoracic vena cava and heart is considered stage III, rather than stage IV, even though outside the abdomen—and it can even be stage II if completely resected en bloc with the nephrectomy specimen.
Lymph node involvement and microscopic residual disease are reported as highly predictive of outcome in patients with stage III FH Wilms tumor.[
Stage IV
In stage IV Wilms tumor (11% of patients), one of the following is present:
- Hematogenous metastases (lung, liver, bone, brain).
- Lymph node metastases outside the abdominopelvic region.
The presence of tumor within the adrenal gland is not interpreted as metastasis and staging depends on all other staging parameters present. According to the criteria described above, the primary tumor is assigned a local stage, which determines local therapy. For example, a patient may have stage IV, local stage III disease.
Stage V (bilateral)
In stage V Wilms tumor (5% of patients), bilateral involvement by tumor is present at diagnosis. The current paradigm treats all patients with bilateral Wilms tumor the same for the first 6 or 12 weeks. After definitive surgery, the treatment is based on the highest stage of the remaining kidneys and the posttreatment pathology.[
Treatment of Wilms Tumor
Treatment option overview for Wilms tumor
Because of the relative rarity of Wilms tumor, all patients with this tumor should consider joining a clinical trial. Treatment planning by a multidisciplinary team of pediatric cancer specialists (surgeon and/or urologist, radiation oncologist, and oncologist) who have experience treating children with Wilms tumor is necessary to determine and implement optimal treatment.
COG and SIOP approaches to Wilms tumor treatment
Most randomized clinical studies for treatment of children with Wilms tumor have been conducted by two large clinical groups (COG RTC and SIOP). Differences between the two groups affect staging and classification. There are two standard approaches to Wilms tumor treatment: the COG RTC uses immediate surgery for all unilateral tumors, and the SIOP uses preoperative chemotherapy as the first step in treatment. Both groups use postoperative chemotherapy, except for selected cases who do not receive chemotherapy, and in advanced stages, radiation therapy is used in a risk-adapted approach.
- COG RTC (includes the previous NWTS group): The NWTS group established standard treatment for Wilms tumor in North America, consisting of initial nephrectomy (when feasible) followed by chemotherapy and, in some patients, radiation therapy.[
219 ,220 ,221 ] This approach allows for early and accurate histological diagnosis, collection of biological materials unaltered by therapy, and staging information, such as the presence of tumor spill or tumor involvement in lymph nodes, before chemotherapy is administered. - SIOP: SIOP is a European consortium whose trials provide preoperative chemotherapy before definitive resection for patients with renal tumors. This results in fewer tumor spills during surgery and lower postoperative stage.[
222 ] When the histological features of Wilms tumors from patients who underwent immediate surgery were compared with the histological features of patients who received preoperative chemotherapy, preoperative chemotherapy was shown to significantly alter the histology, with fewer blastemal and mixed histology types in the tumors. Additionally, there were fewer stage III tumors in the preoperative chemotherapy group.[223 ] - Both SIOP and COG treat infants younger than 6 months with a primary nephrectomy.[
224 ]
This summary focuses on the NWTS (now COG RTC) results and studies.
The major treatment and study conclusions of NWTS-1, NWTS-2, NWTS-3, NWTS-4, and NWTS-5 are as follows:
- Routine, postoperative radiation therapy of the flank is not necessary for children with stage I tumors or stage II tumors with FH when postnephrectomy combination chemotherapy consisting of vincristine and dactinomycin is administered.[
221 ] - The prognosis for patients with stage III FH is best when treatment includes either (a) dactinomycin, vincristine, doxorubicin, and 10.8 Gy of radiation therapy to the flank; or (b) dactinomycin, vincristine, and 20 Gy of radiation therapy to the flank. Whole-abdominal radiation is indicated for extensive intraperitoneal disease or widespread intraperitoneal tumor spill with possible boost to gross residual disease.[
221 ] - The addition of cyclophosphamide at the protocol dose (10 mg/kg/d for 3 days every 6 weeks) to the combination of vincristine, dactinomycin, and doxorubicin does not improve prognosis for patients with stage IV FH tumors.[
221 ] - A single dose of dactinomycin per course (stages I–II FH, stage I anaplastic histology, stage III FH, stages III–IV, or stages I–IV clear cell sarcoma of the kidney) is equivalent to the divided-dose courses, results in the same EFS, achieves greater dose intensity, and is associated with less toxicity and expense.[
225 ] - Eighteen weeks of therapy is adequate for patients with stage I and stage II FH, and stage III and IV patients can be treated with 6 months of therapy instead of 15 months.[
180 ,219 ,225 ,226 ,227 ] - Gain of 1q is associated with inferior survival in patients with unilateral FH Wilms tumor. It is the single most powerful predictor of outcome, and in the presence of 1q gain, neither 1p nor 16q loss is significant. In the absence of 1q gain in unilateral FH Wilms tumor, 1p and/or 16q loss retain some prognostic significance and are associated with a higher risk of recurrence.[
107 ,110 ]
Surgery
The following operative principles have also evolved from NWTS (COG) trials:
- The most important role for the surgeon is to ensure complete tumor removal without rupture and assess the extent of disease. Radical nephrectomy and lymph node sampling via a transabdominal or thoracoabdominal incision is the procedure of choice.[
228 ] A flank incision is not performed because it provides limited exposure to the kidney.For patients with resectable tumors, preoperative biopsy or intraoperative biopsy is not performed because either would upstage the tumor in the current COG staging system.[
228 ] - Routine exploration of the contralateral kidney is not necessary if technically adequate imaging studies do not suggest a bilateral process. If the initial imaging studies suggest bilateral kidney involvement, treatment approaches should facilitate renal-sparing surgery.[
164 ] - About 2% of Wilms tumor cases have ureteral involvement. The presence of gross hematuria, nonfunctioning kidney, or hydronephrosis suggests the tumor may extend into the ureter, and cystoscopy is recommended. En bloc resection to avoid tumor spill is recommended.[
229 ] - The surgeon needs to be aware of the risk of intraoperative spill, especially in patients who have right-sided and large tumors, as noted in a review of cases of intraoperative spill among 1,131 patients registered on COG study AREN03B2 (NCT00898365).[
230 ] - Even if stage IV disease (e.g., pulmonary metastases) is evident on imaging, resection of the renal tumor should be considered. Treatment of local stage I or II Wilms tumor in the setting of distant metastasis does not require local radiation therapy.
Renal-sparing surgery remains controversial and is not supported by the data, except for children with the following:[
- A solitary kidney.
- Predisposition to bilateral tumors. Some children who are predisposed to bilateral tumors and who have very small tumors detected by screening ultrasonography may be considered for renal-sparing surgery to preserve renal tissue.[
231 ] - Horseshoe kidney. Wilms tumor arising in a horseshoe kidney is rare, and accurate preoperative diagnosis is important for planning the operative approach. Primary resection is possible in most cases. Inoperable cases can usually be resected after chemotherapy.[
234 ] - Wilms tumor in infants with Denys-Drash or Frasier syndrome (to delay the need for dialysis).
Renal-sparing surgery does not appear to be feasible for most patients at the time of diagnosis because of the location of the tumor within the kidney, even in patients with very low-risk tumors.[
Wilms tumor rarely invades adjacent organs; therefore, resection of contiguous organs is seldom indicated. There is an increased incidence of complications occurring in more extensive resections that involve removal of additional organs beyond the diaphragm and adrenal gland. This finding has led to the recommendation in current COG protocols that patients in whom nephrectomy will require removal of additional organs should be considered for initial biopsy, neoadjuvant chemotherapy, and then secondary resection.[
Lymph node sampling
Lymph node status is a major long-term predictor of outcome in patients with Wilms tumor.[
- The 5-year OS rate was lower for patients with 0 lymph nodes sampled (87% vs. 91% for 1–5 lymph nodes, 93% for 6–10 lymph nodes, 95% for >10 lymph nodes; P = .005). Multivariate analysis confirmed a survival advantage for patients having 1 to 5 lymph nodes sampled (HR, 0.6; P = .016), 6 to 10 lymph nodes sampled (HR, 0.521; P = .048), and >10 lymph nodes sampled (HR, 0.403; P = .039), compared with patients with 0 lymph nodes examined.[
240 ] - In NWTS-5, failure to sample lymph nodes was associated with higher risk of relapse in stage I and II patients.[
241 ] AREN0532 showed similar results with a poorer EFS observed for patients who did not have a lymph node biopsy. The EFS for stage III patients without lymph node sampling was 84% (n = 148), compared with 89% for those patients who had lymph node sampling (n = 387) (P = .067).[242 ] - Hilar and periaortic lymph node sampling is appropriate even if the nodes appear normal.[
228 ,240 ] Furthermore, any suspicious node basin is sampled. Margins of resection, residual tumor, and any suspicious node basins are marked with titanium clips.
Pleural effusions
The presence of a pleural effusion does not appear to necessitate a change in therapy. In a multi-institutional retrospective review of 1,259 children with newly diagnosed Wilms tumor, 94 (7.5%) had a pleural effusion.[
- Patients with a pleural effusion were older than those without a pleural effusion (median, 4.3 vs. 3.5 years; P = .004) and were more likely to present with advanced-stage disease (local stage III, 83% vs. 51.6%; P = .0001).
- Only 14 patients underwent a thoracentesis, 3 of whom had pulmonary parenchymal metastatic disease at the time of diagnosis.
- Thirty patients with a pleural effusion received chest radiation therapy as part of their therapy, 29 of whom had pulmonary or mediastinal metastases.
- Sixty-four patients with a pleural effusion did not receive chest radiation therapy, 59 of whom did not have associated pulmonary disease at the time of diagnosis.
- Only three patients with a pleural effusion and no history of pulmonary disease later experienced a relapse with thoracic disease.
- For the entire cohort of patients with pleural effusions, the EFS rate was 86.2%, and the OS rate was 91.5%.
Chemotherapy
Preoperative chemotherapy before nephrectomy is indicated in the following situations, which have been listed previously under situations requiring a biopsy:[
- Wilms tumor in a solitary kidney.
- Synchronous bilateral Wilms tumor.
- Extension of tumor thrombus in the inferior vena cava above the level of the hepatic veins. About 4% of Wilms tumor patients present with inferior vena cava or atrial involvement, and 11% of patients present with renal vein involvement. Embolization of a caval thrombus to the pulmonary artery is rare but can be lethal, and the presence of a thrombus must be identified preoperatively to prevent this occurrence and guide treatment.[
165 ,178 ] - Tumor involves contiguous structures whereby the only means of removing the kidney tumor requires removal of the other structures (e.g., spleen, pancreas, or colon but excluding the adrenal gland).
- Inoperable Wilms tumor.
- Pulmonary compromise resulting from extensive pulmonary metastases.
For more information, see the
A large contemporary series included 124 patients with Wilms tumor and intracaval extension who were treated at North American centers. Most patients (82%) received a three-drug neoadjuvant chemotherapy regimen. Neoadjuvant chemotherapy reduced the need for cardiopulmonary bypass and also avoided the complexity of intrahepatic caval thrombus resection.[
- The thrombus level regression rate was 45% overall, with suprahepatic level showing the best response (62%).
- Cardiopulmonary bypass was potentially avoided in 67% of patients.
- The perioperative complication rate was significantly lower after neoadjuvant chemotherapy (25%), compared with up-front surgery (55%; P = .005).
- Overall, the 2-year EFS rate was 93%, and the OS rate was 96%. The rates were higher for patients with FH tumors (98% for FH vs. 82% for unfavorable/anaplastic histology tumors).
- Incomplete resection and viable thrombus cells did not affect EFS or OS.
Preoperative chemotherapy follows a biopsy. The biopsy may be performed through a flank approach.[
In a meta-analysis that investigated the effect of neoadjuvant chemotherapy on thrombus viability for Wilms tumor where intravascular extension was defined as any Wilms tumor with extension beyond the renal vein. Neoadjuvant chemotherapy was found to be effective in achieving thrombus nonviability in around 50% of patients with tumor extension into the inferior vena cava. No added benefit was identified from extended cycles of neoadjuvant chemotherapy. Most patients received chemotherapy consisting of dactinomycin and vincristine with or without doxorubicin.[
In North America, the use of preoperative chemotherapy in patients with evidence of a contained preoperative rupture has been suggested to avoid intraoperative spill, but this is controversial.[
All infants younger than 12 months (including newborns) who will be treated with chemotherapy require a 50% reduction in chemotherapy dose compared with the dose given to older children.[
Liver function tests in children with Wilms tumor are monitored closely during the early course of therapy because severe hepatic toxic effects (including sinusoidal obstructive syndrome, which was previously called veno-occlusive disease) have been reported in these patients.[
Patients who develop renal failure while undergoing therapy can continue chemotherapy with vincristine, dactinomycin, and doxorubicin. Vincristine and doxorubicin can be given at full doses. However, dactinomycin is associated with severe neutropenia. Dose reductions for these agents may not be necessary, but accurate pharmacological and pharmacokinetic studies are needed while the patient is receiving therapy.[
Augmentation of therapy improves EFS for patients with FH Wilms tumor and loss of heterozygosity of 1p and 16q. In the AREN0532 (NCT00352534) and AREN0533 (NCT00379340) trials, patients with stage I and stage II FH Wilms tumor who were treated with the DD-4A regimen (dactinomycin, vincristine, and doxorubicin) demonstrated a 4-year EFS rate of 87.3%, compared with the 4-year EFS rate of 68.8% (P = .042) for stage I and stage II patients treated on the NWTS-5 trial. Patients with stage III and stage IV disease had a 4-year EFS rate of 90.2% when treated with regimen M (see
Postoperative radiation therapy to the tumor bed is required when a biopsy is performed or in the setting of local tumor stage III. In a study of 1,488 patients with Wilms tumors who underwent surgery and radiation therapy, delay in starting radiation therapy after surgery of greater than 14 days was associated with an increased risk of mortality for patients with nonmetastatic Wilms tumor.[
| Regimen Name | Regimen Description |
|---|---|
| Regimen EE-4A[ |
Vincristine, dactinomycin × 18 weeks postnephrectomy |
| Regimen DD-4A[ |
Vincristine, dactinomycin, doxorubicin × 24 weeks; baseline nephrectomy or biopsy with subsequent nephrectomy |
| Regimen I[ |
Vincristine, doxorubicin, cyclophosphamide, etoposide × 24 weeks postnephrectomy |
| Regimen M[ |
Vincristine, dactinomycin, doxorubicin, cyclophosphamide, and etoposide with subsequent radiation therapy |
| Regimen UH1[ |
Vincristine, doxorubicin, cyclophosphamide, carboplatin, and etoposide × 30 weeks + radiation therapy |
| Regimen UH2[ |
Vincristine, doxorubicin, cyclophosphamide, carboplatin, etoposide, vincristine, and irinotecan × 36 weeks + radiation therapy |
Radiation therapy
Radiation therapy is used to improve local control and treat sites of metastatic disease. Radiation therapy has historically been dependent on stage and histology, but more recently is also guided by the tumor molecular signature.[
COG approach
Up-front surgery provides histological confirmation and tumor extent, providing the rationale for adjuvant therapy, including radiation therapy. Besides histology, postoperative risk factors for worse local control include: (1) incomplete resection, (2) positive margins, and (3) nodal involvement. Radiation therapy is not used in patients with stage I or stage II FH Wilms tumor. For patients with FH stage III Wilms tumor, flank or abdominal radiation therapy is used for treatment. In cases of unfavorable histology (focal or diffuse anaplasia), flank or abdominal radiation therapy is indicated for all patients. For more information, see
Results of NWTS (COG RTC) trials have shown the following:
- Flank radiation therapy covers the tumor bed, involved nodal region, and entire adjacent vertebral bodies at 10.8 Gy in 1.8-Gy fractions. The dose of radiation therapy is based on the results of the NWTS-3 study in which there was no increase in abdominal relapse for stage III FH patients receiving 10 Gy versus 20 Gy with DD-4A chemotherapy.[
268 ] - Whole-abdominal radiation therapy is 10.5 Gy in 1.5-Gy fractions and is used to treat diffuse spill or peritoneal metastasis.[
264 ] - In the closed COG AREN0321 (NCT00335556) study, the radiation therapy dose to the tumor bed was 10.8 Gy in 1.8-Gy fractions, with the exception of patients with stage III diffuse anaplasia, where a dose of 19.8 Gy in 1.8-Gy fractions was used. This remains the current standard of treatment.[
267 ] - Results of the early NWTS studies (1 and 2) suggested that a radiation therapy delay of more than 10 days after surgery resulted in worse local control, particularly in unfavorable histology Wilms tumor.[
269 ,270 ] However, no difference in local control was found if radiation therapy was delayed more than 10 days after surgery for patients with stages II to IV FH tumors treated on the NWTS-3 or NWTS-4 trials.[103 ] More recent data from the National Cancer Database confirmed improved survival in patients with nonmetastatic Wilms tumor who received adjuvant radiation therapy less than or equal to 14 days postoperatively.[265 ] - Results from the NWTS-3 and NWTS-4 trials indicated that there was no survival benefit of whole-lung irradiation in the setting of lung metastases seen on CT scan only.[
271 ] Current COG guidelines allow for omission of whole-lung irradiation in cases of FH disease without extrapulmonary metastases, loss of heterozygosity at 1p and 16q, and complete response at 6 weeks after vincristine, dactinomycin, and doxorubicin.[185 ] When whole-lung irradiation is given, a dose of 12 Gy in 1.5-Gy fractions is indicated for children older than 12 months and 10.5 Gy in 1.5-Gy fractions for patients younger than 12 months with pulmonary metastasis. - Other sites of metastatic disease in Wilms tumor are uncommon and may include liver, extra-abdominal nodes, brain, and bone. In the COG AREN0533 (NCT00379340) study, the radiation therapy doses being used for patients younger than 16 years are 19.8 Gy in 1.8-Gy fractions to liver and gross residual nodes, 21.6 Gy in 1.8-Gy fractions to the whole brain with a boost of 10.8 Gy in 1.8-Gy fractions to gross metastatic disease in the brain, and 25.2 Gy in 14 fractions for bone metastasis. For patients older than 16 years, the radiation therapy dose to the whole brain and bone is increased to 30.6 Gy in 1.8-Gy fractions.
| Local/Locoregional Disease | ||||
|---|---|---|---|---|
| XRT = radiation therapy. | ||||
| a Requires whole-abdominal XRT in 1.5 Gy daily fractions. Patients with diffuse unresectable peritoneal implants receive 21 Gy. | ||||
| b Whole-lung irradiation is given in 1.5 Gy daily fractions. | ||||
| c Not all patients receive radiation therapy. | ||||
| d A boost is given for macroscopic disease. | ||||
| Stage I | Stage II | Stage III | Stage III (diffuse spill, peritoneal metastasis, preoperative rupture)a | |
| Favorable histology | No XRT | No XRT | 10.8 Gy | 10.5 Gy |
| Focal anaplasia | 10.8 Gy | 10.8 Gy | 10.8 Gy | 10.5 Gy |
| Diffuse anaplasia | 10.8 Gy | 10.8 Gy | 19.8 Gy | 10.5 Gy + 9 Gy boost |
| Metastatic Disease | ||||
| Stage IV Lung | Stage IV Liver | Stage IV Brain | Stage IV Bone | |
| Favorable histology | 10.5 Gy for age <12 monthsb,c; 12 Gy for age >12 monthsb,c | 19.8 Gy +/- 5.4 to 10.8 Gy boostd | 21.6 Gy + 10.8 Gy boost for age <16 years; 30.6 Gy for age >16 years | 25.2 Gy for age <16 years; 30.6 Gy for age >16 years |
| Focal or diffuse anaplasia | 10.5 Gy for age <12 monthsb; 12 Gy for age >12 monthsb | 19.8 Gy +/- 5.4 to 10.8 Gy boostd | 21.6 Gy + 10.8 Gy boost for age <16 years; 30.6 Gy for age >16 years | 25.2 Gy for age <16 years; 30.6 Gy for age >16 years |
SIOP approach
Based on the experience of previous SIOP trials, children who need radiation therapy undergo postoperative treatment to the flank and/or metastatic sites. The SIOP 1 to 9 trials demonstrated that preoperative radiation therapy or preoperative chemotherapy decreased the proportion of patients who developed tumor spillage, from more than 20% to 5%. The noninferiority of preoperative chemotherapy to preoperative radiation therapy in the SIOP 5 trial, and the concern over secondary malignancies with preoperative radiation therapy, led SIOP to recommend preoperative chemotherapy as the standard initial treatment.[
Abdominal radiation therapy has been omitted for patients with metastatic, local stage III Wilms tumor who had complete necrosis after 6 weeks of preoperative chemotherapy. It was also omitted in patients with stage III Wilms tumor who received 4 weeks of preoperative chemotherapy (n = 19) and had complete necrosis. The outcomes were excellent for both groups of patients with stage III Wilms tumor who had complete necrosis. The 5-year EFS and OS rates were 100% for patients with stage III disease and 95% for patients with metastatic local stage III disease.[
Treatment of stage I Wilms tumor
| Histology | 4-Year RFS or EFS | 4-Year OS | Treatmentb |
|---|---|---|---|
| DA = diffuse anaplastic; EFS = event-free survival; FA = focal anaplastic; FH = favorable histology; LOH = loss of heterozygosity; OS = overall survival; RFS = relapse-free survival; XRT = radiation therapy. | |||
| a Source: Grundy et al.,[ |
|||
| b For chemotherapy regimen descriptions, see |
|||
| c One patient with a pulmonary relapse 4.12 years after diagnosis. | |||
| FH <24 mo/tumor weight <550g | 90% | 100% | Surgery, including lymph node biopsy only |
| FH >24 mo/tumor weight >550g | 94% RFS | 98% | Nephrectomy + lymph node sampling followed by regimen EE-4A |
| FH with LOH 1p/16q (n = 8) | 100% EFS | 100% | Nephrectomy + lymph node sampling followed by regimen DD-4A |
| FA | 100% | 100% (n = 8) | Nephrectomy + lymph node sampling followed by regimen DD-4A and XRT |
| DA | 100%c | 100% (n = 10) | Nephrectomy + lymph node sampling followed by regimen DD-4A and XRT |
Evidence (surgery only for children younger than 2 years at diagnosis with stage I FH tumor that weighed <550 g):
In the AREN0532 (NCT00352534) trial, the COG validated the findings from the NWTS-5 trial that nephrectomy only is appropriate therapy for patients younger than 2 years at diagnosis with stage I FH Wilms tumor that weighed less than 550 g.
- The AREN0532 (NCT00352534) trial was designed to confirm the findings from NWTS-5 that adjuvant chemotherapy could be omitted for children younger than 2 years at diagnosis with stage I FH Wilms tumor that weighed less than 550 g. A total of 116 patients met the criteria for very low-risk Wilms tumor and were enrolled on the study.[
184 ,185 ,275 ]- Twelve patients relapsed.
- The estimated 4-year EFS rate was 89.7%, and the OS rate was 100%.
- 11p15 methylation status was associated with relapse (20% relapse with loss of heterozygosity, 25% relapse with loss of imprinting, and 3.3% relapse with retention of the normal imprinting [P = .011]).
- The risk of developing metachronous Wilms tumor is very low in patients with very low-risk Wilms tumor who lack evidence of an underlying syndrome.
Evidence (treatment of stage I epithelial-predominant FH Wilms tumor):
- The COG reported the outcomes for patients of all ages with stage I FH Wilms tumors showing epithelial-predominant histology. Approximately 20% of stage I FH Wilms tumors registered on AREN03B2 were epithelial predominant. In this group of 177 patients with stage I epithelial-predominant FH Wilms tumors, 117 patients were treated with EE-4A, and 57 patients were classified as having a very low-risk Wilms tumor and were treated with observation only.[
201 ][Level of evidence C1]- The 4-year EFS rate was 96.2%, and the OS rate was 100%.
- There was no statistical difference in EFS and OS based on age at diagnosis (<48 months and >48 months) or treatment (EE-4A vs. observation only).
- There were six events: three patients developed contralateral tumors after their initial diagnosis, and two of these patients had received adjuvant chemotherapy for their initial tumors. Three patients developed metastatic disease, and all of these patients had previously received EE-4A as their primary therapy.
Evidence (treatment of anaplastic stage I Wilms tumor):
- The AREN0321 (NCT00335556) study demonstrated that outcomes for patients with stage I anaplastic Wilms tumor were improved with the addition of doxorubicin and flank radiation therapy to vincristine/dactinomycin therapy.[
274 ]- The 4-year EFS and OS rate estimates were 100% in AREN0321, compared with 70% and 81.5%, respectively, in an updated analysis of 27 patients from NWTS-5 (median follow-up, 13.3 years). One patient with diffuse anaplasia relapsed 4.12 years after diagnosis on the AREN0321 trial.
- The addition of doxorubicin and radiation therapy to AREN0321 was based on the pattern of relapse observed in stage I anaplastic Wilms tumor in the abdomen and distant sites in the NWTS-5 trial.
- Retrospective analysis of all patients with stage I anaplastic Wilms tumor treated on NWTS-1 through NWTS-5 and AREN0321 showed a significant improvement in EFS for patients treated with doxorubicin (4-year EFS rate, 97.2% vs. 77.5%; P = .01), but no difference in EFS according to flank radiation therapy was shown (4-year EFS rate, 91.7% vs. 80.2%; P = .15).
- The rate of local recurrence was low (3.6%) and appeared to be similar for patients who received flank radiation therapy (4%) and patients who did not receive flank radiation therapy (6.2%). Local relapse occurred only in patients with diffuse anaplasia.
Current Clinical Trials
Use our
Treatment of stage II Wilms tumor
| Histology | 4-Year RFS or EFS | 4-Year OS | Treatmentb |
|---|---|---|---|
| DA = diffuse anaplastic; EFS = event-free survival; FA = focal anaplastic; FH = favorable histology; LOH = loss of heterozygosity; OS = overall survival; RFS = relapse-free survival; XRT = radiation therapy. | |||
| a Source: Grundy et al.,[ |
|||
| b For chemotherapy regimen descriptions, see |
|||
| FH | 86% RFS | 98% | Nephrectomy + lymph node sampling followed by regimen EE-4A |
| FH LOH 1p/16q (n = 24) | 83% EFS | 100% | Nephrectomy + lymph node sampling followed by regimen DD-4A |
| FA | 100% EFS | 100% (n = 2) | Nephrectomy + lymph node sampling followed by abdominal XRT and regimen DD-4A |
| DA | 84% EFS | 84% (n = 19) | Nephrectomy + lymph node sampling followed by abdominal XRT and regimen UH1 |
Treatment of stage II patients with intraoperative spill
In a review of 499 patients from the NWTS-4 trial with stage II FH Wilms tumor, 95 of the patients experienced tumor spill. The 8-year RFS and OS rates for patients who experienced intraoperative tumor spill and were treated with vincristine and dactinomycin without flank radiation therapy were lower (75.7% and 90.3%, respectively) than the rates for those who did not experience tumor spill (85% and 95.6%, respectively). None of these differences achieved statistical significance.[
On the NWTS-3, NWTS-4, and NWTS-5 trials, patients with intraoperative spill were divided into two groups: (1) those with diffuse spillage involving the whole abdominal cavity; and (2) those with local spillage confined to the flank. Patients with diffuse spillage were treated with radiation therapy to the entire abdomen and three-drug chemotherapy (vincristine, dactinomycin, and doxorubicin), whereas patients with local spillage were treated with vincristine and dactinomycin only. Based on an analysis of patients treated on NWTS-3 and NWTS-4 indicating that patients with stage II disease and local spillage had inferior OS compared with patients with stage II disease without local spillage, COG studies treat patients with local spillage with doxorubicin and flank radiation.[
Current Clinical Trials
Use our
Treatment of stage III Wilms tumor
For information about patients classified as stage III purely based on local spill, see the
| Histology | 4-Year RFS or EFS | 4-Year OS | Treatmentb |
|---|---|---|---|
| DA = diffuse anaplastic; EFS = event-free survival; FA = focal anaplastic; FH = favorable histology; LOH = loss of heterozygosity; OS = overall survival; RFS = relapse-free survival; XRT = radiation therapy. | |||
| a Source: Grundy et al.,[ |
|||
| b For chemotherapy regimen descriptions, see |
|||
| FH (all patients) | 88% EFS | 97% | Nephrectomy + lymph node sampling followed by abdominal XRT and regimen DD-4A |
| FH (without LOH of 1p and/or 16q) and positive lymph nodes (n = 109) | 82% EFS | 97% | Nephrectomy + lymph node sampling followed by abdominal XRT and regimen DD-4A |
| FH (without LOH of 1p and/or 16q) and negative lymph nodes (n = 169) | 97% EFS | 99% | Nephrectomy + lymph node sampling followed by abdominal XRT and regimen DD-4A |
| FH (with LOH of 1p and 16q) (n = 31) | 87% EFS | 94% | Nephrectomy + lymph node sampling followed by abdominal XRT and regimen M |
| FH (with LOH of 1p and 16q) and negative lymph nodes (n = 12) | 92% | 92% | Nephrectomy + lymph node sampling followed by abdominal XRT and regimen M |
| FH (with LOH of 1p or 16q) and negative lymph nodes (n = 13) | 85% | 92% | Nephrectomy + lymph node sampling followed by abdominal XRT and regimen M |
| FH (with LOH of 1p or 16q) and negative lymph nodes (n = 68) | 87% | 97% | Nephrectomy + lymph node sampling followed by abdominal XRT and regimen DD-4A |
| FH (with LOH of 1p or 16q) and positive lymph nodes (n = 48) | 74% | 92% | Nephrectomy + lymph node sampling followed by abdominal XRT and regimen DD-4A |
| FA | 70% RFS | 79% (n = 10) | Nephrectomy + lymph node sampling followed by abdominal XRT and regimen DD-4A |
| FA (preoperative treatment) | 71% RFS | 71% (n = 7) | Preoperative treatment with regimen DD-4A followed by nephrectomy + lymph node sampling and abdominal XRT |
| DA | 46% EFS | 53% (n = 16) | Preoperative treatment with regimen I followed by nephrectomy + lymph node sampling and abdominal XRT |
| DA | 82% EFS | 91% (n = 23) | Immediate nephrectomy + lymph node sampling followed by abdominal XRT and regimen UH1 |
Radiation therapy
Early initiation of radiation therapy is a critical component of multimodal therapy for patients with nonmetastatic Wilms tumor. In a review of 1,488 patients with Wilms tumor who underwent surgery and radiation therapy, a surgery-to-radiation therapy interval of greater than 14 days was associated with an increased risk of mortality (HR, 2.13; P = .013). This underscores the importance of initiating radiation therapy within 14 days of surgery, which is specified in Wilms tumor treatment protocols.[
Loss of heterozygosity of 1p or 16q
Loss of heterozygosity of 1p or 16q was shown to influence EFS but not OS in 635 patients with stage III FH Wilms tumor enrolled in the COG AREN0532 or AREN03B2 protocols. When combined, a negative lymph node status (related to histology) and a negative loss of heterozygosity status (related to the primary tumor) was a strong predictor of excellent EFS and OS.[
- Patients who had lymph nodes sampled during surgery had improved EFS (4-year EFS rate, 90.3%) relative to patients without lymph nodes sampled (4-year EFS rate, 80.0%; P = .0037). No difference was seen in OS whether lymph nodes were sampled (4-year OS rates, 97.0% for patients with lymph nodes sampled vs. 94.9% for those without lymph nodes sampled; P = .078).
- Patients with positive lymph nodes at nephrectomy had worse EFS than those with negative lymph nodes (83.5% vs. 94.2% at 4-years; HR, 2.78; P = .00017). A borderline significant effect was observed for OS (4-year OS rates, 95.1% vs. 98.2%; HR, 2.50; P = .054).
- An analysis of patients with stage III Wilms tumor according to lymph node status (positive or negative) and singular loss of heterozygosity of 1p or 16q status (positive for one or negative for both) is shown in
Table 7 .Table 7. An analysis of Patients with Stage III Wilms Tumor According to Lymph Node Status and Singular LOH 1p or 16q Status Lymph Node Status LOH 1p or 16q 4-Year EFS Ratea HRb - = negative; + = positive; EFS = event-free survival; HR = hazard ratio; LOH = loss of heterozygosity. a Compared with patients both negative for lymph node and singular LOH, a significant difference in EFS across the groups was observed (log-rankP< .0001). b Overall survival did not reach statistical significance between groups. - - 96.2% - (n = 212) - +1p or 16q 89.5% 3.04 (n = 72) + +1p 73.9% 6.33 (n = 37) + +16q 79.3% + - 86.0% 3.57 (n = 104)
Loss of heterozygosity of 1p and 16q
Therapy was augmented for patients with loss of heterozygosity of 1p and 16q who were enrolled in the AREN0533 (NCT00379340) trial. Patients with stage III and stage IV Wilms tumor with loss of heterozygosity were treated with regimen M. The 4-year EFS rate was 90.2%, and the OS rate was 96.1%, compared with a 4-year EFS rate of 61.3% (P = .001) and a 4-year OS rate of 86.0% (P = .087) for patients in the NWTS-5 trial. The study suggested an improvement in survival, but it was not powered to detect differences in survival.[
Current Clinical Trials
Use our
Treatment of stage IV Wilms tumor
| Histology | 4-Year RFS or EFS | 4-Year OS | Treatmentb |
|---|---|---|---|
| CR = complete response; DA = diffuse anaplasia; EFS = event-free survival; FA = focal anaplasia; FH = favorable histology; LOH = loss of heterozygosity; OS = overall survival; RFS = relapse-free survival; XRT = radiation therapy. | |||
| a Source: Grundy et al.,[ |
|||
| b For chemotherapy regimen descriptions, see |
|||
| c Abdominal XRT is planned according to local stage of renal tumor. | |||
| d Pulmonary XRT is reserved for patients with chest x-ray/chest computed tomography evidence of pulmonary metastases. | |||
| e For more information, see theAREN0533 (NCT00379340)andAREN0321 (NCT00335556)studies. | |||
| FH (with isolated lung nodules) | 85% EFS | 96% | Nephrectomy + lymph node sampling, followed by abdominal XRT,c +/- bilateral pulmonary XRT,d and regimen DD-4A or regimen Me |
| FH (no LOH of 1p and 16q) with isolated lung nodules with CR to DD-4A | 80% EFS | 96% | Nephrectomy + lymph node sampling, followed by abdominal XRTc and regimen DD-4A |
| FH (no LOH of 1p and 16q) with isolated lung nodules with incomplete response to DD-4A | 99% EFS | 95% | Nephrectomy + lymph node sampling, followed by abdominal XRTc and bilateral pulmonary XRTd and regimen M |
| FH (LOH of 1p and 16q) with isolated lung nodules (n = 18) | 100% | 100% | Nephrectomy + lymph node sampling followed by abdominal XRTc and bilateral pulmonary XRTd and regimen M |
| FH (with LOH of 1p and/or 16q) (n = 20) | 95% EFS | 100% | Nephrectomy + lymph node sampling, abdominal XRTc radiation to sites of metastases, and regimen M |
| FH with extrapulmonary metastases, with or without lung metastases | 76% EFS | 89% | Nephrectomy + lymph node sampling followed by abdominal XRTc, regimen M, and local control of other metastatic sites; if lung metastases are present, bilateral pulmonary XRTd |
| FA | 80% EFS | 80% (n = 5) | Nephrectomy + lymph node sampling, followed by abdominal XRT,c radiation to sites of metastases, bilateral pulmonary XRT,d and regimen UH1/revised UH1 (AREN0321, consisting of vincristine, doxorubicin, cyclophosphamide, carboplatin, and etoposide) |
| DA | 33% EFS | 33% (n = 10) | Immediate nephrectomy + lymph node sampling followed by abdominal XRT,c radiation to sites of metastases, whole-lung XRT,d and regimen I |
| DA (preoperative treatment) | 60% EFS | 70% (n = 10) | Preoperative treatment with regimen UH2 followed by nephrectomy + lymph node sampling, followed by abdominal XRT,c radiation to sites of metastases, and whole-lung XRTd |
Stage IV disease is defined by the presence of hematogenous metastases to the lung, liver, bone, brain, or other sites, with the lung being the most common site. The presence of liver metastases at diagnosis is not an independent adverse prognostic factor in patients with stage IV Wilms tumor.[
Chromosome 1q gain
In the AREN0533 (NCT00379340) trial, 30% of patients with stage IV pulmonary disease had 1q gain. These patients trended toward worse EFS, regardless of lung response and whether they received regimen DD-4A or M.[
Treatment of pulmonary nodules and metastases
Historically, chest x-rays were used to detect pulmonary metastases. The introduction of CT created controversy because many patients had lung nodules detected by chest CT scans that were not seen on chest x-rays. Management of newly diagnosed patients with FH Wilms tumor who have lung nodules detected only by CT scans (with negative chest x-ray) has elicited controversy as to whether they need to be treated with additional intensive treatment that is accompanied by acute and late toxicities.
Evidence (treatment of pulmonary nodules detected by chest CT scan only):
- A retrospective review of 186 patients from NWTS-4 and NWTS-5 with CT-only–detected lung nodules reported on the use of doxorubicin, vincristine, and dactinomycin versus the use of two drugs.[
280 ]- Patients who received doxorubicin, vincristine, and dactinomycin with or without lung irradiation had a 5-year EFS rate of 80% versus an EFS rate of 56% for patients receiving only two drugs (P = .004).
- There was no difference in EFS according to whether the lung was irradiated.
- There was no difference in the 5-year OS rate (87% vs. 86%).
Retrospective studies from Europe have examined the impact of omitting pulmonary radiation in patients with pulmonary metastases diagnosed by chest x-ray. European investigators omitted radiation from the treatment of most patients with Wilms tumor and pulmonary metastases as identified on chest x-ray who were treated on the SIOP-93-01 (NCT00003804) trial. The European approach to renal tumors differs from the approach used in North America. All patients who were shown to have a renal tumor by imaging underwent 9 weeks of prenephrectomy chemotherapy consisting of vincristine, dactinomycin, and doxorubicin.
Evidence (omission of pulmonary irradiation):
- In a retrospective SIOP study, 234 newly diagnosed patients with Wilms tumor presenting with pulmonary metastases were treated according to the response of the pulmonary metastases to the prenephrectomy chemotherapy.[
281 ]- Patients who were in complete remission (67%) after 6 weeks of therapy continued with the same chemotherapy and did not require radiation to their lungs.
- The 5-year EFS rate was 77%, and the OS rate was 88%.
- Patients who had residual pulmonary metastases were evaluated for metastasectomy.
- Thirty-seven patients (17%) obtained complete remission with surgery, and their outcome was similar to that of the group of patients who were treated with chemotherapy. Tumor viability in the resected pulmonary metastases was not a factor for omitting radiation therapy.
- The 5-year EFS rate was 84%, and the OS rate was 92%.
- Patients with residual pulmonary metastases that were incompletely resected or inoperable received more aggressive chemotherapy consisting of ifosfamide/anthracycline alternating with carboplatin/etoposide for 9 weeks.
- Patients showing a complete remission at that time were spared pulmonary radiation and continued with chemotherapy, whereas patients with residual pulmonary metastases continued with additional chemotherapy (to complete 34 weeks) and pulmonary irradiation. The 5-year OS rate was 48%, compared with the OS rates for patients who responded to chemotherapy alone (88%) and those who underwent metastasectomy (92%) (P < .001).
- Patients with high-risk histologies, such as anaplastic Wilms tumor, were treated with more aggressive chemotherapy but had a poorer outcome, compared with that of patients with nonanaplastic histologies (5-year OS rate, 33% vs. 87%; P < .001).
- Patients who were in complete remission (67%) after 6 weeks of therapy continued with the same chemotherapy and did not require radiation to their lungs.
- Based on the European experience, the COG AREN0533 (NCT00379340) study applied a new strategy for patients with FH Wilms tumor and isolated lung metastases to improve EFS while reducing exposure to lung irradiation. Therapy was adjusted based on lung nodule response and tumor-specific loss of heterozygosity at 1p and 16q.[
266 ][Level of evidence C2]- Of the 292 patients enrolled in the study, 133 patients (42%) showed a complete lung nodule response after 6 weeks of DD-4A (vincristine, dactinomycin, doxorubicin) and continued receiving the same chemotherapy without lung radiation therapy. The 4-year EFS rate was 80%, and OS rate was 96%.
- Patients who had an incomplete lung nodule response (n = 145) or loss of heterozygosity at 1p/16q (n = 18) received lung radiation therapy and four cycles of cyclophosphamide/etoposide in addition to the DD-4A drugs (regimen M). The 4-year EFS rate was 89%, and the OS rate was 95% for the incomplete lung nodule response group without loss of heterozygosity. Of the patients with pulmonary metastases only and loss of heterozygosity, the 4-year EFS and OS rates were 100%.
- In a post hoc analysis of 1q gain in 212 patients enrolled in AREN0533 who had DNA available, patients with lung nodule complete remission with 1q gain had a significantly worse 4-year EFS rate (57% vs. 86%, P = .001) and trend toward inferior OS rates (89% vs. 97%). Relapses were predominantly pulmonary. There was no difference in outcome for patients with incomplete lung nodule response based on 1q gain.
- Regimen M has a higher potential for late effects (increased risk of secondary leukemias and risk of infertility related to a cumulative dose of cyclophosphamide of 8.8 g/m2).
- COG showed that initial lung radiation therapy could be avoided in approximately 40% of patients. OS was excellent; however, there was a trend toward more events than expected (expected, 15% and observed, 20%; P = .052).
Although fewer patients were spared pulmonary radiation when treated in the COG trial than in the European trials, it is important to note several differences between the studies and why the studies cannot be directly compared.[
Liver metastases at diagnosis
The liver is an infrequent site of metastases at diagnosis for patients with stage IV FH Wilms tumor, but it is the most common site after the lung. In 634 patients with stage IV FH Wilms tumor from the NWTS-4 and -5 studies, 96 (15%) presented with liver involvement.[
The impact of liver metastases at diagnosis on patient care management has not been studied in a prospective manner. Most reported experiences come from single institutions and/or retrospective studies that do not include more modern surgical approaches or contemporary risk stratification parameters. In aggregate, there are not sufficient data to support liver metastases as an unfavorable site for metastatic disease, although there were some early SIOP studies that conflicted with this finding.[
NWTS
A retrospective analysis of 742 patients with stage IV Wilms tumor who were treated on the NWTS-4 and -5 trials examined the outcomes of patients with and without lung metastases alone. The study also investigated survival outcomes for these patients according to whether they underwent a resection of their hepatic tumors.[
- Twenty-two patients underwent primary liver resections (18 wedge resections and 4 lobectomies). After chemotherapy and/or radiation therapy, 13 patients underwent liver resections (7 wedge resections, 5 lobectomies, and 1 trisegmentectomy).
- Seventy-one patients (67%) did not undergo surgery for their liver disease. In 14 patients, the liver disease responded completely to chemotherapy alone.
- Eighty-two patients received abdominal radiation therapy.
- The EFS rate was 75% for patients with metastatic FH Wilms tumor. For patients with lung-only metastases (n = 513), the EFS rate was 76%. For patients with liver metastases without lung metastases (n = 34), the EFS rate was 76%. The EFS rate was 70% (n = 62) for patients with both liver and lung metastases, compared with 64% for patients (n = 25) with other sites of metastases. There were no significant differences between the stage IV groups of patients (P = .60).
- The EFS rate was 86% for patients who underwent primary resection of the liver metastases (n = 22), compared with 68% for patients who did not have a primary resection of the liver metastases (P = .09).
- No significant difference in EFS was observed for patients treated with chemotherapy alone compared with patients treated with chemotherapy and radiation therapy (P = .63). The EFS rates were 64% for patients who did not receive abdominal radiation therapy, 77% for patients who received abdominal radiation therapy without a boost, and 72% for patients who received abdominal radiation therapy with a boost (P = .05).
- Adult patients comprised part of the study population (<10% were older than 12 years and 3% were older than 16 years).
- Based on these data, the authors concluded that liver metastasis at diagnosis is not an independent adverse prognostic factor for patients with stage IV disease. They also suggested that there is no reason to perform hepatic resection in the up-front setting. These two studies occurred between the 1960s and 1990s. During this time, hepatic resection techniques and approaches changed based on a better understanding of the liver anatomy. Furthermore, contemporary risk stratification approaches that include tumor cytogenetics were not evaluated in these studies nor were more conventional radiation techniques such as IMRT, which has the potential to provide better protection to normal tissues.[
287 ]
SIOP
The SIOP/Gesellschaft für Pädiatrische Hämatologie und Onkologie (GPOH) group has recommended a more aggressive surgical approach. This recommendation was based on the reported outcomes of 29 patients treated on the SIOP93-01/GPOH and SIOP2001/GPOH trials that enrolled 1,365 patients between 1994 and 2004. Two of the patients had diffuse anaplasia.[
- The 5-year OS rate was 62.6% for patients with hepatic metastases, whereas the OS rate was 76.3% for all patients with stage IV disease. Within the group of patients with liver metastases, 14 did not undergo liver resections. Four of the patients (13%) responded to chemotherapy and were still alive at the time of the publication.
- Fifteen patients underwent liver surgery (11 primary liver resections, 6 complete resections, and 5 incomplete resections). All six patients who had a complete resection survived.
- As a result of these findings, the authors recommended an aggressive initial surgical approach for patients with liver disease. However, it is important to remember that SIOP patients receive preoperative chemotherapy before nephrectomy and/or resection of metastatic lesions.
The French cohort enrolled on the SIOP2001 trial included 131 patients with stage IV FH Wilms tumor. Of these patients, 18 (14%) had liver metastases at diagnosis, including 4 (3%) with isolated liver metastases.[
- The 5-year EFS rate was 83%, and the OS rate was 88% for this cohort of patients with liver metastases.
- The authors concluded that liver involvement does not appear to be an adverse prognostic marker in patients with metastatic Wilms tumors, although there was a small number of patients in this study.
Treatment of stage IV diffuse anaplastic Wilms tumor
In the AREN0321 (NCT00335556) study, the combination of vincristine and irinotecan (VI) was tested in an up-front window for patients with diffuse anaplastic Wilms tumor and measurable disease.[
- Fourteen patients with stage IV diffuse anaplastic Wilms tumor with measurable disease received the window therapy; one patient achieved a complete response (CR), ten patients achieved partial responses (PRs), and no patients had stable disease. This resulted in a CR and PR rate of 79%.
- Patients who responded to VI in the window therapy had VI incorporated into their regimen (UH2).
- Because of the observed cardiac/pulmonary toxicities encountered in this trial, the study was interrupted and amended with reduced doses of doxorubicin, cyclophosphamide, and etoposide (when combined with carboplatin).
- Further study of the modified regimen is planned in patients with newly diagnosed diffuse anaplastic Wilms tumor.
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- AREN1921 (NCT04322318) (A Study of Combination Chemotherapy for Patients With Newly Diagnosed Diffuse Anaplastic Wilms Tumors and Relapsed Favorable-Histology Wilms Tumors): This clinical trial aims to improve survival by intensifying treatment for patients with diffuse anaplastic Wilms tumors. This trial will expand on the overall improvement observed with the revised UH1/UH2 regimen from the AREN0321 trial, which was modified along with the addition of vincristine/irinotecan.
Current Clinical Trials
Use our
Treatment of stage V Wilms tumor and those predisposed to developing bilateral Wilms tumor
Currently, there is not a standard approach for the treatment of
Management of a child with bilateral Wilms tumor is very challenging. The goals of therapy are to eradicate all tumor and to preserve as much normal renal tissue as possible, with the hope of decreasing the risk of chronic renal failure among these children.[
Historically, based on the NWTS-4 and NWTS-5 trials and trials performed in Europe, patients with bilateral Wilms tumor have had a lower EFS and OS than have patients with localized Wilms tumor. The NWTS-4 study reported that the 8-year EFS rate for patients with bilateral FH Wilms tumor was 74%, and the OS rate was 89%; for patients with anaplastic histology, the EFS rate was 40%, and the OS rate was 45%.[
Treatment options for stage V (bilateral) Wilms tumor include the following:
-
Preoperative chemotherapy and resection . -
Renal transplant .
Preoperative chemotherapy and resection
For patients with bilateral Wilms tumor, the goal of therapy is to preserve as much renal tissue as possible without compromising overall outcome. This approach is used to avoid the late effect of end-stage renal disease, which can be caused by underlying germline genetic aberrations and treatment-related loss of functional renal tissue. End-stage renal disease occurs more frequently in patients with bilateral Wilms tumor (12% nonsyndromic) than in patients with unilateral Wilms tumor (<1%). Functional renal outcome is considerably better after bilateral nephron-sparing surgery than after other types of surgery.[
Traditionally, patients have undergone bilateral renal biopsies, with staging of each kidney followed by preoperative chemotherapy. In the first prospective multi-institutional treatment trial (COG AREN0534 [NCT00945009]), pretreatment biopsies were not required if results of imaging tests were consistent with Wilms tumor.[
For patients who are treated with preoperative chemotherapy, the tumor pathology needs to be evaluated after 4 to 8 weeks. For patients not treated in a clinical trial, the ideal time to perform a biopsy or resection is unknown because minimal shrinkage may reflect chemotherapy-induced differentiation or anaplastic histology. A planned attempt at resection or biopsy of apparently unresectable tumor is undertaken no later than 12 weeks from diagnosis. Continuing therapy without evaluating tumor pathology in a patient with bilateral Wilms tumor may miss anaplastic histology or chemotherapy-induced differentiation (including rhabdomyomatous differentiation) and thus increase toxicity for the patient without providing additional benefit for tumor control. Anaplastic histology occurs in 10% of patients with bilateral Wilms tumor, and these tumors respond poorly to chemotherapy.[
Once the diagnosis is confirmed, a complete resection is performed. Histological confirmation of the diagnosis is not straightforward. In a series of 27 patients from the NWTS-4 study, discordant pathology (unilateral anaplastic tumor) was seen in 20 cases (74%), which highlights the need to obtain tissue from both kidneys. Seven children who were later diagnosed with diffuse anaplastic tumors had core biopsies performed to establish the diagnosis; however, anaplasia was not found. Anaplasia was identified in only three of the nine patients when an open-wedge biopsy was performed and in seven of nine patients who had a partial or complete nephrectomy.[
The decision to administer chemotherapy and/or radiation therapy after biopsy or a second-look operation is dependent on the tumor's response to initial therapy. More aggressive therapy is required for patients with inadequate response to initial therapy observed at the second procedure or in the setting of anaplasia.[
End-stage renal disease is the most clinically significant morbidity in patients with bilateral Wilms tumor and can be caused by underlying germline genetic aberrations, as well as treatment-related loss of functional renal tissue. Long-term monitoring of renal function is required after treatment for bilateral disease.
Evidence (preoperative chemotherapy and resection for bilateral Wilms tumor):
- The first prospective study in bilateral Wilms tumor (AREN0534 [NCT00945009]) aimed to improve EFS and OS while preserving renal tissue by intensifying preoperative chemotherapy (using three drugs—vincristine, dactinomycin, and doxorubicin), completing definitive surgery by 12 weeks from diagnosis, and modifying postoperative chemotherapy based on histological response.[
176 ]; [292 ][Level of evidence C2]- For the arm that treated children with bilateral Wilms tumor, results showed that central review of imaging, surgical resection within 12 weeks of diagnosis, and response-based and histology-based postoperative therapy improved EFS and OS, when compared with the historical outcomes of children with bilateral Wilms tumor.
- For the 180 patients with bilateral Wilms tumor, the 4-year EFS rate was 81% (95% confidence interval [CI], 74%–87%), and the OS rate was 95% (95% CI, 91%–99%). The above-described approach with risk assignment for treatment based on both staging and postoperative histopathology yielded excellent outcomes for patients with bilateral Wilms tumors but did not improve outcome for patients with diffuse anaplasia. Seven patients who had completely necrotic tumors had a 4-year EFS rate of 100%. Of 118 patients who had tumors with intermediate-risk histopathology, the 4-year EFS rate was 82% and the OS rate was 97%. Fourteen patients with blastemal-type tumors had a 4-year EFS rate of 79% and an OS rate of 93%. Patients who had complete necrosis were assigned to the low-risk category and those with blastemal-type histopathology to the high-risk category for subsequent treatments. For 18 patients who had diffuse anaplasia, the 4-year EFS rate was 61% and the OS rate was 72%. For seven patients who had focal anaplasia, the EFS rate was 71% and the OS rate was 100%.[
292 ][Level of evidence C2] Because biopsy was not performed before treatment in this series, some of the patients enrolled may have had only nephrogenic rests and not a true Wilms tumor. This finding may have improved these survival figures over historical controls. - Participants in the AREN0534 trial were followed for up to 10 years. The overall 8-year EFS rate was 75% (95% CI, 69%–83%), with a median follow-up of 8.6 years. The highest risk of an EFS event was immediately after diagnosis, with a declining rate over the following 2 years. Over 25% of first recurrences occurred more than 2 years after diagnosis. These late events tended to occur in year 3, with a smaller number of events after year 6. The risk of late recurrences was higher in female patients, patients with anaplastic histology tumors, and patients with negative lymph nodes or margins. Distant relapses in the lungs, liver, or abdomen tended to occur early, while relapses after 2 years were usually in kidney tissue (80% of recurrences). Based on this study, the investigators suggested that observation of patients with bilateral Wilms tumor continue beyond 4 years after diagnosis.[
293 ] - One of the aims of the study was that 75% of patients undergo definitive surgery by 12 weeks. After induction chemotherapy, 163 of 189 patients (84%) underwent definitive surgical treatment in at least one kidney by 12 weeks, and 39% of patients retained parts of both kidneys.
- Chemotherapy after surgery was tailored according to histological response. The 4-year EFS rate was 84.1% for FH tumors, 58.2% for anaplastic histology tumors, and 82% for blastemal-type tumors.
- Because of the higher risk of renal failure in patients with bilateral Wilms tumor than in patients with unilateral Wilms tumor, one of the goals of the study was that 50% of the patients undergo bilateral nephron-sparing surgery. This threshold was not met, with only 39% of patients successfully treated with bilateral nephron-sparing surgery.
- Based on the above study, the recommendation was to continue with three-drug preoperative chemotherapy for 6 to 12 weeks followed by nephron-sparing surgery whenever possible. After resection, postoperative therapy is based on the histology of the resected specimen. The disappointing use of nephron-sparing surgery in this study may have been because of the level of experience of the surgeons in this multi-institutional study.
- Outcomes of patients with anaplasia and bilateral Wilms tumor were examined in the AREN0534 study. Twenty-seven patients were enrolled (17 had diffuse anaplasia and 10 had focal anaplasia). Twenty-six patients had bilateral disease. One patient presented with unilateral disease (within the patient's solitary kidney).[
294 ]- Twenty-one of the 26 patients with bilateral disease had discordant pathology. This suggests that if a biopsy is needed, both kidneys should be sampled.
- The 4-year and 8-year EFS rates were both 53% for patients with diffuse anaplasia. The 4-year EFS rate was 80%, and the 8-year EFS rate was 70% for patients with focal anaplasia.
- The EFS and OS did not differ by margin status.
- All children who died experienced prior relapse or progression within 18 months of study enrollment.
- In a retrospective review of 49 patients with Wilms tumor who received preoperative therapy according to the SIOP-93-01 (NCT00003804) guidelines, the timing of surgery was determined when there was no longer imaging evidence of tumor regression. The mean treatment duration was 80 days before renal-sparing surgery.[
295 ]- The 5-year EFS rate was 83.4%, and the OS rate was 89.5%.
- All but one of the patients had renal-sparing surgery in at least one kidney.
- Despite the good survival, 14% of the patients developed end-stage renal disease.
- In a retrospective review from St. Jude Children's Research Hospital, investigators described their experience with preoperative chemotherapy followed by renal-sparing procedures in children with bilateral FH Wilms tumor.[
296 ]- In one series, 39 of 42 patients with bilateral FH Wilms tumor underwent successful bilateral renal-sparing procedures after receiving preoperative chemotherapy. Three patients underwent unilateral nephrectomy with contralateral nephron-sparing surgery. Three patients required early (within 4 months) repeat nephron-sparing surgery for residual tumor. In the long term, seven patients had local tumor recurrence, and three patients had intestinal obstruction.
- The OS rate was 86% (mean follow-up, 4.1 years). Of the six patients who died, five had diffuse anaplastic histology.
- All of the patients had an estimated glomerular filtration rate of more than 60 mL/min/1.73m2 at the last follow-up; none of the patients developed end-stage renal disease.
- The authors concluded that bilateral renal-sparing surgery is almost always feasible and can be done safely with good oncologic outcomes in patients with synchronous, bilateral Wilms tumor. It should be considered even if preoperative imaging studies suggest that the lesions are unresectable. Sparing of renal parenchyma is likely to help preserve renal function in children who are at significant risk of chronic renal insufficiency. Careful long-term follow-up is required to fully assess the potential progression of renal dysfunction.
- A follow-up review of these patients revealed the following: 8 of 36 patients underwent repeat nephron-sparing surgery, and an additional two patients required a third nephron-sparing surgery. Six of these patients were alive without disease at the 4.5-year follow-up. The two patients who died had blastemal-predominant histology.[
297 ]
For information about recurrent disease, see the
Treatment of patients with multicentric or bilaterally predisposed unilateral Wilms tumors
Based on an identified subpopulation of patients with Wilms tumor who are at risk for metachronous disease, coupled with an increased risk of end-stage renal disease, the COG conducted the largest prospective study (AREN0534 [NCT00945009]) of these patients. The goal of this study was to preserve renal tissue while maintaining excellent overall outcomes.[
Patients were identified by the treating institution as having a predisposition syndrome. Induction chemotherapy was determined by the presence of localized or metastatic disease found on imaging (and histology if a biopsy had been performed) at the time of diagnosis. Surgery, including renal-sparing surgery, was based on the radiographic response at 6 or 12 weeks, and additional chemotherapy was determined by histology. Patients with favorable histology and stage III or IV disease or any patient with anaplasia received radiation therapy.[
- Thirty-four patients were enrolled with the following underlying conditions: Beckwith-Wiedemann syndrome (n = 9), hemihypertrophy (n = 9), multicentric tumors (n = 10), WAGR syndrome (n = 2), solitary kidney (n = 2), Denys-Drash syndrome (n = 1), and Simpson-Golabi-Behmel syndrome (n = 1).
- The 4-year EFS and OS rates were 94% and 100%, respectively, with a median follow-up of 4.49 years. Two patients relapsed (one in the tumor bed and one in the abdomen); none of the deaths occurred during induction.
- Prenephrectomy chemotherapy facilitated renal preservation in 22 of 34 patients (65%). Eleven partial nephrectomies were performed after two cycles of chemotherapy, and nine partial nephrectomies were performed after four cycles of chemotherapy. Two of the tumors completely resolved after treatment with chemotherapy and required no subsequent surgery.
- There were 22 patients with a known predisposition syndrome for which routine ultrasonography screening would have been expected. Sixteen of these patients had stage I disease, three had stage II disease, and three had stage III disease. Thirteen of the tumors were detected through routine ultrasonography.
- These results suggest that a standardized treatment approach that includes preoperative chemotherapy, surgical resection within 6 to 12 weeks, and histology-based postoperative chemotherapy results in excellent EFS, OS, and preservation of renal parenchyma.
WAGR syndrome and Wilms tumor outcomes
The comprehensive International WAGR Syndrome Association survey was used to analyze tumor characteristics, treatment, congenital risk factors, and kidney function in children with WAGR syndrome and Wilms tumors. Of 145 children with WAGR syndrome, 64 developed Wilms tumors (44%).[
- Of patients with Wilms tumors, three had relapsed disease and one died. Most patients presented with low-stage disease (stages I–II) and favorable histology.
- Chronic kidney disease developed in five children with WAGR syndrome and no Wilms tumors (6.2%) and in 34 children with WAGR syndrome and Wilms tumors (53.1%). Two children with Wilms tumors required hemodialysis, and one child underwent a kidney transplant.
- Compared with total nephrectomy, nephron-sparing surgery appeared protective against chronic kidney disease, but it was only used in 37.5% of cases.
- By univariate analysis, chronic kidney disease at any stage was associated with complete nephrectomy (P < .0001), chemotherapy duration longer than 12 months, and three-drug therapy. By multivariate analysis, prior nephrectomy was the only significant variable (P = .0002).
Renal transplant
Renal transplant for children with stage V Wilms tumor is usually delayed until 1 to 2 years have passed without evidence of malignancy because most relapses occur within 2 years of diagnosis.[
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the
Current Clinical Trials
Use our
Treatment and outcomes of recurrent Wilms tumor
Patients with recurrent Wilms tumor should consider enrolling in phase I and phase II clinical trials. Other treatment options for recurrent Wilms tumor are discussed below.
Palliative care remains a central focus of management regardless of whether disease-directed therapy is pursued at the time of progression. This ensures that quality of life is maximized while attempting to reduce symptoms and stress related to the terminal illness.
Prognosis, prognostic factors, and risk categories for recurrent Wilms tumor
Approximately 15% of patients with FH Wilms tumor and 50% of patients with anaplastic histology Wilms tumor experience recurrence.[
About 95% of first Wilms tumor recurrences occur within 2 years of initial diagnosis. Relapse more than 5 years after diagnosis is considered a late recurrence and is rare. In the largest retrospective study of more than 1,300 children enrolled in various Wilms tumor trials, the median time to late recurrence after first recurrence was 13 years (range, 5–17 years).[
A number of potential prognostic features influencing postrecurrence outcomes have been analyzed, but it is difficult to determine whether these factors are independent of each other. Also, the following prognostic factors appear to be changing as therapy for primary and recurrent Wilms tumor evolves:
- Anaplastic histology.[
306 ] - Advanced tumor stage.[
306 ] - Sex. Sex was predictive of outcome, with males faring worse than females.[
301 ,307 ]
The NWTS-5 trial showed that time to recurrence and site of recurrence are no longer prognostically significant.[
Based on these results, the following three risk categories have been identified:
- Standard risk: Patients with FH Wilms tumor who relapse after therapy with only vincristine and/or dactinomycin. These patients account for approximately 30% of recurrences and are expected to have EFS rates of 70% to 80%.[
302 ] - High risk: Patients with FH Wilms tumor who relapse after therapy with three or more agents. These patients account for 45% to 50% of children with Wilms tumor who relapse and have survival rates in the 40% to 50% range.[
302 ] - Very high risk: Patients with recurrent anaplastic or blastemal-predominant Wilms tumor. These patients account for 10% to 15% of all Wilms tumor relapses and are expected to have survival rates in the 10% range.[
183 ,302 ]
Treatment of standard-risk relapsed Wilms tumor
In children who had small stage I Wilms tumor and were treated with surgery alone, the EFS rate was 84%. All but one child who relapsed was salvaged with treatment tailored to the site of recurrence.[
Successful retreatment can be accomplished for Wilms tumor patients whose initial therapy consisted of immediate nephrectomy followed by chemotherapy with vincristine and dactinomycin and who relapse.
Treatment options for standard-risk relapsed Wilms tumor include the following:
-
Surgery, radiation therapy, and chemotherapy .
Surgery, radiation therapy, and chemotherapy
Evidence (surgery, radiation therapy, and chemotherapy):
- Fifty-eight patients were treated on the NWTS-5 relapse protocol with surgical excision when feasible, radiation therapy, and courses of vincristine, doxorubicin, and cyclophosphamide alternating with etoposide and cyclophosphamide.[
307 ]- The 4-year EFS rate after relapse was 71%, and the OS rate was 82%.
- For patients whose site of relapse was only the lungs, the 4-year EFS rate was 68%, and the OS rate was 81%.
- The SIOP Renal Tumor Study Group (SIOP-RTSG) analyzed the outcomes of low- and intermediate-risk patients (n = 109) who relapsed after treatment with vincristine and dactinomycin for primary Wilms tumor from their SIOP 93-01 and SIOP 2001 studies.[
309 ][Level of evidence: C1]- The postrelapse 5-year EFS rate was 72.3%, and the OS rate was 79.3%.
- Patients treated with vincristine, dactinomycin, and doxorubicin (VAD) did not fare worse than patients treated with more intensive therapies (such as cyclophosphamide, carboplatin, etoposide, and doxorubicin or ifosfamide, carboplatin, and etoposide [ICE] backbones; HR for EFS, 0.611; HR for OS, 0.438).
- There was no survival difference between patients treated with VAD compared with ICE-based regimens.
- The type of alkylating agent (ifosfamide or cyclophosphamide) used in more intensive treatment regimens did not affect survival rates.
- Incorporation of high-dose hematopoietic stem cell transplant (HSCT) to consolidate relapse treatment did not improve the outcome for standard-risk relapse patients.
- Future studies may be able to identify patients who may benefit from VAD treatment rather than more intensive treatment with combination chemotherapy including cyclophosphamide, carboplatin, etoposide, and doxorubicin.
Treatment of high-risk and very high-risk relapsed Wilms tumor
Treatment options for high-risk and very high-risk relapsed Wilms tumor include the following:
-
Chemotherapy, surgery, and/or radiation therapy . -
HSCT .
Chemotherapy, surgery, and/or radiation therapy
Evidence (chemotherapy, surgery, and/or radiation therapy):
- Approximately 50% of unilateral Wilms tumor patients who relapse or progress after initial treatment with vincristine, dactinomycin, and doxorubicin and radiation therapy can be successfully re-treated. Sixty patients with unilateral Wilms tumor were treated on the NWTS-5 relapse protocol with alternating courses of cyclophosphamide/etoposide and carboplatin/etoposide, surgery, and radiation therapy.[
301 ][Level of evidence B4]- The 4-year EFS rate for patients with high-risk Wilms tumor was 42%, and the OS rate was 48%.
- High-risk patients who relapsed in the lungs only had a 4-year EFS rate of 49% and an OS rate of 53%.
Patients with stage II, stage III, and stage IV anaplastic tumors at diagnosis have a very poor prognosis upon recurrence.[
HSCT
High-dose chemotherapy followed by autologous HSCT has been used for recurrent high-risk patients.[
Evidence (HSCT):
- Investigators used the European Blood and Marrow Transplantation Registry to examine the outcomes of children with Wilms tumor (n = 69) who received high-dose chemotherapy with autologous HSCT as consolidation during first or second remission. Different high-dose chemotherapy regimens were used, containing either melphalan (n = 34) or thiotepa (n = 14).[
315 ][Level of evidence C1]- The 5-year OS and EFS probabilities were 0.67 (± 0.06) and 0.63 (± 0.06), respectively (median observation time, 7.8 years).
- Children who underwent transplant in first remission had 5-year EFS and OS probabilities of 0.69 and 0.72, respectively.
- The use of melphalan alone for high-dose chemotherapy led to noninferior survival rates, compared with other drugs or drug combinations, and to better engraftment, compared with thiotepa-containing regimens.
- Similar outcome results were reported in a series of 54 patients with Wilms tumor who received high-dose chemotherapy with autologous stem cell rescue (first-line therapy, n = 13; recurrence therapy, n = 41).[
314 ][Level of evidence C1]- For patients treated at the time of recurrence, the 5-year EFS and OS rates were 57% and 69%, respectively (median follow-up, 7 years).
- The outcomes of 253 patients with relapsed Wilms tumor who received high-dose chemotherapy followed by autologous HSCT between 1990 and 2013 were reported to and reviewed by the Center for International Blood and Marrow Transplantation Research.[
316 ]- The 5-year estimate for EFS was 36%, and the 5-year estimate for OS was 45%.
- Relapse of primary disease was the cause of death in 81% of the population.
No randomized trials comparing chemotherapy and transplant have been reported, and case series suffer from selection bias.
Patients in whom such salvage attempts fail should be offered treatment on phase I or phase II clinical trials.
Treatment options under clinical evaluation for recurrent Wilms tumor
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- AREN1921 (NCT04322318) (A Study of Combination Chemotherapy for Patients With Newly Diagnosed Diffuse Anaplastic Wilms Tumor and Relapsed Favorable-Histology [FH] Wilms Tumor): Patients with standard-risk relapsed FH Wilms tumor (pretreated with two drugs) will receive chemotherapy consisting of vincristine/doxorubicin/cyclophosphamide alternating with cyclophosphamide/carboplatin/etoposide alternating with vincristine/irinotecan. Patients with high-risk relapsed FH Wilms tumor (pretreated with three drugs) and very high-risk relapsed FH Wilms tumor (pretreated with ≥ four drugs) will be treated with cyclophosphamide and topotecan added to a backbone of ifosfamide, carboplatin, and etoposide.
Follow-up after treatment
For patients who have completed therapy for Wilms tumor and exhibit features consistent with genetic predisposition, such as bilateral Wilms tumor, screening involves renal ultrasonography examination every 3 months for metachronous tumors during the risk period for that particular syndrome (5 years for WT1-related syndromes; 8 years for Beckwith-Wiedemann syndrome).
Late effects after Wilms tumor therapy
Children treated for Wilms tumor are at increased risk of developing the following:
- Premature mortality after Wilms tumor diagnosis. Long-term morbidity and mortality among unilateral, nonsyndromic Wilms tumor survivors were evaluated in the Childhood Cancer Survivor Study (CCSS). Among 2,008 survivors, 142 deaths occurred (standardized mortality ratio [SMR], 2.9 [95% CI, 2.5–3.5]; 35-year cumulative incidence of death, 7.8% [95% CI, 6.3%–9.2%]). The most frequent causes of death were subsequent malignant neoplasms (SMNs) (n = 42), Wilms tumor relapse (n = 30), and cardiac related (n = 9). Survivors who were treated with vincristine and dactinomycin (VA) alone had comparable risk for all-cause mortality (SMR,1.0) and health-related late mortality (SMR, 1.5) relative to the general population.[
317 ] - Chronic health conditions. Findings from the same CCSS study are as follows:[
317 ]- Of unilateral Wilms tumor survivors (n = 2,008), the 35-year cumulative incidence of any grades 3 to 5 chronic health condition was 34.1% among Wilms tumor survivors and 14.8% among siblings.
- Survivors treated with VA alone had a modestly increased risk of developing grades 3 to 5 chronic health conditions compared with siblings (RR, 1.5).
- Risks of grades 3 to 5 chronic health conditions, including intestinal obstruction (8.1%), kidney failure (2.4%), premature ovarian insufficiency (7.3%) and heart failure (4.0%), increased by treatment intensity in a dose-dependent manner and by exposure to doxorubicin and/or radiation therapy. This risk was most pronounced among survivors with high-risk disease, including those with early relapse (<5 years).
- Compared with siblings, survivors treated with VA showed a higher risk of intestinal obstruction (RR, 9.4) and kidney failure (RR, 11.9) but the magnitude of the risk was lower than that for more intensive treatment groups.
- Both whole-lung and whole-abdomen radiation therapy were associated with increased risks of SMNs, heart failure, and intestinal obstruction, and the magnitude of risk increased with rising doses.
- Whole-abdomen or flank radiation therapy higher than 20 Gy was associated with an increased risk of intestinal obstruction compared with no radiation therapy, whereas radiation therapy doses of 20 Gy or less were not associated with any chronic health condition.
- SMNs.[
318 ,319 ] In the CCSS study of Wilms tumor survivors (n = 2,008), 82 developed SMNs (standardized incidence ratio [SIR], 4.1), representing a cumulative incidence rate of 6.1%. Breast (SIR, 6.9), thyroid (SIR, 4.7) and intestinal/colorectal (SIR, 12.0) were the most frequently reported cancers.[317 ] The cumulative incidence of invasive breast cancer in Wilms tumor survivors who had received pulmonary radiation for metastatic Wilms tumor is nearly 15% by age 40 years.[320 ] - Congestive heart failure. The risk of congestive heart failure is influenced by dose of doxorubicin received, radiation to the heart, and female sex.[
321 ] CCSS investigators reported that cumulative doxorubicin doses of 250 mg/m2 or higher were associated with a nearly 5-fold rate of development of heart failure, compared with no doxorubicin exposure.[317 ] - Infertility for patients who received whole-abdomen irradiation and/or cyclophosphamide.[
322 ,323 ] The 35-year cumulative incidence rate of premature ovarian insufficiency was 7.3%. Whole-abdomen radiation therapy was associated with an increased risk of premature ovarian insufficiency, with a greater risk with increasing dose (≤20 Gy: RR, 13.1; >20 Gy: RR, 36.5).[317 ] Female survivors of Wilms tumor diagnosed before age 40 years from a St. Jude Lifetime Cohort Study experienced increased premature ovarian insufficiency compared with controls (9.3% vs. 0.6%; P < .01), likely because of abdominal irradiation that exposed the ovaries. None of the females exposed to hemiabdominal radiation therapy experienced premature ovarian insufficiency.[319 ] - Complications of pregnancy.[
324 ] One study examined the effects of radiation therapy to the abdomen (partial or whole abdomen) on pregnancy and pregnancy outcomes in patients with a history of Wilms tumor. The investigators reviewed hypertension complicating pregnancy, early or threatened labor, malposition of the fetus, premature rupture of membranes, obstructed labor, abnormality of forces of labor, and umbilical cord complications. There were 1,021 pregnancies (with a duration of 20 weeks or longer), including 955 live births, from which there were 700 sets of maternal and offspring medical records. Female survivors of Wilms tumor who received radiation therapy are at an increased risk of hypertension complicating pregnancy, early or threatened labor, and fetal malposition. The offspring of these patients were more likely to be premature and have low birth weights.[324 ] For more information, see theReproduction section in Late Effects of Treatment for Childhood Cancer. - Late kidney failure. Based on the CCSS study, the cumulative incidence of late kidney failure among survivors was 2.4% (approximately a 10-fold increased rate relative to siblings). No single chemotherapy or radiation therapy–related exposure was associated with kidney failure, which suggests that nephrectomy may be the primary risk factor.[
317 ] Rates of late kidney failure increase with advancing age. This finding highlights the importance of long-term monitoring of kidney function in these patients, particularly when the patients have other comorbidities. The cumulative incidence of end-stage renal disease caused by chronic renal failure at 20 years from diagnosis of Wilms tumor is 3.1% for patients with bilateral Wilms tumor.[213 ] - Neurocognitive impairment. Among 264 long-term Wilms tumor survivors participating in a St. Jude Lifetime Cohort Study who completed neurocognitive testing, 19.7% had grades 2 to 3 executive function impairment, compared with 12% of controls (P < .01). In survivors, processing speed (19.7% vs. 8.4%) and memory (20.9% vs. 9.9%) showed a higher prevalence of moderate to severe impairments, compared with controls (P < .01). Only impairments in processing speed were associated with the presence of grades 2 to 4 cardiovascular disease.[
319 ] - Physical performance. In the CCSS study, survivors were more likely to report poor physical functioning (OR, 2.9) than siblings.[
317 ] In a St. Jude Lifetime Cohort Study, 270 long-term Wilms tumor survivors completed function testing and were observed to have excess impairments in aerobic function, mobility, strength, endurance and flexibility, compared with controls. There were no clear associations between these outcomes and treatment exposures.[319 ]
Late renal effects in patients with Wilms and underlying genetic abnormalities include the following:
- Children with WAGR syndrome or other germline WT1 pathogenic variants are monitored throughout their lives because they are at increased risk of developing hypertension, nephropathy, and renal failure.[
213 ] - Patients with Wilms tumor and aniridia without genitourinary abnormalities are at lower risk but are monitored for nephropathy or renal failure.[
325 ] - Children with Wilms tumor and any genitourinary anomalies are also at increased risk of late renal failure and are monitored. Features associated with germline WT1 pathogenic variants that increase the risk of developing renal failure include the following:[
213 ]- Stromal predominant histology.
- Bilateral disease.
- Intralobar nephrogenic rests.
- Wilms tumor diagnosed before age 2 years.
For a full discussion of the late effects of cancer treatment in children and adolescents, see
References:
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute.
Available online . Last accessed February 25, 2025. - Howlader N, Noone AM, Krapcho M, et al.: SEER Cancer Statistics Review (CSR) 1975-2016. Bethesda, Md: National Cancer Institute, 2019.
Available online . Last accessed August 8, 2022. - Breslow N, Olshan A, Beckwith JB, et al.: Ethnic variation in the incidence, diagnosis, prognosis, and follow-up of children with Wilms' tumor. J Natl Cancer Inst 86 (1): 49-51, 1994.
- Breslow N, Olshan A, Beckwith JB, et al.: Epidemiology of Wilms tumor. Med Pediatr Oncol 21 (3): 172-81, 1993.
- Horner MJ, Ries LA, Krapcho M, et al.: SEER Cancer Statistics Review, 1975-2006. National Cancer Institute, 2009.
Also available online . Last accessed August 21, 2023. - Scott RH, Stiller CA, Walker L, et al.: Syndromes and constitutional chromosomal abnormalities associated with Wilms tumour. J Med Genet 43 (9): 705-15, 2006.
- Narod SA, Hawkins MM, Robertson CM, et al.: Congenital anomalies and childhood cancer in Great Britain. Am J Hum Genet 60 (3): 474-85, 1997.
- Vujanić GM, Apps JR, Moroz V, et al.: Nephrogenic rests in Wilms tumors treated with preoperative chemotherapy: The UK SIOP Wilms Tumor 2001 Trial experience. Pediatr Blood Cancer 64 (11): , 2017.
- Dumoucel S, Gauthier-Villars M, Stoppa-Lyonnet D, et al.: Malformations, genetic abnormalities, and Wilms tumor. Pediatr Blood Cancer 61 (1): 140-4, 2014.
- Gracia Bouthelier R, Lapunzina P: Follow-up and risk of tumors in overgrowth syndromes. J Pediatr Endocrinol Metab 18 (Suppl 1): 1227-35, 2005.
- Lapunzina P: Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am J Med Genet C Semin Med Genet 137 (1): 53-71, 2005.
- Treger TD, Chowdhury T, Pritchard-Jones K, et al.: The genetic changes of Wilms tumour. Nat Rev Nephrol 15 (4): 240-251, 2019.
- Duffy KA, Trout KL, Gunckle JM, et al.: Results From the WAGR Syndrome Patient Registry: Characterization of WAGR Spectrum and Recommendations for Care Management. Front Pediatr 9: 733018, 2021.
- Clericuzio CL: Clinical phenotypes and Wilms tumor. Med Pediatr Oncol 21 (3): 182-7, 1993.
- Fischbach BV, Trout KL, Lewis J, et al.: WAGR syndrome: a clinical review of 54 cases. Pediatrics 116 (4): 984-8, 2005.
- Breslow NE, Norris R, Norkool PA, et al.: Characteristics and outcomes of children with the Wilms tumor-Aniridia syndrome: a report from the National Wilms Tumor Study Group. J Clin Oncol 21 (24): 4579-85, 2003.
- Hol JA, Jongmans MCJ, Sudour-Bonnange H, et al.: Clinical characteristics and outcomes of children with WAGR syndrome and Wilms tumor and/or nephroblastomatosis: The 30-year SIOP-RTSG experience. Cancer 127 (4): 628-638, 2021.
- Tracy ET, Leraas H, Olson L, et al.: Wilms tumor characteristics, surgical management, outcomes, and chronic kidney disease in children with WAGR syndrome: A report from the International WAGR Syndrome Association survey. Pediatr Blood Cancer 71 (9): e31172, 2024.
- Barbosa AS, Hadjiathanasiou CG, Theodoridis C, et al.: The same mutation affecting the splicing of WT1 gene is present on Frasier syndrome patients with or without Wilms' tumor. Hum Mutat 13 (2): 146-53, 1999.
- Koziell AB, Grundy R, Barratt TM, et al.: Evidence for the genetic heterogeneity of nephropathic phenotypes associated with Denys-Drash and Frasier syndromes. Am J Hum Genet 64 (6): 1778-81, 1999.
- Royer-Pokora B, Beier M, Henzler M, et al.: Twenty-four new cases of WT1 germline mutations and review of the literature: genotype/phenotype correlations for Wilms tumor development. Am J Med Genet A 127 (3): 249-57, 2004.
- Pelletier J, Bruening W, Kashtan CE, et al.: Germline mutations in the Wilms' tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 67 (2): 437-47, 1991.
- Mueller RF: The Denys-Drash syndrome. J Med Genet 31 (6): 471-7, 1994.
- Barbaux S, Niaudet P, Gubler MC, et al.: Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet 17 (4): 467-70, 1997.
- Connolly GK, Harris RD, Shumate C, et al.: Pediatric cancer incidence among individuals with overgrowth syndromes and overgrowth features: A population-based assessment in seven million children. Cancer 130 (3): 467-475, 2024.
- Porteus MH, Narkool P, Neuberg D, et al.: Characteristics and outcome of children with Beckwith-Wiedemann syndrome and Wilms' tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol 18 (10): 2026-31, 2000.
- Rump P, Zeegers MP, van Essen AJ: Tumor risk in Beckwith-Wiedemann syndrome: A review and meta-analysis. Am J Med Genet A 136 (1): 95-104, 2005.
- Choufani S, Shuman C, Weksberg R: Molecular findings in Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 163C (2): 131-40, 2013.
- Eggermann T, Algar E, Lapunzina P, et al.: Clinical utility gene card for: Beckwith-Wiedemann Syndrome. Eur J Hum Genet 22 (3): , 2014.
- Ibrahim A, Kirby G, Hardy C, et al.: Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clin Epigenetics 6 (1): 11, 2014.
- Brioude F, Lacoste A, Netchine I, et al.: Beckwith-Wiedemann syndrome: growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Horm Res Paediatr 80 (6): 457-65, 2013.
- Mussa A, Russo S, Larizza L, et al.: (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome: a paradigm for genomic medicine. Clin Genet 89 (4): 403-415, 2016.
- Green DM, Breslow NE, Beckwith JB, et al.: Screening of children with hemihypertrophy, aniridia, and Beckwith-Wiedemann syndrome in patients with Wilms tumor: a report from the National Wilms Tumor Study. Med Pediatr Oncol 21 (3): 188-92, 1993.
- DeBaun MR, Siegel MJ, Choyke PL: Nephromegaly in infancy and early childhood: a risk factor for Wilms tumor in Beckwith-Wiedemann syndrome. J Pediatr 132 (3 Pt 1): 401-4, 1998.
- DeBaun MR, Tucker MA: Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr 132 (3 Pt 1): 398-400, 1998.
- Hol JA, Kuiper RP, van Dijk F, et al.: Prevalence of (Epi)genetic Predisposing Factors in a 5-Year Unselected National Wilms Tumor Cohort: A Comprehensive Clinical and Genomic Characterization. J Clin Oncol 40 (17): 1892-1902, 2022.
- Milani D, Pezzani L, Tabano S, et al.: Beckwith-Wiedemann and IMAGe syndromes: two very different diseases caused by mutations on the same gene. Appl Clin Genet 7: 169-75, 2014.
- Morris MR, Astuti D, Maher ER: Perlman syndrome: overgrowth, Wilms tumor predisposition and DIS3L2. Am J Med Genet C Semin Med Genet 163C (2): 106-13, 2013.
- Astuti D, Morris MR, Cooper WN, et al.: Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet 44 (3): 277-84, 2012.
- Golabi M, Leung A, Lopez C: Simpson-Golabi-Behmel Syndrome Type 1. In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp.
Available online . Last accessed December 22, 2022. - Peterman CM, Fevurly RD, Alomari AI, et al.: Sonographic screening for Wilms tumor in children with CLOVES syndrome. Pediatr Blood Cancer 64 (12): , 2017.
- Fagali C, Kok F, Nicola P, et al.: MLPA analysis in 30 Sotos syndrome patients revealed one total NSD1 deletion and two partial deletions not previously reported. Eur J Med Genet 52 (5): 333-6, 2009 Sep-Oct.
- Isidor B, Bourdeaut F, Lafon D, et al.: Wilms' tumor in patients with 9q22.3 microdeletion syndrome suggests a role for PTCH1 in nephroblastomas. Eur J Hum Genet 21 (7): 784-7, 2013.
- Cayrol J, Nightingale M, Challis J, et al.: Wilms Tumor Associated With the 9q22.3 Microdeletion Syndrome: 2 New Case Reports and a Review of The Literature. J Pediatr Hematol Oncol 41 (8): e517-e520, 2019.
- Cairney AE, Andrews M, Greenberg M, et al.: Wilms tumor in three patients with Bloom syndrome. J Pediatr 111 (3): 414-6, 1987.
- Hartley AL, Birch JM, Tricker K, et al.: Wilms' tumor in the Li-Fraumeni cancer family syndrome. Cancer Genet Cytogenet 67 (2): 133-5, 1993.
- Bourdeaut F, Guiochon-Mantel A, Fabre M, et al.: Alagille syndrome and nephroblastoma: Unusual coincidence of two rare disorders. Pediatr Blood Cancer 50 (4): 908-11, 2008.
- Russell B, Johnston JJ, Biesecker LG, et al.: Clinical management of patients with ASXL1 mutations and Bohring-Opitz syndrome, emphasizing the need for Wilms tumor surveillance. Am J Med Genet A 167A (9): 2122-31, 2015.
- Bonaïti-Pellié C, Chompret A, Tournade MF, et al.: Genetics and epidemiology of Wilms' tumor: the French Wilms' tumor study. Med Pediatr Oncol 20 (4): 284-91, 1992.
- Winther JF, Sankila R, Boice JD, et al.: Cancer in siblings of children with cancer in the Nordic countries: a population-based cohort study. Lancet 358 (9283): 711-7, 2001.
- Breslow NE, Olson J, Moksness J, et al.: Familial Wilms' tumor: a descriptive study. Med Pediatr Oncol 27 (5): 398-403, 1996.
- Li FP, Williams WR, Gimbrere K, et al.: Heritable fraction of unilateral Wilms tumor. Pediatrics 81 (1): 147-9, 1988.
- Rahman N, Arbour L, Tonin P, et al.: Evidence for a familial Wilms' tumour gene (FWT1) on chromosome 17q12-q21. Nat Genet 13 (4): 461-3, 1996.
- McDonald JM, Douglass EC, Fisher R, et al.: Linkage of familial Wilms' tumor predisposition to chromosome 19 and a two-locus model for the etiology of familial tumors. Cancer Res 58 (7): 1387-90, 1998.
- Halliday BJ, Fukuzawa R, Markie DM, et al.: Germline mutations and somatic inactivation of TRIM28 in Wilms tumour. PLoS Genet 14 (6): e1007399, 2018.
- Grundy P, Koufos A, Morgan K, et al.: Familial predisposition to Wilms' tumour does not map to the short arm of chromosome 11. Nature 336 (6197): 374-6, 1988.
- Little SE, Hanks SP, King-Underwood L, et al.: Frequency and heritability of WT1 mutations in nonsyndromic Wilms' tumor patients: a UK Children's Cancer Study Group Study. J Clin Oncol 22 (20): 4140-6, 2004.
- Hanks S, Perdeaux ER, Seal S, et al.: Germline mutations in the PAF1 complex gene CTR9 predispose to Wilms tumour. Nat Commun 5: 4398, 2014.
- Scott RH, Douglas J, Baskcomb L, et al.: Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet 40 (11): 1329-34, 2008.
- Grønskov K, Olsen JH, Sand A, et al.: Population-based risk estimates of Wilms tumor in sporadic aniridia. A comprehensive mutation screening procedure of PAX6 identifies 80% of mutations in aniridia. Hum Genet 109 (1): 11-8, 2001.
- Clericuzio C, Hingorani M, Crolla JA, et al.: Clinical utility gene card for: WAGR syndrome. Eur J Hum Genet 19 (4): , 2011.
- Kalish JM, Biesecker LG, Brioude F, et al.: Nomenclature and definition in asymmetric regional body overgrowth. Am J Med Genet A 173 (7): 1735-1738, 2017.
- Shuman C, Smith AC, Steele L, et al.: Constitutional UPD for chromosome 11p15 in individuals with isolated hemihyperplasia is associated with high tumor risk and occurs following assisted reproductive technologies. Am J Med Genet A 140 (14): 1497-503, 2006.
- Shanske AL: Trisomy 18 in a second 20-year-old woman. Am J Med Genet A 140 (9): 966-7, 2006.
- Reid S, Renwick A, Seal S, et al.: Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumour. J Med Genet 42 (2): 147-51, 2005.
- Hirsch B, Shimamura A, Moreau L, et al.: Association of biallelic BRCA2/FANCD1 mutations with spontaneous chromosomal instability and solid tumors of childhood. Blood 103 (7): 2554-9, 2004.
- Reid S, Schindler D, Hanenberg H, et al.: Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet 39 (2): 162-4, 2007.
- Rios P, Bauer H, Schleiermacher G, et al.: Environmental exposures related to parental habits in the perinatal period and the risk of Wilms' tumor in children. Cancer Epidemiol 66: 101706, 2020.
- Coorens THH, Treger TD, Al-Saadi R, et al.: Embryonal precursors of Wilms tumor. Science 366 (6470): 1247-1251, 2019.
- Gadd S, Huff V, Walz AL, et al.: A Children's Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet 49 (10): 1487-1494, 2017.
- Wegert J, Wittmann S, Leuschner I, et al.: WTX inactivation is a frequent, but late event in Wilms tumors without apparent clinical impact. Genes Chromosomes Cancer 48 (12): 1102-11, 2009.
- Ruteshouser EC, Robinson SM, Huff V: Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer 47 (6): 461-70, 2008.
- Walz AL, Ooms A, Gadd S, et al.: Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 27 (2): 286-97, 2015.
- Wegert J, Ishaque N, Vardapour R, et al.: Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 27 (2): 298-311, 2015.
- Rakheja D, Chen KS, Liu Y, et al.: Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun 2: 4802, 2014.
- Torrezan GT, Ferreira EN, Nakahata AM, et al.: Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nat Commun 5: 4039, 2014.
- Dome JS, Huff V: Wilms Tumor Predisposition. In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp.
Available online . Last accessed August 16, 2022. - Mahamdallie SS, Hanks S, Karlin KL, et al.: Mutations in the transcriptional repressor REST predispose to Wilms tumor. Nat Genet 47 (12): 1471-4, 2015.
- Huff V: Wilms tumor genetics. Am J Med Genet 79 (4): 260-7, 1998.
- Scott RH, Murray A, Baskcomb L, et al.: Stratification of Wilms tumor by genetic and epigenetic analysis. Oncotarget 3 (3): 327-35, 2012.
- Corbin M, de Reyniès A, Rickman DS, et al.: WNT/beta-catenin pathway activation in Wilms tumors: a unifying mechanism with multiple entries? Genes Chromosomes Cancer 48 (9): 816-27, 2009.
- Maiti S, Alam R, Amos CI, et al.: Frequent association of beta-catenin and WT1 mutations in Wilms tumors. Cancer Res 60 (22): 6288-92, 2000.
- Gadd S, Huff V, Huang CC, et al.: Clinically relevant subsets identified by gene expression patterns support a revised ontogenic model of Wilms tumor: a Children's Oncology Group Study. Neoplasia 14 (8): 742-56, 2012.
- Breslow NE, Beckwith JB, Perlman EJ, et al.: Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr Blood Cancer 47 (3): 260-7, 2006.
- Fukuzawa R, Heathcott RW, More HE, et al.: Sequential WT1 and CTNNB1 mutations and alterations of beta-catenin localisation in intralobar nephrogenic rests and associated Wilms tumours: two case studies. J Clin Pathol 60 (9): 1013-6, 2007.
- Perlman EJ, Gadd S, Arold ST, et al.: MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology Wilms tumours. Nat Commun 6: 10013, 2015.
- Diller L, Ghahremani M, Morgan J, et al.: Constitutional WT1 mutations in Wilms' tumor patients. J Clin Oncol 16 (11): 3634-40, 1998.
- Perlman EJ, Grundy PE, Anderson JR, et al.: WT1 mutation and 11P15 loss of heterozygosity predict relapse in very low-risk wilms tumors treated with surgery alone: a children's oncology group study. J Clin Oncol 29 (6): 698-703, 2011.
- Lipska BS, Ranchin B, Iatropoulos P, et al.: Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int 85 (5): 1169-78, 2014.
- Lehnhardt A, Karnatz C, Ahlenstiel-Grunow T, et al.: Clinical and molecular characterization of patients with heterozygous mutations in wilms tumor suppressor gene 1. Clin J Am Soc Nephrol 10 (5): 825-31, 2015.
- Marakhonov AV, Vasilyeva TA, Voskresenskaya AA, et al.: LMO2 gene deletions significantly worsen the prognosis of Wilms' tumor development in patients with WAGR syndrome. Hum Mol Genet 28 (19): 3323-3326, 2019.
- Scott RH, Walker L, Olsen ØE, et al.: Surveillance for Wilms tumour in at-risk children: pragmatic recommendations for best practice. Arch Dis Child 91 (12): 995-9, 2006.
- Koesters R, Ridder R, Kopp-Schneider A, et al.: Mutational activation of the beta-catenin proto-oncogene is a common event in the development of Wilms' tumors. Cancer Res 59 (16): 3880-2, 1999.
- Koesters R, Niggli F, von Knebel Doeberitz M, et al.: Nuclear accumulation of beta-catenin protein in Wilms' tumours. J Pathol 199 (1): 68-76, 2003.
- Major MB, Camp ND, Berndt JD, et al.: Wilms tumor suppressor WTX negatively regulates WNT/beta-catenin signaling. Science 316 (5827): 1043-6, 2007.
- Rivera MN, Kim WJ, Wells J, et al.: An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 315 (5812): 642-5, 2007.
- Fukuzawa R, Anaka MR, Weeks RJ, et al.: Canonical WNT signalling determines lineage specificity in Wilms tumour. Oncogene 28 (8): 1063-75, 2009.
- Jenkins ZA, van Kogelenberg M, Morgan T, et al.: Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nat Genet 41 (1): 95-100, 2009.
- Grohmann A, Tanneberger K, Alzner A, et al.: AMER1 regulates the distribution of the tumor suppressor APC between microtubules and the plasma membrane. J Cell Sci 120 (Pt 21): 3738-47, 2007.
- Satoh Y, Nakadate H, Nakagawachi T, et al.: Genetic and epigenetic alterations on the short arm of chromosome 11 are involved in a majority of sporadic Wilms' tumours. Br J Cancer 95 (4): 541-7, 2006.
- Algar EM, St Heaps L, Darmanian A, et al.: Paternally inherited submicroscopic duplication at 11p15.5 implicates insulin-like growth factor II in overgrowth and Wilms' tumorigenesis. Cancer Res 67 (5): 2360-5, 2007.
- Lennerz JK, Timmerman RJ, Grange DK, et al.: Addition of H19 'loss of methylation testing' for Beckwith-Wiedemann syndrome (BWS) increases the diagnostic yield. J Mol Diagn 12 (5): 576-88, 2010.
- Mussa A, Molinatto C, Baldassarre G, et al.: Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J Pediatr 176: 142-149.e1, 2016.
- Bliek J, Gicquel C, Maas S, et al.: Epigenotyping as a tool for the prediction of tumor risk and tumor type in patients with Beckwith-Wiedemann syndrome (BWS). J Pediatr 145 (6): 796-9, 2004.
- Bjornsson HT, Brown LJ, Fallin MD, et al.: Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst 99 (16): 1270-3, 2007.
- Fukuzawa R, Breslow NE, Morison IM, et al.: Epigenetic differences between Wilms' tumours in white and east-Asian children. Lancet 363 (9407): 446-51, 2004.
- Gratias EJ, Dome JS, Jennings LJ, et al.: Association of Chromosome 1q Gain With Inferior Survival in Favorable-Histology Wilms Tumor: A Report From the Children's Oncology Group. J Clin Oncol 34 (26): 3189-94, 2016.
- Chagtai T, Zill C, Dainese L, et al.: Gain of 1q As a Prognostic Biomarker in Wilms Tumors (WTs) Treated With Preoperative Chemotherapy in the International Society of Paediatric Oncology (SIOP) WT 2001 Trial: A SIOP Renal Tumours Biology Consortium Study. J Clin Oncol 34 (26): 3195-203, 2016.
- Gadd S, Huff V, Skol AD, et al.: Genetic changes associated with relapse in favorable histology Wilms tumor: A Children's Oncology Group AREN03B2 study. Cell Rep Med 3 (6): 100644, 2022.
- Grundy PE, Breslow NE, Li S, et al.: Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol 23 (29): 7312-21, 2005.
- Messahel B, Williams R, Ridolfi A, et al.: Allele loss at 16q defines poorer prognosis Wilms tumour irrespective of treatment approach in the UKW1-3 clinical trials: a Children's Cancer and Leukaemia Group (CCLG) Study. Eur J Cancer 45 (5): 819-26, 2009.
- Spreafico F, Gamba B, Mariani L, et al.: Loss of heterozygosity analysis at different chromosome regions in Wilms tumor confirms 1p allelic loss as a marker of worse prognosis: a study from the Italian Association of Pediatric Hematology and Oncology. J Urol 189 (1): 260-6, 2013.
- Gratias EJ, Jennings LJ, Anderson JR, et al.: Gain of 1q is associated with inferior event-free and overall survival in patients with favorable histology Wilms tumor: a report from the Children's Oncology Group. Cancer 119 (21): 3887-94, 2013.
- Hohenstein P, Pritchard-Jones K, Charlton J: The yin and yang of kidney development and Wilms' tumors. Genes Dev 29 (5): 467-82, 2015.
- Foulkes WD, Priest JR, Duchaine TF: DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer 14 (10): 662-72, 2014.
- Wu MK, Sabbaghian N, Xu B, et al.: Biallelic DICER1 mutations occur in Wilms tumours. J Pathol 230 (2): 154-64, 2013.
- Palculict TB, Ruteshouser EC, Fan Y, et al.: Identification of germline DICER1 mutations and loss of heterozygosity in familial Wilms tumour. J Med Genet 53 (6): 385-8, 2016.
- Chang HM, Triboulet R, Thornton JE, et al.: A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature 497 (7448): 244-8, 2013.
- Alessandri JL, Cuillier F, Ramful D, et al.: Perlman syndrome: report, prenatal findings and review. Am J Med Genet A 146A (19): 2532-7, 2008.
- Bardeesy N, Falkoff D, Petruzzi MJ, et al.: Anaplastic Wilms' tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet 7 (1): 91-7, 1994.
- el Bahtimi R, Hazen-Martin DJ, Re GG, et al.: Immunophenotype, mRNA expression, and gene structure of p53 in Wilms' tumors. Mod Pathol 9 (3): 238-44, 1996.
- Wallkamm V, Dörlich R, Rahm K, et al.: Live imaging of Xwnt5A-ROR2 complexes. PLoS One 9 (10): e109428, 2014.
- Ooms AH, Gadd S, Gerhard DS, et al.: Significance of TP53 Mutation in Wilms Tumors with Diffuse Anaplasia: A Report from the Children's Oncology Group. Clin Cancer Res 22 (22): 5582-5591, 2016.
- Williams RD, Al-Saadi R, Chagtai T, et al.: Subtype-specific FBXW7 mutation and MYCN copy number gain in Wilms' tumor. Clin Cancer Res 16 (7): 2036-45, 2010.
- Mahamdallie S, Yost S, Poyastro-Pearson E, et al.: Identification of new Wilms tumour predisposition genes: an exome sequencing study. Lancet Child Adolesc Health 3 (5): 322-331, 2019.
- Armstrong AE, Gadd S, Huff V, et al.: A unique subset of low-risk Wilms tumors is characterized by loss of function of TRIM28 (KAP1), a gene critical in early renal development: A Children's Oncology Group study. PLoS One 13 (12): e0208936, 2018.
- Diets IJ, Hoyer J, Ekici AB, et al.: TRIM28 haploinsufficiency predisposes to Wilms tumor. Int J Cancer 145 (4): 941-951, 2019.
- Hol JA, Diets IJ, de Krijger RR, et al.: TRIM28 variants and Wilms' tumour predisposition. J Pathol 254 (4): 494-504, 2021.
- Garavelli L, Piemontese MR, Cavazza A, et al.: Multiple tumor types including leiomyoma and Wilms tumor in a patient with Gorlin syndrome due to 9q22.3 microdeletion encompassing the PTCH1 and FANC-C loci. Am J Med Genet A 161A (11): 2894-901, 2013.
- Cajaiba MM, Bale AE, Alvarez-Franco M, et al.: Rhabdomyosarcoma, Wilms tumor, and deletion of the patched gene in Gorlin syndrome. Nat Clin Pract Oncol 3 (10): 575-80, 2006.
- Williams RD, Chagtai T, Alcaide-German M, et al.: Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget 6 (9): 7232-43, 2015.
- Fievet A, Belaud-Rotureau MA, Dugay F, et al.: Involvement of germline DDX1-MYCN duplication in inherited nephroblastoma. Eur J Med Genet 56 (12): 643-7, 2013.
- Jiménez Martín O, Schlosser A, Furtwängler R, et al.: MYCN and MAX alterations in Wilms tumor and identification of novel N-MYC interaction partners as biomarker candidates. Cancer Cell Int 21 (1): 555, 2021.
- Martins AG, Pinto AT, Domingues R, et al.: Identification of a novel CTR9 germline mutation in a family with Wilms tumor. Eur J Med Genet 61 (5): 294-299, 2018.
- Parsons DW, Janeway KA, Patton DR, et al.: Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol 40 (20): 2224-2234, 2022.
- Argani P, Tickoo SK, Matoso A, et al.: Adult Wilms Tumor: Genetic Evidence of Origin of a Subset of Cases From Metanephric Adenoma. Am J Surg Pathol 46 (7): 988-999, 2022.
- Choueiri TK, Cheville J, Palescandolo E, et al.: BRAF mutations in metanephric adenoma of the kidney. Eur Urol 62 (5): 917-22, 2012.
- Wobker SE, Matoso A, Pratilas CA, et al.: Metanephric Adenoma-Epithelial Wilms Tumor Overlap Lesions: An Analysis of BRAF Status. Am J Surg Pathol 43 (9): 1157-1169, 2019.
- Charlton J, Irtan S, Bergeron C, et al.: Bilateral Wilms tumour: a review of clinical and molecular features. Expert Rev Mol Med 19: e8, 2017.
- Beckwith JB, Kiviat NB, Bonadio JF: Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms' tumor. Pediatr Pathol 10 (1-2): 1-36, 1990.
- Hu M, Fletcher J, McCahon E, et al.: Bilateral Wilms tumor and early presentation in pediatric patients is associated with the truncation of the Wilms tumor 1 protein. J Pediatr 163 (1): 224-9, 2013.
- Murphy AJ, Davidoff AM: Bilateral Wilms Tumor: A Surgical Perspective. Children (Basel) 5 (10): , 2018.
- Kalish JM, Doros L, Helman LJ, et al.: Surveillance Recommendations for Children with Overgrowth Syndromes and Predisposition to Wilms Tumors and Hepatoblastoma. Clin Cancer Res 23 (13): e115-e122, 2017.
- Mussa A, Duffy KA, Carli D, et al.: The effectiveness of Wilms tumor screening in Beckwith-Wiedemann spectrum. J Cancer Res Clin Oncol 145 (12): 3115-3123, 2019.
- Teplick A, Kowalski M, Biegel JA, et al.: Educational paper: screening in cancer predisposition syndromes: guidelines for the general pediatrician. Eur J Pediatr 170 (3): 285-94, 2011.
- Hol JA, Jewell R, Chowdhury T, et al.: Wilms tumour surveillance in at-risk children: Literature review and recommendations from the SIOP-Europe Host Genome Working Group and SIOP Renal Tumour Study Group. Eur J Cancer 153: 51-63, 2021.
- Ehrlich PF, Chi YY, Chintagumpala MM, et al.: Results of Treatment for Patients With Multicentric or Bilaterally Predisposed Unilateral Wilms Tumor (AREN0534): A report from the Children's Oncology Group. Cancer 126 (15): 3516-3525, 2020.
- Brioude F, Kalish JM, Mussa A, et al.: Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol 14 (4): 229-249, 2018.
- Lima Cunha D, Arno G, Corton M, et al.: The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye. Genes (Basel) 10 (12): , 2019.
- Hingorani M, Hanson I, van Heyningen V: Aniridia. Eur J Hum Genet 20 (10): 1011-7, 2012.
- van Heyningen V, Hoovers JM, de Kraker J, et al.: Raised risk of Wilms tumour in patients with aniridia and submicroscopic WT1 deletion. J Med Genet 44 (12): 787-90, 2007.
- Russell B, Tan W-H, Graham JM: Bohring-Opitz Syndrome. In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp.
Available online . Last accessed November 9, 2022. - Fernandes C, Paúl A, Venâncio MM, et al.: Simpson-Golabi-Behmel syndrome: One family, same mutation, different outcome. Am J Med Genet A 185 (8): 2502-2506, 2021.
- Greene AK, Kieran M, Burrows PE, et al.: Wilms tumor screening is unnecessary in Klippel-Trenaunay syndrome. Pediatrics 113 (4): e326-9, 2004.
- Schultz KAP, Williams GM, Kamihara J, et al.: DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin Cancer Res 24 (10): 2251-2261, 2018.
- Mitchell SG, Pencheva B, Porter CC: Germline Genetics and Childhood Cancer: Emerging Cancer Predisposition Syndromes and Psychosocial Impacts. Curr Oncol Rep 21 (10): 85, 2019.
- Schoettler PJ, Smith CC, Nishitani M, et al.: Anaplastic sarcoma of the kidney (DICER1-sarcoma of the kidney): A report from the International Pleuropulmonary Blastoma/DICER1 Registry. Pediatr Blood Cancer 71 (8): e31090, 2024.
- Maciaszek JL, Oak N, Nichols KE: Recent advances in Wilms' tumor predisposition. Hum Mol Genet 29 (R2): R138-R149, 2020.
- Cullinan N, Villani A, Mourad S, et al.: An eHealth decision-support tool to prioritize referral practices for genetic evaluation of patients with Wilms tumor. Int J Cancer 146 (4): 1010-1017, 2020.
- Green DM: Wilms' tumor. In: Greem DM: Diagnosis and Management of Malignant Solid Tumors in Infants and Children. Martinus Nijhoff Publishing, 1985, pp 129-86.
- Artunduaga M, Eklund M, van der Beek JN, et al.: Imaging of pediatric renal tumors: A COG Diagnostic Imaging Committee/SPR Oncology Committee White Paper focused on Wilms tumor and nephrogenic rests. Pediatr Blood Cancer 70 (Suppl 4): e30004, 2023.
- Khanna G, Naranjo A, Hoffer F, et al.: Detection of preoperative wilms tumor rupture with CT: a report from the Children's Oncology Group. Radiology 266 (2): 610-7, 2013.
- McDonald K, Duffy P, Chowdhury T, et al.: Added value of abdominal cross-sectional imaging (CT or MRI) in staging of Wilms' tumours. Clin Radiol 68 (1): 16-20, 2013.
- Ritchey ML, Shamberger RC, Hamilton T, et al.: Fate of bilateral renal lesions missed on preoperative imaging: a report from the National Wilms Tumor Study Group. J Urol 174 (4 Pt 2): 1519-21; discussion 1521, 2005.
- Khanna G, Rosen N, Anderson JR, et al.: Evaluation of diagnostic performance of CT for detection of tumor thrombus in children with Wilms tumor: a report from the Children's Oncology Group. Pediatr Blood Cancer 58 (4): 551-5, 2012.
- Sandberg JK, Chi YY, Smith EA, et al.: Imaging Characteristics of Nephrogenic Rests Versus Small Wilms Tumors: A Report From the Children's Oncology Group Study AREN03B2. AJR Am J Roentgenol 214 (5): 987-994, 2020.
- Watson T, Oostveen M, Rogers H, et al.: The role of imaging in the initial investigation of paediatric renal tumours. Lancet Child Adolesc Health 4 (3): 232-241, 2020.
- Servaes SE, Hoffer FA, Smith EA, et al.: Imaging of Wilms tumor: an update. Pediatr Radiol 49 (11): 1441-1452, 2019.
- Al-Hadidi A, Rinehardt HN, Sutthatarn P, et al.: Incidence and management of pleural effusions in patients with Wilms tumor: A Pediatric Surgical Oncology Research Collaborative study. Int J Cancer 151 (10): 1696-1702, 2022.
- Begent J, Sebire NJ, Levitt G, et al.: Pilot study of F(18)-Fluorodeoxyglucose Positron Emission Tomography/computerised tomography in Wilms' tumour: correlation with conventional imaging, pathology and immunohistochemistry. Eur J Cancer 47 (3): 389-96, 2011.
- Callaghan MU, Wong TE, Federici AB: Treatment of acquired von Willebrand syndrome in childhood. Blood 122 (12): 2019-22, 2013.
- Charlebois J, Rivard GÉ, St-Louis J: Management of acquired von Willebrand syndrome. Transfus Apher Sci 57 (6): 721-723, 2018.
- Shamberger RC, Guthrie KA, Ritchey ML, et al.: Surgery-related factors and local recurrence of Wilms tumor in National Wilms Tumor Study 4. Ann Surg 229 (2): 292-7, 1999.
- Hamilton TE, Green DM, Perlman EJ, et al.: Bilateral Wilms' tumor with anaplasia: lessons from the National Wilms' Tumor Study. J Pediatr Surg 41 (10): 1641-4, 2006.
- Servaes S, Khanna G, Naranjo A, et al.: Comparison of diagnostic performance of CT and MRI for abdominal staging of pediatric renal tumors: a report from the Children's Oncology Group. Pediatr Radiol 45 (2): 166-72, 2015.
- Ehrlich P, Chi YY, Chintagumpala MM, et al.: Results of the First Prospective Multi-institutional Treatment Study in Children With Bilateral Wilms Tumor (AREN0534): A Report From the Children's Oncology Group. Ann Surg 266 (3): 470-478, 2017.
- Othersen HB, DeLorimer A, Hrabovsky E, et al.: Surgical evaluation of lymph node metastases in Wilms' tumor. J Pediatr Surg 25 (3): 330-1, 1990.
- Shamberger RC, Ritchey ML, Haase GM, et al.: Intravascular extension of Wilms tumor. Ann Surg 234 (1): 116-21, 2001.
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Green DM, Breslow NE, Beckwith JB, et al.: Effect of duration of treatment on treatment outcome and cost of treatment for Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 16 (12): 3744-51, 1998.
- Kalapurakal JA, Dome JS, Perlman EJ, et al.: Management of Wilms' tumour: current practice and future goals. Lancet Oncol 5 (1): 37-46, 2004.
- Ehrlich PF: Wilms tumor: progress and considerations for the surgeon. Surg Oncol 16 (3): 157-71, 2007.
- Dome JS, Cotton CA, Perlman EJ, et al.: Treatment of anaplastic histology Wilms' tumor: results from the fifth National Wilms' Tumor Study. J Clin Oncol 24 (15): 2352-8, 2006.
- Shamberger RC, Anderson JR, Breslow NE, et al.: Long-term outcomes for infants with very low risk Wilms tumor treated with surgery alone in National Wilms Tumor Study-5. Ann Surg 251 (3): 555-8, 2010.
- Fernandez CV, Perlman EJ, Mullen EA, et al.: Clinical Outcome and Biological Predictors of Relapse After Nephrectomy Only for Very Low-risk Wilms Tumor: A Report From Children's Oncology Group AREN0532. Ann Surg 265 (4): 835-840, 2017.
- Hol JA, Lopez-Yurda MI, Van Tinteren H, et al.: Prognostic significance of age in 5631 patients with Wilms tumour prospectively registered in International Society of Paediatric Oncology (SIOP) 93-01 and 2001. PLoS One 14 (8): e0221373, 2019.
- Qian DC, Sykes-Martin KD, Tobillo R, et al.: Impact of Age on Overall Survival Among Children With Wilms Tumor: A Population-based Registry Analysis. Am J Clin Oncol 46 (5): 213-218, 2023.
- Chan GJ, Stohr BA, Osunkoya AO, et al.: Wilms Tumor: An Unexpected Diagnosis in Adult Patients. Arch Pathol Lab Med 148 (6): 722-727, 2024.
- Mitry E, Ciccolallo L, Coleman MP, et al.: Incidence of and survival from Wilms' tumour in adults in Europe: data from the EUROCARE study. Eur J Cancer 42 (14): 2363-8, 2006.
- Ali AN, Diaz R, Shu HK, et al.: A Surveillance, Epidemiology and End Results (SEER) program comparison of adult and pediatric Wilms' tumor. Cancer 118 (9): 2541-51, 2012.
- Walker JP, Saltzman AF, Kessler ER, et al.: Adult Wilms Tumor During Pregnancy: Case Report and Literature Review. Urology 129: 200-205, 2019.
- Brown JT, Harik LR, Barbee MS, et al.: Multidisciplinary Care of Adult Wilms' Tumor During Pregnancy: A Case Report and Review of the Literature. Clin Genitourin Cancer 18 (1): e1-e4, 2020.
- Chen I, Pasalic D, Fischer-Valuck B, et al.: Disparity in Outcomes for Adolescent and Young Adult Patients Diagnosed With Pediatric Solid Tumors Across 4 Decades. Am J Clin Oncol 41 (5): 471-475, 2018.
- Saltzman AF, Carrasco A, Amini A, et al.: Patterns of Care and Survival Comparison of Adult and Pediatric Wilms Tumor in the United States: A Study of the National Cancer Database. Urology 135: 50-56, 2020.
- Kalapurakal JA, Nan B, Norkool P, et al.: Treatment outcomes in adults with favorable histologic type Wilms tumor-an update from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys 60 (5): 1379-84, 2004.
- Arrigo S, Beckwith JB, Sharples K, et al.: Better survival after combined modality care for adults with Wilms' tumor. A report from the National Wilms' Tumor Study. Cancer 66 (5): 827-30, 1990.
- Byrd RL, Evans AE, D'Angio GJ: Adult Wilms tumor: effect of combined therapy on survival. J Urol 127 (4): 648-51, 1982.
- de Vries-Brilland M, Sionneau B, Dutriaux C, et al.: Successful Treatment of Metastatic Adult Wilms Tumor With Anti-BRAF Treatment: A Case Report and a Brief Review of the Literature. Clin Genitourin Cancer 17 (4): e721-e723, 2019.
- Segers H, van den Heuvel-Eibrink MM, Pritchard-Jones K, et al.: Management of adults with Wilms' tumor: recommendations based on international consensus. Expert Rev Anticancer Ther 11 (7): 1105-13, 2011.
- Perlman EJ: Pediatric renal tumors: practical updates for the pathologist. Pediatr Dev Pathol 8 (3): 320-38, 2005 May-Jun.
- Parsons LN, Mullen EA, Geller JI, et al.: Outcome analysis of stage I epithelial-predominant favorable-histology Wilms tumors: A report from Children's Oncology Group study AREN03B2. Cancer 126 (12): 2866-2871, 2020.
- Popov SD, Sebire NJ, Pritchard-Jones K, et al.: Renal tumors in children aged 10-16 Years: a report from the United Kingdom Children's Cancer and Leukaemia Group. Pediatr Dev Pathol 14 (3): 189-93, 2011 May-Jun.
- Indolfi P, Jenkner A, Terenziani M, et al.: Synchronous bilateral Wilms tumor: a report from the Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP). Cancer 119 (8): 1586-92, 2013.
- Hamilton TE, Ritchey ML, Haase GM, et al.: The management of synchronous bilateral Wilms tumor: a report from the National Wilms Tumor Study Group. Ann Surg 253 (5): 1004-10, 2011.
- Williams RD, Al-Saadi R, Natrajan R, et al.: Molecular profiling reveals frequent gain of MYCN and anaplasia-specific loss of 4q and 14q in Wilms tumor. Genes Chromosomes Cancer 50 (12): 982-95, 2011.
- Vujanić GM, Harms D, Sandstedt B, et al.: New definitions of focal and diffuse anaplasia in Wilms tumor: the International Society of Paediatric Oncology (SIOP) experience. Med Pediatr Oncol 32 (5): 317-23, 1999.
- Faria P, Beckwith JB, Mishra K, et al.: Focal versus diffuse anaplasia in Wilms tumor--new definitions with prognostic significance: a report from the National Wilms Tumor Study Group. Am J Surg Pathol 20 (8): 909-20, 1996.
- Beckwith JB: Precursor lesions of Wilms tumor: clinical and biological implications. Med Pediatr Oncol 21 (3): 158-68, 1993.
- Hennigar RA, O'Shea PA, Grattan-Smith JD: Clinicopathologic features of nephrogenic rests and nephroblastomatosis. Adv Anat Pathol 8 (5): 276-89, 2001.
- Fukuzawa R, Reeve AE: Molecular pathology and epidemiology of nephrogenic rests and Wilms tumors. J Pediatr Hematol Oncol 29 (9): 589-94, 2007.
- Vuononvirta R, Sebire NJ, Dallosso AR, et al.: Perilobar nephrogenic rests are nonobligate molecular genetic precursor lesions of insulin-like growth factor-II-associated Wilms tumors. Clin Cancer Res 14 (23): 7635-44, 2008.
- Perlman EJ, Faria P, Soares A, et al.: Hyperplastic perilobar nephroblastomatosis: long-term survival of 52 patients. Pediatr Blood Cancer 46 (2): 203-21, 2006.
- Lange J, Peterson SM, Takashima JR, et al.: Risk factors for end stage renal disease in non-WT1-syndromic Wilms tumor. J Urol 186 (2): 378-86, 2011.
- Coppes MJ, Arnold M, Beckwith JB, et al.: Factors affecting the risk of contralateral Wilms tumor development: a report from the National Wilms Tumor Study Group. Cancer 85 (7): 1616-25, 1999.
- Cooke A, Deshpande AV, La Hei ER, et al.: Ectopic nephrogenic rests in children: the clinicosurgical implications. J Pediatr Surg 44 (12): e13-6, 2009.
- Wilms' tumor: status report, 1990. By the National Wilms' Tumor Study Committee. J Clin Oncol 9 (5): 877-87, 1991.
- Green DM, Breslow NE, D'Angio GJ, et al.: Outcome of patients with Stage II/favorable histology Wilms tumor with and without local tumor spill: a report from the National Wilms Tumor Study Group. Pediatr Blood Cancer 61 (1): 134-9, 2014.
- Ehrlich PF, Anderson JR, Ritchey ML, et al.: Clinicopathologic findings predictive of relapse in children with stage III favorable-histology Wilms tumor. J Clin Oncol 31 (9): 1196-201, 2013.
- D'Angio GJ, Breslow N, Beckwith JB, et al.: Treatment of Wilms' tumor. Results of the Third National Wilms' Tumor Study. Cancer 64 (2): 349-60, 1989.
- Jereb B, Burgers JM, Tournade MF, et al.: Radiotherapy in the SIOP (International Society of Pediatric Oncology) nephroblastoma studies: a review. Med Pediatr Oncol 22 (4): 221-7, 1994.
- Green DM: The treatment of stages I-IV favorable histology Wilms' tumor. J Clin Oncol 22 (8): 1366-72, 2004.
- Graf N, Tournade MF, de Kraker J: The role of preoperative chemotherapy in the management of Wilms' tumor. The SIOP studies. International Society of Pediatric Oncology. Urol Clin North Am 27 (3): 443-54, 2000.
- Vujanić GM, D'Hooghe E, Popov SD, et al.: The effect of preoperative chemotherapy on histological subtyping and staging of Wilms tumors: The United Kingdom Children's Cancer Study Group (UKCCSG) Wilms tumor trial 3 (UKW3) experience. Pediatr Blood Cancer 66 (3): e27549, 2019.
- van den Heuvel-Eibrink MM, Hol JA, Pritchard-Jones K, et al.: Position paper: Rationale for the treatment of Wilms tumour in the UMBRELLA SIOP-RTSG 2016 protocol. Nat Rev Urol 14 (12): 743-752, 2017.
- Green DM, Breslow NE, Beckwith JB, et al.: Comparison between single-dose and divided-dose administration of dactinomycin and doxorubicin for patients with Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 16 (1): 237-45, 1998.
- D'Angio GJ, Evans AE, Breslow N, et al.: The treatment of Wilms' tumor: Results of the national Wilms' tumor study. Cancer 38 (2): 633-46, 1976.
- D'Angio GJ, Evans A, Breslow N, et al.: The treatment of Wilms' tumor: results of the Second National Wilms' Tumor Study. Cancer 47 (9): 2302-11, 1981.
- Kieran K, Anderson JR, Dome JS, et al.: Lymph node involvement in Wilms tumor: results from National Wilms Tumor Studies 4 and 5. J Pediatr Surg 47 (4): 700-6, 2012.
- Ritchey M, Daley S, Shamberger RC, et al.: Ureteral extension in Wilms' tumor: a report from the National Wilms' Tumor Study Group (NWTSG). J Pediatr Surg 43 (9): 1625-9, 2008.
- Gow KW, Barnhart DC, Hamilton TE, et al.: Primary nephrectomy and intraoperative tumor spill: report from the Children's Oncology Group (COG) renal tumors committee. J Pediatr Surg 48 (1): 34-8, 2013.
- McNeil DE, Langer JC, Choyke P, et al.: Feasibility of partial nephrectomy for Wilms' tumor in children with Beckwith-Wiedemann syndrome who have been screened with abdominal ultrasonography. J Pediatr Surg 37 (1): 57-60, 2002.
- Scalabre A, Bergeron C, Brioude F, et al.: Is Nephron Sparing Surgery Justified in Wilms Tumor With Beckwith-Wiedemann Syndrome or Isolated Hemihypertrophy? Pediatr Blood Cancer 63 (9): 1571-7, 2016.
- Auber F, Jeanpierre C, Denamur E, et al.: Management of Wilms tumors in Drash and Frasier syndromes. Pediatr Blood Cancer 52 (1): 55-9, 2009.
- Neville H, Ritchey ML, Shamberger RC, et al.: The occurrence of Wilms tumor in horseshoe kidneys: a report from the National Wilms Tumor Study Group (NWTSG). J Pediatr Surg 37 (8): 1134-7, 2002.
- Ferrer FA, Rosen N, Herbst K, et al.: Image based feasibility of renal sparing surgery for very low risk unilateral Wilms tumors: a report from the Children's Oncology Group. J Urol 190 (5): 1846-51, 2013.
- Ritchey ML: Renal sparing surgery for Wilms tumor. J Urol 174 (4 Pt 1): 1172-3, 2005.
- Cozzi DA, Zani A: Nephron-sparing surgery in children with primary renal tumor: indications and results. Semin Pediatr Surg 15 (1): 3-9, 2006.
- Ritchey ML, Kelalis PP, Breslow N, et al.: Surgical complications after nephrectomy for Wilms' tumor. Surg Gynecol Obstet 175 (6): 507-14, 1992.
- Ehrlich PF, Ferrer FA, Ritchey ML, et al.: Hepatic metastasis at diagnosis in patients with Wilms tumor is not an independent adverse prognostic factor for stage IV Wilms tumor: a report from the Children's Oncology Group/National Wilms Tumor Study Group. Ann Surg 250 (4): 642-8, 2009.
- Zhuge Y, Cheung MC, Yang R, et al.: Improved survival with lymph node sampling in Wilms tumor. J Surg Res 167 (2): e199-203, 2011.
- Ehrlich PF, Ritchey ML, Hamilton TE, et al.: Quality assessment for Wilms' tumor: a report from the National Wilms' Tumor Study-5. J Pediatr Surg 40 (1): 208-12; discussion 212-3, 2005.
- Fernandez CV, Mullen EA, Chi YY, et al.: Outcome and Prognostic Factors in Stage III Favorable-Histology Wilms Tumor: A Report From the Children's Oncology Group Study AREN0532. J Clin Oncol 36 (3): 254-261, 2018.
- Ritchey ML: Primary nephrectomy for Wilms' tumor: approach of the National Wilms' Tumor Study Group. Urology 47 (6): 787-91, 1996.
- Ritchey ML, Pringle KC, Breslow NE, et al.: Management and outcome of inoperable Wilms tumor. A report of National Wilms Tumor Study-3. Ann Surg 220 (5): 683-90, 1994.
- Ritchey ML, Shamberger RC, Haase G, et al.: Surgical complications after primary nephrectomy for Wilms' tumor: report from the National Wilms' Tumor Study Group. J Am Coll Surg 192 (1): 63-8; quiz 146, 2001.
- Naik-Mathuria B, Utria AF, Ehrlich PF, et al.: Management and Outcomes of Wilms Tumor With Suprarenal Intravascular Extension: A Pediatric Surgical Oncology Research Collaborative Study. Ann Surg 279 (3): 528-535, 2024.
- Tournade MF, Com-Nougué C, Voûte PA, et al.: Results of the Sixth International Society of Pediatric Oncology Wilms' Tumor Trial and Study: a risk-adapted therapeutic approach in Wilms' tumor. J Clin Oncol 11 (6): 1014-23, 1993.
- Oberholzer HF, Falkson G, De Jager LC: Successful management of inferior vena cava and right atrial nephroblastoma tumor thrombus with preoperative chemotherapy. Med Pediatr Oncol 20 (1): 61-3, 1992.
- Saarinen UM, Wikström S, Koskimies O, et al.: Percutaneous needle biopsy preceding preoperative chemotherapy in the management of massive renal tumors in children. J Clin Oncol 9 (3): 406-15, 1991.
- Dykes EH, Marwaha RK, Dicks-Mireaux C, et al.: Risks and benefits of percutaneous biopsy and primary chemotherapy in advanced Wilms' tumour. J Pediatr Surg 26 (5): 610-2, 1991.
- Thompson WR, Newman K, Seibel N, et al.: A strategy for resection of Wilms' tumor with vena cava or atrial extension. J Pediatr Surg 27 (7): 912-5, 1992.
- Szavay P, Luithle T, Semler O, et al.: Surgery of cavoatrial tumor thrombus in nephroblastoma: a report of the SIOP/GPOH study. Pediatr Blood Cancer 43 (1): 40-5, 2004.
- Powis M, Messahel B, Hobson R, et al.: Surgical complications after immediate nephrectomy versus preoperative chemotherapy in non-metastatic Wilms' tumour: findings from the 1991-2001 United Kingdom Children's Cancer Study Group UKW3 Trial. J Pediatr Surg 48 (11): 2181-6, 2013.
- Boam TD, Gabriel M, Shukla R, et al.: Impact of neoadjuvant chemotherapy on thrombus viability in patients with Wilms tumour and caval extension: systematic review with meta-analysis. BJS Open 5 (3): , 2021.
- Rutigliano DN, Kayton ML, Steinherz P, et al.: The use of preoperative chemotherapy in Wilms' tumor with contained retroperitoneal rupture. J Pediatr Surg 42 (9): 1595-9, 2007.
- Brisse HJ, Schleiermacher G, Sarnacki S, et al.: Preoperative Wilms tumor rupture: a retrospective study of 57 patients. Cancer 113 (1): 202-13, 2008.
- Corn BW, Goldwein JW, Evans I, et al.: Outcomes in low-risk babies treated with half-dose chemotherapy according to the Third National Wilms' Tumor Study. J Clin Oncol 10 (8): 1305-9, 1992.
- Morgan E, Baum E, Breslow N, et al.: Chemotherapy-related toxicity in infants treated according to the Second National Wilms' Tumor Study. J Clin Oncol 6 (1): 51-5, 1988.
- Green DM, Norkool P, Breslow NE, et al.: Severe hepatic toxicity after treatment with vincristine and dactinomycin using single-dose or divided-dose schedules: a report from the National Wilms' Tumor Study. J Clin Oncol 8 (9): 1525-30, 1990.
- Raine J, Bowman A, Wallendszus K, et al.: Hepatopathy-thrombocytopenia syndrome--a complication of dactinomycin therapy for Wilms' tumor: a report from the United Kingdom Childrens Cancer Study Group. J Clin Oncol 9 (2): 268-73, 1991.
- Oosterom N, Gooskens SLM, Renfro LA, et al.: Severe Hepatopathy in National Wilms Tumor Studies 3-5: Prevalence, Clinical Features, and Outcomes After Reintroduction of Chemotherapy. J Clin Oncol 41 (26): 4247-4256, 2023.
- Feusner JH, Ritchey ML, Norkool PA, et al.: Renal failure does not preclude cure in children receiving chemotherapy for Wilms tumor: a report from the National Wilms Tumor Study Group. Pediatr Blood Cancer 50 (2): 242-5, 2008.
- Veal GJ, English MW, Grundy RG, et al.: Pharmacokinetically guided dosing of carboplatin in paediatric cancer patients with bilateral nephrectomy. Cancer Chemother Pharmacol 54 (4): 295-300, 2004.
- Dix DB, Fernandez CV, Chi YY, et al.: Augmentation of Therapy for Combined Loss of Heterozygosity 1p and 16q in Favorable Histology Wilms Tumor: A Children's Oncology Group AREN0532 and AREN0533 Study Report. J Clin Oncol 37 (30): 2769-2777, 2019.
- Stokes CL, Stokes WA, Kalapurakal JA, et al.: Timing of Radiation Therapy in Pediatric Wilms Tumor: A Report From the National Cancer Database. Int J Radiat Oncol Biol Phys 101 (2): 453-461, 2018.
- Dix DB, Seibel NL, Chi YY, et al.: Treatment of Stage IV Favorable Histology Wilms Tumor With Lung Metastases: A Report From the Children's Oncology Group AREN0533 Study. J Clin Oncol 36 (16): 1564-1570, 2018.
- Daw NC, Chi YY, Kalapurakal JA, et al.: Activity of Vincristine and Irinotecan in Diffuse Anaplastic Wilms Tumor and Therapy Outcomes of Stage II to IV Disease: Results of the Children's Oncology Group AREN0321 Study. J Clin Oncol 38 (14): 1558-1568, 2020.
- Thomas PR, Tefft M, Compaan PJ, et al.: Results of two radiation therapy randomizations in the third National Wilms' Tumor Study. Cancer 68 (8): 1703-7, 1991.
- Tefft M, D'Angio GJ, Beckwith B, et al.: Patterns of intra-abdominal relapse (IAR) in patients with Wilms' tumor who received radiation: analysis by histopathology. A report of National Wilms' Tumor Studies 1 and 2 (NWTS-1 & 2). Int J Radiat Oncol Biol Phys 6 (6): 663-7, 1980.
- Thomas PR, Tefft M, Farewell VT, et al.: Abdominal relapses in irradiated second National Wilms' Tumor Study patients. J Clin Oncol 2 (10): 1098-101, 1984.
- Meisel JA, Guthrie KA, Breslow NE, et al.: Significance and management of computed tomography detected pulmonary nodules: a report from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys 44 (3): 579-85, 1999.
- Dávila Fajardo R, Furtwängler R, van Grotel M, et al.: Outcome of Stage IV Completely Necrotic Wilms Tumour and Local Stage III Treated According to the SIOP 2001 Protocol. Cancers (Basel) 13 (5): , 2021.
- Vujanić GM, Graf N, D'Hooghe E, et al.: Omission of adjuvant chemotherapy in patients with completely necrotic Wilms tumor stage I and radiotherapy in stage III: The 30-year SIOP-RTSG experience. Pediatr Blood Cancer 71 (3): e30852, 2024.
- Daw NC, Chi YY, Kim Y, et al.: Treatment of stage I anaplastic Wilms' tumour: a report from the Children's Oncology Group AREN0321 study. Eur J Cancer 118: 58-66, 2019.
- Green DM, Breslow NE, Beckwith JB, et al.: Treatment with nephrectomy only for small, stage I/favorable histology Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 19 (17): 3719-24, 2001.
- Armstrong AE, Daw NC, Renfro LA, et al.: Treatment of focal anaplastic Wilms tumor: A report from the Children's Oncology Group AREN0321 and AREN03B2 studies. Cancer 131 (2): e35713, 2025.
- Kalapurakal JA, Li SM, Breslow NE, et al.: Intraoperative spillage of favorable histology wilms tumor cells: influence of irradiation and chemotherapy regimens on abdominal recurrence. A report from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys 76 (1): 201-6, 2010.
- Evageliou N, Renfro LA, Geller J, et al.: Prognostic impact of lymph node involvement and loss of heterozygosity of 1p or 16q in stage III favorable histology Wilms tumor: A report from Children's Oncology Group Studies AREN03B2 and AREN0532. Cancer 130 (5): 792-802, 2024.
- Benedetti DJ, Varela CR, Renfro LA, et al.: Treatment of children with favorable histology Wilms tumor with extrapulmonary metastases: A report from the COG studies AREN0533 and AREN03B2 and NWTSG study NWTS-5. Cancer 130 (6): 947-961, 2024.
- Grundy PE, Green DM, Dirks AC, et al.: Clinical significance of pulmonary nodules detected by CT and Not CXR in patients treated for favorable histology Wilms tumor on national Wilms tumor studies-4 and -5: a report from the Children's Oncology Group. Pediatr Blood Cancer 59 (4): 631-5, 2012.
- Verschuur A, Van Tinteren H, Graf N, et al.: Treatment of pulmonary metastases in children with stage IV nephroblastoma with risk-based use of pulmonary radiotherapy. J Clin Oncol 30 (28): 3533-9, 2012.
- Varan A, Büyükpamukçu N, Cağlar M, et al.: Prognostic significance of metastatic site at diagnosis in Wilms' tumor: results from a single center. J Pediatr Hematol Oncol 27 (4): 188-91, 2005.
- Szavay P, Luithle T, Graf N, et al.: Primary hepatic metastases in nephroblastoma--a report of the SIOP/GPOH Study. J Pediatr Surg 41 (1): 168-72; discussion 168-72, 2006.
- Fuchs J, Szavay P, Luithle T, et al.: Surgical implications for liver metastases in nephroblastoma--data from the SIOP/GPOH study. Surg Oncol 17 (1): 33-40, 2008.
- Aronson DC, Maharaj A, Sheik-Gafoor MH, et al.: The results of treatment of children with metastatic Wilms tumours (WT) in an African setting: do liver metastases have a negative impact on survival? Pediatr Blood Cancer 59 (2): 391-4, 2012.
- Liné A, Sudour-Bonnange H, Languillat-Fouquet V, et al.: Liver metastasis at diagnosis in children with nephroblastoma enrolled in SIOP2001 protocol: A French multicentric study. Pediatr Blood Cancer 67 (6): e28201, 2020.
- Kalapurakal JA, Pokhrel D, Gopalakrishnan M, et al.: Advantages of whole-liver intensity modulated radiation therapy in children with Wilms tumor and liver metastasis. Int J Radiat Oncol Biol Phys 85 (3): 754-60, 2013.
- Breslow NE, Collins AJ, Ritchey ML, et al.: End stage renal disease in patients with Wilms tumor: results from the National Wilms Tumor Study Group and the United States Renal Data System. J Urol 174 (5): 1972-5, 2005.
- Sudour-Bonnange H, van Tinteren H, Ramírez-Villar GL, et al.: Characteristics and outcome of synchronous bilateral Wilms tumour in the SIOP WT 2001 Study: Report from the SIOP Renal Tumour Study Group (SIOP-RTSG). Br J Cancer 131 (6): 972-981, 2024.
- Zuppan CW, Beckwith JB, Weeks DA, et al.: The effect of preoperative therapy on the histologic features of Wilms' tumor. An analysis of cases from the Third National Wilms' Tumor Study. Cancer 68 (2): 385-94, 1991.
- Ehrlich PF: Bilateral Wilms' tumor: the need to improve outcomes. Expert Rev Anticancer Ther 9 (7): 963-73, 2009.
- Chintagumpala MM, Perlman EJ, Tornwall B, et al.: Outcomes based on histopathologic response to preoperative chemotherapy in children with bilateral Wilms tumor: A prospective study (COG AREN0534). Cancer 128 (13): 2493-2503, 2022.
- Murphy AJ, Brzezinski J, Renfro LA, et al.: Long-term outcomes and patterns of relapse in patients with bilateral Wilms tumor or bilaterally predisposed unilateral Wilms tumor, a report from the COG AREN0534 study. Int J Cancer 155 (10): 1824-1831, 2024.
- Romao RLP, Aldrink JH, Renfro LA, et al.: Bilateral Wilms tumor with anaplasia: A report from the Children's Oncology Group Study AREN0534. Pediatr Blood Cancer 71 (7): e30981, 2024.
- Sudour H, Audry G, Schleimacher G, et al.: Bilateral Wilms tumors (WT) treated with the SIOP 93 protocol in France: epidemiological survey and patient outcome. Pediatr Blood Cancer 59 (1): 57-61, 2012.
- Davidoff AM, Interiano RB, Wynn L, et al.: Overall Survival and Renal Function of Patients With Synchronous Bilateral Wilms Tumor Undergoing Surgery at a Single Institution. Ann Surg 262 (4): 570-6, 2015.
- Kieran K, Williams MA, McGregor LM, et al.: Repeat nephron-sparing surgery for children with bilateral Wilms tumor. J Pediatr Surg 49 (1): 149-53, 2014.
- Kist-van Holthe JE, Ho PL, Stablein D, et al.: Outcome of renal transplantation for Wilms' tumor and Denys-Drash syndrome: a report of the North American Pediatric Renal Transplant Cooperative Study. Pediatr Transplant 9 (3): 305-10, 2005.
- Venkatramani R, Chi YY, Coppes MJ, et al.: Outcome of patients with intracranial relapse enrolled on national Wilms Tumor Study Group clinical trials. Pediatr Blood Cancer 64 (7): , 2017.
- Iaboni DSM, Chi YY, Kim Y, et al.: Outcome of Wilms tumor patients with bone metastasis enrolled on National Wilms Tumor Studies 1-5: A report from the Children's Oncology Group. Pediatr Blood Cancer 66 (1): e27430, 2019.
- Malogolowkin M, Cotton CA, Green DM, et al.: Treatment of Wilms tumor relapsing after initial treatment with vincristine, actinomycin D, and doxorubicin. A report from the National Wilms Tumor Study Group. Pediatr Blood Cancer 50 (2): 236-41, 2008.
- Reinhard H, Schmidt A, Furtwängler R, et al.: Outcome of relapses of nephroblastoma in patients registered in the SIOP/GPOH trials and studies. Oncol Rep 20 (2): 463-7, 2008.
- Schneller N, Daw N, Throckmorton W, et al.: Outcomes of relapsed favorable-histology Wilms tumor in non-clinical trial setting. Pediatr Blood Cancer 72 (1): e31347, 2025.
- Malogolowkin M, Spreafico F, Dome JS, et al.: Incidence and outcomes of patients with late recurrence of Wilms' tumor. Pediatr Blood Cancer 60 (10): 1612-5, 2013.
- Radhakrishnan V, Mishra S, Raja A, et al.: Relapse of Wilms tumor after 20 years: a rare presentation and review of literature. Pediatric Hematology Oncology Journal 1 (4): 86-8, 2016.
Also available online . Last accessed December 22, 2022. - Grundy P, Breslow N, Green DM, et al.: Prognostic factors for children with recurrent Wilms' tumor: results from the Second and Third National Wilms' Tumor Study. J Clin Oncol 7 (5): 638-47, 1989.
- Green DM, Cotton CA, Malogolowkin M, et al.: Treatment of Wilms tumor relapsing after initial treatment with vincristine and actinomycin D: a report from the National Wilms Tumor Study Group. Pediatr Blood Cancer 48 (5): 493-9, 2007.
- Warmann SW, Furtwängler R, Blumenstock G, et al.: Tumor biology influences the prognosis of nephroblastoma patients with primary pulmonary metastases: results from SIOP 93-01/GPOH and SIOP 2001/GPOH. Ann Surg 254 (1): 155-62, 2011.
- Groenendijk A, van Tinteren H, Jiang Y, et al.: Outcome of SIOP patients with low- or intermediate-risk Wilms tumour relapsing after initial vincristine and actinomycin-D therapy only - the SIOP 93-01 and 2001 protocols. Eur J Cancer 163: 88-97, 2022.
- Abu-Ghosh AM, Krailo MD, Goldman SC, et al.: Ifosfamide, carboplatin and etoposide in children with poor-risk relapsed Wilms' tumor: a Children's Cancer Group report. Ann Oncol 13 (3): 460-9, 2002.
- Garaventa A, Hartmann O, Bernard JL, et al.: Autologous bone marrow transplantation for pediatric Wilms' tumor: the experience of the European Bone Marrow Transplantation Solid Tumor Registry. Med Pediatr Oncol 22 (1): 11-4, 1994.
- Pein F, Michon J, Valteau-Couanet D, et al.: High-dose melphalan, etoposide, and carboplatin followed by autologous stem-cell rescue in pediatric high-risk recurrent Wilms' tumor: a French Society of Pediatric Oncology study. J Clin Oncol 16 (10): 3295-301, 1998.
- Rossoff J, Tse WT, Duerst RE, et al.: High-dose chemotherapy and autologous hematopoietic stem-cell rescue for treatment of relapsed and refractory Wilms tumor: Re-evaluating outcomes. Pediatr Hematol Oncol 35 (5-6): 316-321, 2018 Aug - Sep.
- Delafoy M, Verschuur A, Scheleirmacher G, et al.: High-dose chemotherapy followed by autologous stem cell rescue in Wilms tumors: French report on toxicity and efficacy. Pediatr Blood Cancer 69 (3): e29431, 2022.
- Spreafico F, Dalissier A, Pötschger U, et al.: High dose chemotherapy and autologous hematopoietic cell transplantation for Wilms tumor: a study of the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant 55 (2): 376-383, 2020.
- Malogolowkin MH, Hemmer MT, Le-Rademacher J, et al.: Outcomes following autologous hematopoietic stem cell transplant for patients with relapsed Wilms' tumor: a CIBMTR retrospective analysis. Bone Marrow Transplant 52 (11): 1549-1555, 2017.
- Weil BR, Murphy AJ, Liu Q, et al.: Late Health Outcomes Among Survivors of Wilms Tumor Diagnosed Over Three Decades: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 41 (14): 2638-2650, 2023.
- Wong KF, Reulen RC, Winter DL, et al.: Risk of Adverse Health and Social Outcomes Up to 50 Years After Wilms Tumor: The British Childhood Cancer Survivor Study. J Clin Oncol 34 (15): 1772-9, 2016.
- Foster KL, Salehabadi SM, Green DM, et al.: Clinical Assessment of Late Health Outcomes in Survivors of Wilms Tumor. Pediatrics 150 (5): , 2022.
- Lange JM, Takashima JR, Peterson SM, et al.: Breast cancer in female survivors of Wilms tumor: a report from the national Wilms tumor late effects study. Cancer 120 (23): 3722-30, 2014.
- Green DM, Grigoriev YA, Nan B, et al.: Congestive heart failure after treatment for Wilms' tumor: a report from the National Wilms' Tumor Study group. J Clin Oncol 19 (7): 1926-34, 2001.
- Practice Committee of the American Society for Reproductive Medicine. Electronic address: asrm@asrm.org: Fertility preservation in patients undergoing gonadotoxic therapy or gonadectomy: a committee opinion. Fertil Steril 112 (6): 1022-1033, 2019.
- Green DM, Liu W, Kutteh WH, et al.: Cumulative alkylating agent exposure and semen parameters in adult survivors of childhood cancer: a report from the St Jude Lifetime Cohort Study. Lancet Oncol 15 (11): 1215-23, 2014.
- Green DM, Lange JM, Peabody EM, et al.: Pregnancy outcome after treatment for Wilms tumor: a report from the national Wilms tumor long-term follow-up study. J Clin Oncol 28 (17): 2824-30, 2010.
- Breslow NE, Takashima JR, Ritchey ML, et al.: Renal failure in the Denys-Drash and Wilms' tumor-aniridia syndromes. Cancer Res 60 (15): 4030-2, 2000.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
Page Footer
I want to...
Audiences
Secure Member Sites
The Cigna Group Information
Disclaimer
Individual and family medical and dental insurance plans are insured by Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc., and Cigna HealthCare of Texas, Inc. Group health insurance and health benefit plans are insured or administered by CHLIC, Connecticut General Life Insurance Company (CGLIC), or their affiliates (see
All insurance policies and group benefit plans contain exclusions and limitations. For availability, costs and complete details of coverage, contact a licensed agent or Cigna sales representative. This website is not intended for residents of New Mexico.